

Summary
Cells that suffer substantial inhibition of DNA replication halt their cell cycle via a checkpoint response mediated by the PI3 kinases ATM and ATR. It is unclear how cells cope with milder replication insults, which are under the threshold for ATM and ATR activation. A third PI3 kinase, DNA-dependent protein kinase (DNA-PK), is also activated following replication inhibition, but the role DNA-PK might play in response to perturbed replication is unclear since this kinase does not activate the signaling cascades involved in the S-phase checkpoint. Here we report that mild, transient drug-induced perturbation of DNA replication rapidly induced DNA breaks that promptly disappeared in cells that contained a functional DNA-PK whereas such breaks persisted in cells that were deficient in DNA-PK activity. After the initial transient burst of DNA breaks, cells with a functional DNA-PK did not halt replication and continued to synthesize DNA at a slow pace in the presence of replication inhibitors. In contrast, DNA-PK deficient cells subject to low levels of replication inhibition halted cell cycle progression via an ATR-mediated S-phase checkpoint. The ATM kinase was dispensable for the induction of the initial DNA breaks. These observations suggest that DNA-PK is involved in setting a high threshold for the ATR-Chk1-mediated S-phase checkpoint by promptly repairing DNA breaks that appear immediately following inhibition of DNA replication.
Keywords: DNA-PK, replication arrest, non-homologous end joining, aphidicolin, DNA damage S-phase checkpoint
Introduction
Cells are constantly exposed to environmental and metabolic insults such as radiation, chemical agents and perturbation of DNA replication. Such exposure may generate DNA lesions that lead to mutations and DNA breaks and cause genomic instability. Potentially genotoxic lesions are recognized by damage-sensor kinases that are members of the phosphatidylinositol 3-kinase family: ataxia telangiectasia mutated (ATM), ATM- and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK) 1; 2. Replication-mediated DNA breaks are predominantly recognized by the ATM and ATR kinases, which induce a DNA damage S-phase checkpoint 3; 4; 5. The third kinase, DNA-PK, is primarily involved in the response to double strand DNA breaks (DSBs) induced by replication independent lesions (for a recent review, see 6). In contrast to ATM and ATR, DNA-PK is not directly involved in the activation of the S-phase checkpoint. However, cells deficient in the catalytic subunit of DNA-PK are hypersensitive to replication inhibition by hydroxyurea (HU) 7, suggesting that DNA-PK plays a role in the response to replication perturbation. The role of DNA-PK in the response to DSBs at replication forks has yet to be elucidated.
DNA-PK consists of a catalytic subunit (DNA-PKcs) and of the Ku heterodimer (Ku70/Ku80) regulatory subunit 8. The DNA-PK complex plays a major role in activating nonhomologous end-joining (NHEJ) repair in mammalian cells 8; 9; 10 and is involved in induction of programmed cell death, telomere maintenance, and innate immunity 6; 9. The Ku subunit first binds to DNA ends and then recruits DNA-PKcs 11, which can tether broken DNA ends together. The assembled DNA-PK can phosphorylate the histone H2AX in the absence of ATM, forming foci of phosphorylated H2AX (γ-H2AX) in a manner akin to that described for ATM and ATR 12; 13 (for a review see 14). The assembly of Ku and DNA-PKcs at the sites of DSBs is followed by recruitment of the DNA ligase IV-XRCC4 complex and ligation of the two DNA ends.
Mammalian cells have two distinct DNA DSB repair pathways: homologous recombination (HR) and NHEJ. HR requires sequence homology at the sites of DNA breaks and functions at late S-phase and G2 phase when sister chromatids are present. In contrast, NHEJ plays a role at all phases of the cell cycle. HR is the predominant pathway that repairs replication-mediated DSBs 7; 15 and plays an important role in the repair of stalled replication forks 16; 17. However, in both human fibroblasts and Chinese hamster ovary cells, the NHEJ pathway recognized DSBs earlier than the HR pathway 18; 19. Interestingly, HR- or NHEJ (DNA-PKcs)-deficient Chinese hamster ovary cells are sensitive to HU but only HR-deficient cells are sensitive to thymidine 7. These observations suggest that the roles of HR and NHEJ in the recognition and repair of lesions caused by replication perturbations may differ depending on the replication stress.
To study the role of DNA-PK in the response to replication arrest, we used the DNA replication inhibitor aphidicolin (APH). APH, a mycotoxin isolated from Cephalosporium aphidicola, inhibits DNA replication by interacting with the replicating DNA polymerase α(pol α). APH specifically inhibits the activity of replicating DNA polymerases in eukaryotic cells while not affecting other metabolic pathways, such as RNA, protein, and nucleotide biosynthesis 20; 21; 22. APH forms a pol α-DNA-APH ternary complex 23 that does not inhibit the primase activity of the pol α-primase complex but inhibits the elongation step of DNA pol α, δ, and ε24; 25. APH preferentially blocks dCTP incorporation 22; 26; 27. APH inhibits S-phase progression but allows cells in G2, M, and G1 to continue their growth cycle. High levels of APH completely inhibit DNA replication and induce a DNA damage S-phase checkpoint that requires the activation of Chk1 28. However, lower levels of APH decrease the rate of fork progression without activating checkpoints.
We investigated the role of DNA-PK in response to replication inhibition by APH. Here we report that all cells, regardless of DNA-PK status, induced a surge of DNA breaks after a short exposure to APH. When APH levels were low, cells that contained DNA-PK rapidly repaired DNA breaks generated by APH and did not activate an S-phase checkpoint. In the absence of DNA-PKcs, DNA breaks were not repaired and the cells activated a Chk1-mediated DNA damage S-phase checkpoint that required ATR. In these cells, checkpoint activation led to a complete halting of replication fork progression and to the activation of the HR pathway.
Results
Hypersensitivity to low doses of APH in cells deficient in DNA-PKcs
To determine the role of DNA-PK in the response to replication perturbation, we examined the sensitivity of cells with an active DNA-PK and cells deficient in DNA-PKcs- to APH with respect to inhibition of DNA synthesis. For the initial experiments we used a pair of glioma cell lines, M059K and M059J. Both cell lines were derived from the same tumor but M059K has an active DNA-PK whereas M059J has an inactive DNA-PK 29. We determined the rate of DNA replication in the presence or absence of APH by measuring the incorporation of the nucleotide analog bromodeoxyuridine (BrdU) into DNA. Fluorescence-activated cell sorting (FACS) analysis revealed that BrdU incorporation was reduced in a dose-dependent manner in both cell lines. Notably, low APH doses (below 1 μg/ml) sharply suppressed DNA synthesis in cells deficient in DNA-PKcs whereas the suppression was milder in cells with an active DNA-PK. DNA synthesis was completely suppressed in both cell lines at the highest dose of APH (10 μg/ml; Figure 1).
Figure 1.
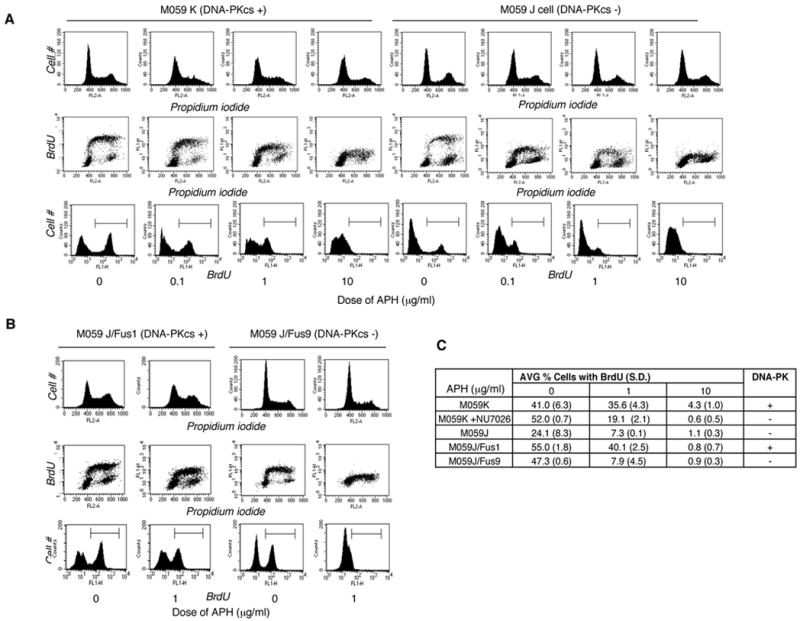
DNA synthesis after treatment with APH. (A) Cell cycle distribution after exposure of M059K and M059J cells to APH for 30 minutes. (B) Cell cycle distribution after exposure of M059J/Fus1 and M059J/Fus9 cells to APH for 30 minutes. The cell cycle distribution was determined by FACS. DNA content was determined by propidium iodide counterstaining and BrdU incorporation was detected with an anti-BrdU antibody (see Materials and Methods). (C) Average percentage of BrdU-positive cells in APH-treated M059K cells, M059K cells treated with 20 μM of the DNA-PK inhibitor Nu7026 30 minutes prior to exposure to APH, M059J cells, M059J/Fus1 and M059J/Fus9 cells. M059K cells have an active DNA-PK), M059J cells are derived from the same tumor as M059K but are deficient in DNA-PKcs, M059J/Fus1 are complemented with a copy of an active DNA-PKcs and M059J/Fus9 cells are M059J cells transfected with an empty vector and therefore, DNA-PKcs deficient. Experiments were repeated at least 3 times with independent samples. Standard deviations are shown in parentheses.
Although the M059K and M059J cells were originally derived from the same tumor, they harbor other differences unrelated to the DNA-PK deficiency. (For example, as shown in Figure 1A and 1C, the frequency of M059K cells in S-phase was consistently higher than the frequency of M059J cells in S-phase. M059J cells exhibit reduced ATM signaling, which is cross-regulated with DNAPK 30. For an example of other documented differences, see 31. ) We applied two tests to examine whether the hypersensitivity to low doses of APH was due to the DNA-PKcs deficiency and not to other differences between the two cell lines. First, we inhibited DNA-PK in M059K cells using NU7026 32. When cells with an active DNA-PK were treated with NU7026 before the addition of APH (Figure 1C), DNA synthesis was strongly suppressed, similar to the suppression observed in DNA-PKcs-deficient cells. Second, we tested whether sensitivity to low doses of APH reflected the different status of DNA-PKcs by comparing M059J/Fus1 cells (complemented with a fragment of human chromosome 8 containing the gene for DNA-PK) with M059J/Fus9 cells (M059J cells that were transfected with an empty vector and therefore remained DNA-PKcs deficient). Importantly, the frequency of cells in S-phase was similar in both cell lines (Figure 1B, 1C). As shown in Figure 1B and 1C, the DNA-PK complemented M059J/Fus1 cells were less sensitive to low doses of APH than the DNA-PK deficient M059J/Fus9. These results demonstrated that sensitivity to low doses of APH significantly increased in the absence of DNA-PKcs.
Phosphorylation of DNA-PK after treatment with APH
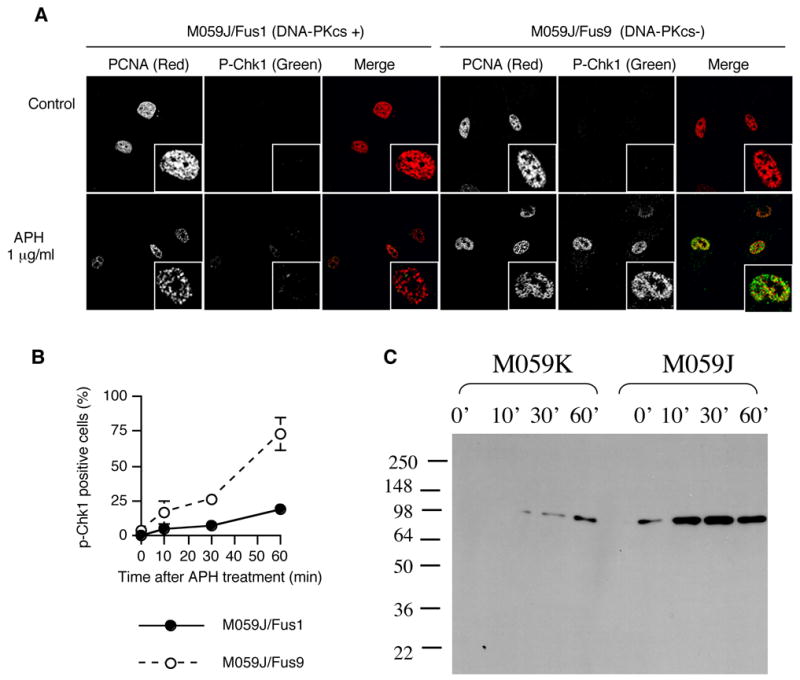
Because the DNA-PK status affected the response of cells to APH, we investigated directly whether DNA-PK was activated by APH. DNA-PKcs is autophosphorylated at several serine and threonine residues after DNA damage 33; 34. It can also be phosphorylated by ATM 35 and ATR 36. Although the residues undergoing phosphorylation vary and correlate with the nature of the triggering damage, DNA-Pkcs phosphorylation on threonine 2609, which reflects either autophosphorylation or phosphorylation by ATM or ATR, correlates with an active enzyme 35; 37. We used antibodies against phospho-DNA-PKcs-T2609 to detect the active form. Cells were also immunostained with proliferating cell nuclear antigen (PCNA), a marker for cells in S-phase. As shown in Figure 2, phospho-DNA-PKcs was absent or very low in untreated cells, but these cells exhibited a marked phosphorylation of DNA-PKcs 10 minutes after exposure to 1 μg/ml APH during S-phase (Figure 2A, left panels, 2B and 2C). As expected, phospho-DNA-PKcs was not observed even after treatment with APH in DNA-PKcs-deficient cells (Figure 2A, right panels). Phospho-DNA-PKcs was also absent from cells with an active DNA-PKcs in the presence of the DNA PK inhibitor NU7026, consistent with the notion that DNA-PK undergoes autophosphorylation. Phosphorylation of DNA-PKcs was not inhibited by UCN-01, an inhibitor of Chk1, suggesting that DNA-PK is activated independent of Chk1 (Figure 2B).
Figure 2.
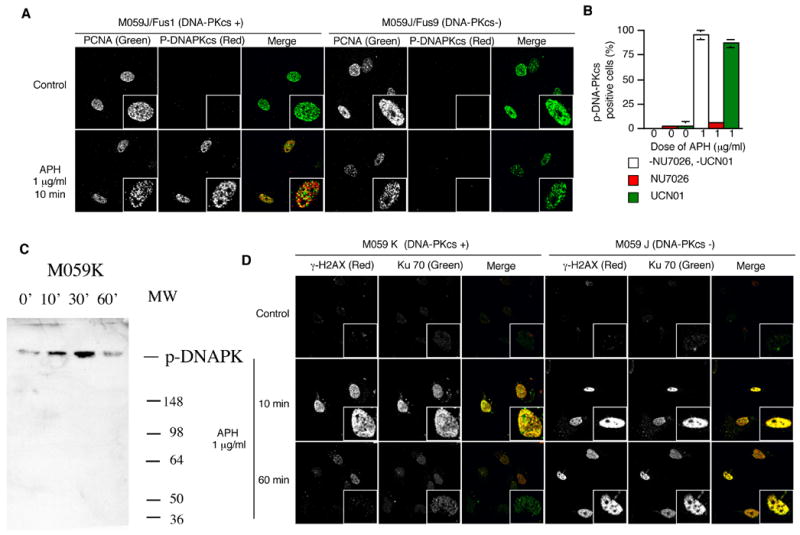
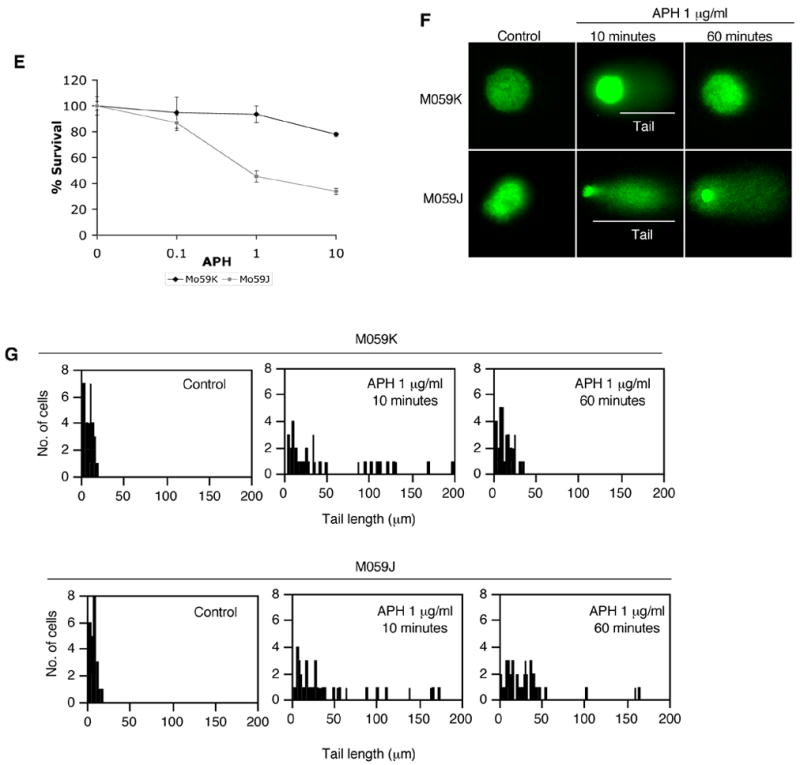
Phosphorylation of DNA-PK and formation of DSBs after treatment with low levels of APH. (A-C). Cells were treated with 1 μg/ml of APH for the indicated times and then immunostained with PCNA, p-DNA-PKcs, γ-H2AX, and Ku70. The images shown are from double staining experiments, but similar data were obtained with single staining with each antibody to control for possible “bleed through” between fluorescent channels. (A) Images of PCNA (green) and p-DNA-PKcs (red) in untreated cells and APH-treated cells. (B) The average percentage of p-DNA-PKcs positive, PCNA positive M059K cells after treatment with 1 μg/ml APH with and without NU7026 and UCN-01. 20 μM NU7026 and 300 nM UCN-01 were added in medium 30 minutes before APH treatment. We scored the number of p-DNA-PKcs-positive cells in 100 PCNA positive nuclei; the experiment was repeated 3 times with independent samples. Error bars indicate standard deviations. (C) Western blot analysis of nuclear proteins with M059K cells treated with APH at the indicated times and probed with an antibody against phosphorylated DNA-PK. (D) Images of γ-H2AX (red) and Ku70 (green) in untreated cells and APH-treated cells. Magnified images are inserted in (A) and (C). (E) colony forming assay of M059K and M059J cells after exposure to 1 μg/μl APH for 2 hours. (F–G) Double stranded DNA breaks formation after APH treatment evaluated by COMET assay. (F) Images of neutral comet assays after treatment with APH. (G) The distribution of tail length. For each data set, we scored 50 nuclei from two independent experiments.
Although M059J cells do not have an active DNA-PKcs, they do have Ku, the regulatory subunit of DNA-PK, which binds DNA before recruiting the catalytic subunit 6; 8; 38. The phosphorylation of DNA-PKcs might have indicated that double stranded DNA ends were formed following exposure to APH. To investigate whether such structures were also formed in DNA-PK deficient cells, and to inquire whether the Ku subunit would recognize DNA ends following exposure to APH in the absence of DNA-PK activity, we examined the localization of Ku70 and γ-H2AX 39; 40. γ-H2AX and Ku70 foci appeared shortly after exposure to low levels of APH (10 minutes after exposure to 1 μg/ml APH).
Ku70 accumulated at the sites of DSBs regardless of the DNA-PK status of the cells. Interestingly, in cells that contain active DNA-PK the intensity of γ-H2AX and Ku70 staining decreased after prolonged APH treatment (60 minutes after the initial exposure). In contrast, high levels of γ-H2AX and Ku70 foci persisted in cells deficient in DNA-PKcs (Figure 2D). These results suggest that a low dose of APH rapidly induced DNA breaks that were recognized by Ku70. In the presence of DNA-PKcs, such breaks were repaired, presumably by NHEJ, and the Ku protein dissociated from DNA ends. By contrast, in the absence of DNA-PK, Ku persisted at sites of unrepaired DNA breaks.
The focal patterns of Ku70 and γ-H2AX implied that APH treatment formed DSBs, which might be toxic to cells. Since APH is not generally toxic, we tested whether cells that are deficient in DNA-PK exhibit an increased sensitivity to low doses of APH. As shown in Figure 2E, a 2-hour exposure to 1 mg/ml APH significantly decreased the survival of Mo59J cells but not M059K cells. Treatment with 10 mg/ml APH affected colony forming abilities of both cell lines but the effect on M059J cells was more prominent. To investigate directly whether exposure to APH produced DSBs, we tested for the presence of broken DNA using a neutral COMET assay. Neutral COMET assays primarily detect double-stranded DNA strands, although single stranded breaks might also possibly be detected 41. As shown in Figure 3, F and G, untreated cells do not show a “tail” in a COMET assay whereas cells treated with APH for 10 minutes exhibited a marked increase in DNA breaks. A COMET tail was not observed after exposure of the cells to hydrogen peroxide, which induces single stranded DNA break, although the same cells exhibited a COMET tail under alkaline conditions (data not shown). These observations suggested that the DNA breaks we observed were double stranded. After 60 minutes, DNA breaks were significantly more abundant in cells deficient in DNA-PKcs, consistent with the patterns exhibited by Ku70 and γ-H2AX.
Figure 3.
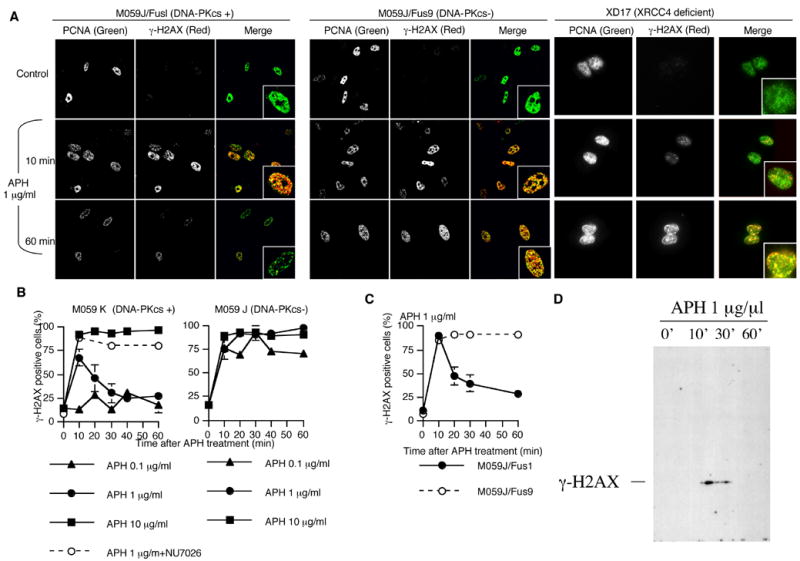
APH-induced γ-H2AX. Cells were treated with of the indicated doses of APH for the indicated times and then immunostained with PCNA and γ-H2AX. (A) Patterns of PCNA (green) and γ-H2AX (red) in untreated cells and in cells treated with 1 μg/ml APH. Magnified images are inserted. (B) The average percentage of γ-H2AX-positive cells in PCNA positive M059K and M059J cells. Empty circles: 20 μM Nu7026 was added 30 minutes before APH treatment. (C) Frequency of γ-H2AX-positive cells in PCNA positive M059J/Fus1 cells and M059J/Fus9 cells after treatment with 1 μg/ml of APH. For (B) and (C), we scored the number of γ-H2AX-positive cells in 100 nuclei of PCNA positive cells; the experiment was repeated 3 times with independent samples. Error bars represent standard deviations. (D) Western blot analysis of nuclear proteins from M059K cells treated with APH for the indicated times and probed with an antibody against γ–H2AX.
Persistent γ-H2AX in DNA-PKcs–deficient cells after treatment with APH
To learn more about the dynamics of the initial surge of DSBs, we determined the kinetics of γ-H2AX formation after APH treatment in the presence and absence of DNA-PK activity. We have also tested the formation of γ-H2AX foci in XR17 cells42, derivatives of XR-1, which are Chinese hamster cells deficient in the activity of XRCC443. Cells were immunostained with γ-H2AX at the indicated time. Cells were also immunostained with PCNA to identify S-phase cells as described. In cells that exhibited DNA-PK and XRCC4 activity, γ-H2AX appeared after 10 minutes of treatment with 1 μg/ml APH and disappeared after 30 minutes of treatment, whereas γ-H2AX was still observed 30 and 60 minutes after treatment in DNA-PKcs and XRCC4 deficient cells (Figure 3, A–C). The frequency of γ-H2AX was only determined in S-phase (PCNA-positive) cells; cells in other stages of the cell cycle did not induce γ-H2AX foci (data not shown). Cells with an active DNA-PK in S-phase did not induce γ-H2AX foci after exposure to 0.1 μg/ml APH, showed a transient induction of γ-H2AX after treatment with 1 μg/ml APH, and showed a persistent induction of γ-H2AX after treatment with 10 μg/ml APH (Figure 3B, left). The same pattern of transient induction of γ-H2AX in S-phase was observed in normal human fibroblasts (GM0037) treated with 1 μg/ml APH (data not shown). In contrast, DNA-PKcs-deficient cells maintained a high frequency of γ-H2AX 60 minutes after exposure to all APH doses (from 0.1 to 10 μg/ml APH) (Figure 3B, right). To test whether DNA-PK was required for the disappearance of γ-H2AX foci after APH treatment, we used a specific DNA-PK inhibitor, NU7026. The disappearance of γ-H2AX in cells with an active DNA-PK after treatment with 1 μg/ml APH was inhibited by NU7026 (Figure 3B, left). We have also determined the kinetics of γ-H2AX foci formation in M059J/Fus1 cells, which were complemented by DNA-PKcs. These cells exhibited a reduction in γ-H2AX foci after APH treatment (Figure 3C) whereas no reduction was observed in the non-complemented M059J/Fus9 cells. These results suggested that the disappearance of γ-H2AX after treatment with low doses of APH depended on DNA-PK.
We next investigated whether the checkpoint kinases, ATM, ATR, and Chk1 were involved in the phosphorylation of H2AX in response to lower levels of APH. As shown in Figure 4A, caffeine, an inhibitor of the ATM and ATR kinases, abrogated the formation of γ-H2AX after 1 μg/ml APH treatment in M059K cells, suggesting that ATM and/or ATR phosphorylates H2AX. In contrast, UCN-01, an inhibitor of Chk1, did not affect the induction of γ-H2AX after 1 μg/ml APH treatment, suggesting that Chk1 is not involved in the induction of γ-H2AX. To distinguish between ATR and ATM, we used cells that expressed a conditional, doxycycline-induced dominant negative form of ATR (ATR kinase dead; ATRkd) 44. As shown in Figure 4B, cells pretreated with 2 μg/ml doxycycline for 2 days to activate ATRkd did not induce γ-H2AX after APH treatment. These results indicate that γ-H2AX was produced by ATR, in agreement with previous reports 45.
Figure 4.
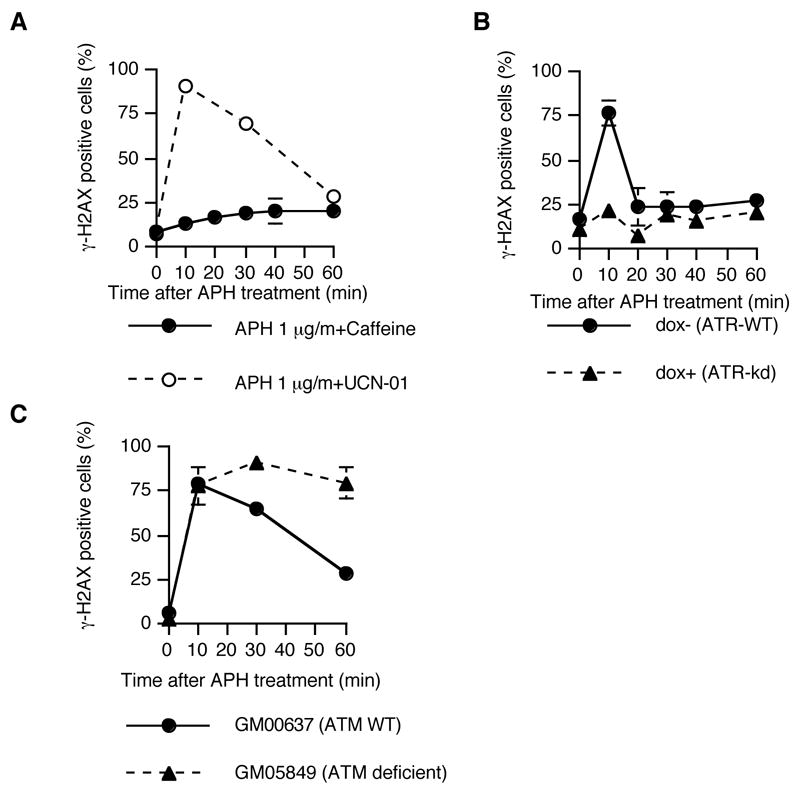
ATR-dependent phosphorylation of H2AX after treatment with APH. Cells were treated with 1 μg/ml of APH for the indicated times and then immunostained with PCNA and γ-H2AX. We scored the number of γ-H2AX-positive cells in 100 PCNA positive nuclei; the experiment was repeated 3 times with independent samples. (A) The frequency of γ-H2AX-positive cells that have PCNA in M059K cells after treatment with caffeine and UCN-01. 3mM caffeine and 300 nM UCN-01 were added 30 minutes before APH treatment. (B) The frequency of γ-H2AX-positive cells that have PCNA in ATRkd cells. To activate the ATRkd, cells were pretreated with 2μg/ml doxycycline for 2 days. (C) The frequency of γ-H2AX-positive cells in human fibroblasts that contain wild-type ATM (GM00637) and fibroblasts that are deficient in ATM (GM05849). Error bars represent standard deviations.
Because the ATM kinase might also play a role in the S-phase checkpoint, we investigated whether this kinase was necessary for the formation of DNA breaks following exposure to low levels of APH. As shown in Figure 4C, cells that were deficient in ATM exhibited transient DNA breaks induced with similar kinetics as cells with wild type ATM. In cells deficient in ATM, the breaks were induced but were not repaired, in line with the observation that the phosphorylation of DNA-PK is partially ATM-dependent 46, 35.
Checkpoint activation after treatment with a low dose of APH occurs in the absence of DNA-PK
The DNA-damage S-phase checkpoint in response to high levels of APH (50 μg/ml) involves the activation of Chk1, a kinase downstream of ATR 28. To examine whether low levels of APH activated Chk1 in cells proficient in DNA-PK activity and DNA-PKcs-deficient cells, we measured the levels of activated Chk1 at various times after treatment. Because Chk1 is phosphorylated after DNA damage 47, the active form of Chk1 can be identified by antibodies against phospho-Chk1 (serine 317). We identified S-phase cells by PCNA staining as described. As shown in Figure 5A and 5B, phosphorylated Chk1 appeared 60 minutes after treatment of cells with an active DNA-PK with 1 μg/ml APH and 10 minutes after treatment of DNA-PKcs-deficient cells. Phosphorylated Chk1 was also detected in DNA-PKcs-deficient cells, but not in cells with an active DNA-PK, after treatment with 0.1 μg/ml APH (Figure 5B). A Western blot analysis confirmed that the levels of Chk1 are higher and more persistent in cells deficient in DNA-PK although some phoshphorylated Chk1 was detected after exposure of DNA-Pkcs positive cells to APH for 60 minutes (Figure 5C). A high dose of APH (10 μg/ml) caused Chk1 phosphorylation in both cells with an active DNA-PK and DNA-PKcs-deficient cells (Figure 5B). These results suggested that active DNA-PK prevented Chk1 phosphorylation after exposure to low doses of APH, probably averting the activation of the Chk1-mediated S-phase checkpoint.
Figure 5.
Checkpoint activation after treatment with APH. Cells were treated the indicated doses of APH for the indicated times and then immunostained with PCNA and p-Chk1 (serine 317). (A) Images of PCNA (red) and p-Chk1 (green) in untreated cells and cells treated with 1 μg/ml of APH. Magnified images are inserted. (B) The frequency of p-Chk1-positive cells in PCNA positive M059J/Fus1 and M059J/Fus cells treated with 10 μg/ml APH. We scored the number of p-Chk1 positive cells in 100 nuclei of cells with PCNA; the experiment was repeated 3 times with independent samples. Error bars represent standard deviations. (C) Western blot analysis of nuclear proteins from M059K and M059J cells treated with APH for the indicated times and probed with antibodies against phosphorylated Chk1.
To elucidate the molecular mechanism of APH action, we used a DNA fiber assay 48 to inquire how low levels of APH affected the initiation and the elongation stages of DNA replication. Cells were pulse labeled with 5-Iodo-2′-deoxyuridine (IdU) before treatment with APH (detected by Cy3; red signal) and labeled with 5-chloro-2′-deoxyuridine (CldU) after treatment (detected by Alexa 488; green signal). Initiation of DNA replication during the first labeling period (before APH treatment) was detected as green-red-green tracks (G-R-G, red signal flanked by two green signals), initiation during the second labeling period (in the presence of APH) was detected as green-only tracks (G), elongation of replication forks that initiated before IdU labeling was detected as unidirectional red-green tracks (R-G), whereas red - only tracks (R) and rare R-G-R tracks suggested termination events. We estimated the frequency of new origin firing, ongoing replication forks, and stalled replication forks after treatment with APH (Figure 6, B–D). Low doses of APH suppressed initiation of DNA replication (measured by the abundance of G-only fibers – Figure 6B) in DNA-PKcs-deficient cells. However, in cells with an active DNA-PK, low APH levels did not inhibit initiation of DNA replication (the abundance of G-only fibers remained constant) whereas replicating DNA tracks (CldU signals in R-G tracks, Figure 6A) were significantly shorter (the average length of the green signals in R-G tracks was reduced from 1.93 μm (standard deviation: 0.4) to 0.45 μm (standard deviation: 0.14)). These data indicated that replication fork progression was suppressed after APH treatment, and that most replication forks continued to progress when exposed to APH doses at or below 1 μg/ml (Figure 6C, 6D). In contrast, in DNA-PKcs-deficient cells, most replication forks exhibited R-only tracks (stalled replication forks) even after treatment with APH doses as low as 0.1 μg/ml (Figure 6A, 6D). These results demonstrated that hypersensitivity to low doses of APH in DNA-PKcs-deficient cells was caused by suppressing the firing of replication origins and stalling the progress of replication forks, hallmarks of the DNA damage-induced S-phase checkpoint. The stalling of replication forks was not affected by the presence or absence of the ATM kinase (compare GM00637 cells and GM05849 cells in Figure 6A).
Figure 6.
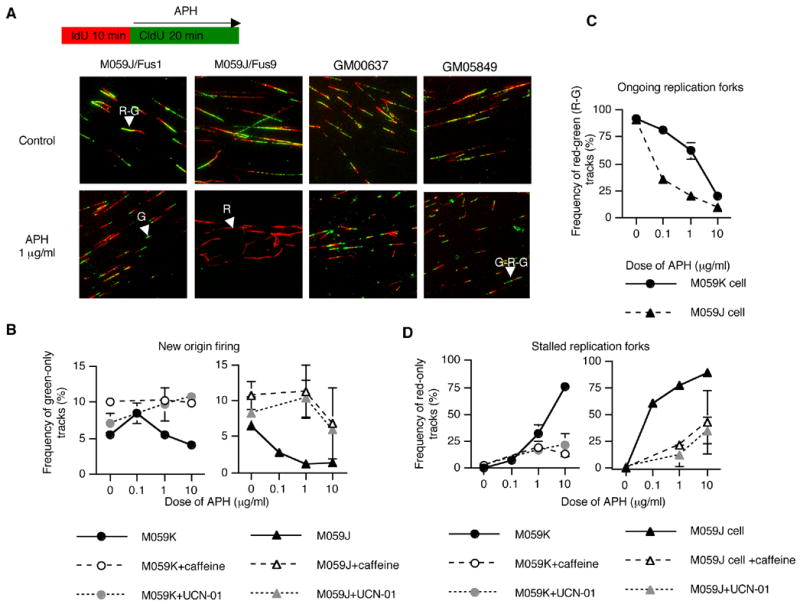
Suppression of origin firing and stalled replication forks after treatment with APH. The rate of replication fork progression in M059J/Fus1 (cells with an active DNA-PK), M059J/Fus9 cells (DNA-PKcs deficient), GM00637 (cells containing wild-type ATM) and GM05849 (cells deficient in ATM) was measured by a DNA fiber assay. Cells were first labeled with IdU for 10 minutes. IdU was then washed out and the cells were exposed to APH concomitant with the addition CldU for 20 min. IdU was immunodetected by Cy-3–labeled antibodies (red color; see Material and Methods). CldU was detected by Alexa488-labeled antibodies (green color). (A) Images of labeled tracks. Red-green tracks (R-G), red-only tracks (R), green-only tracks (G), and green-red-green tracks (G-R-G) are indicated by arrowheads. (B) Abundance of G tracks in M059J and M059J cells (green tracks represent new origin firing). (C) Abundance of R-G tracks (ongoing replication forks). (D) Abundance of R tracks (stalled replication forks). 3mM caffeine and 300 nM UCN-01 were added 30 minutes before APH treatment. For each data point, we counted 100 tracks. Each experiment was repeated twice from independent DNA preparations. Error bars represent standard deviations.
The data presented above suggest that APH-induced DSBs triggered the DNA damage S-phase checkpoint only in the DNA-PKcs-deficient cells. To test this possibility, we treated cells with the checkpoint inhibitors caffeine (inhibitor of phosphatidylinositol 3 kinases such as ATM and ATR) and UCN-01 (inhibitor of Chk1). Consistent with the above hypothesis, both inhibitors abrogated the suppression of origin firing and the stalling of replication forks in DNA-PKcs-deficient cells (Figure 6B and 6D).
Finally, we investigated the effect of DNA-PK deficiency on the activation of Rad51, which is an essential factor for homologous recombination (HR). HR is the major repair pathway activated after replication perturbation 7; 15. As shown in Figure 7A, Rad51 foci were not induced after treatment with 1 μg/ml APH in cells with an active DNA-PK. In contrast, many Rad51 foci appeared 60 minutes after treatment with the same dose of APH in DNA-PKcs-deficient cells (Figure 7A). The distribution of Rad51 foci was constant after treatment with APH in cells with an active DNA-PK but significantly changed 60 minutes after treatment in DNA-PKcs-deficient cells (Figure 7B). These results suggested that HR was activated by APH-induced DSBs when the damage was not repaired by DNA-PK.
Figure 7.
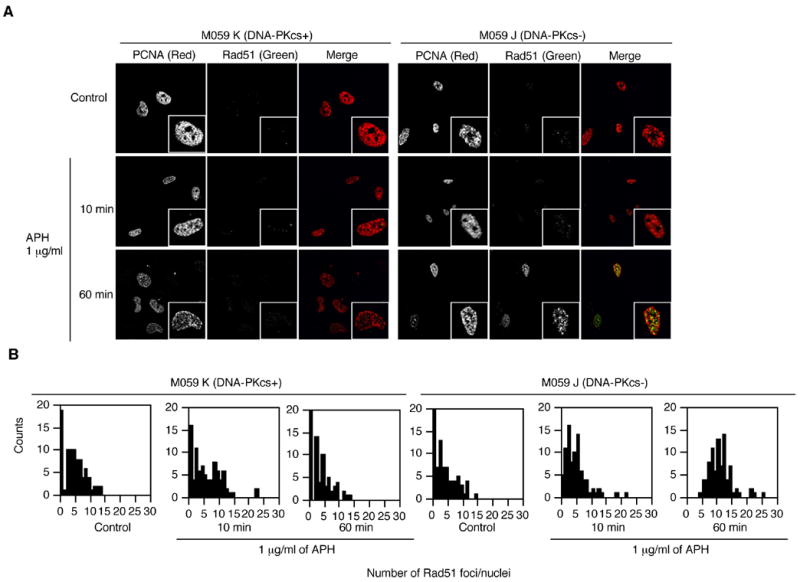
Rad51 patterns after treatment with APH. Cells were treated with 1 μg/ml of APH for the indicated time and then immunostained with PCNA and Rad51. (A) Images of PCNA (red) and Rad51 (green) in untreated cells and in cells treated with 1 μg/ml APH. Magnified images are inserted. (B) The distribution of Rad51 foci after treatment with APH in M059K and M059J cells. We scored the number of Rad51 foci in 30 PCNA positive nuclei. The experiments were repeated 3 times with independent samples.
Discussion
Long treatments with APH are known to generate DSBs 49; 50; 51 and activate fragile site expression 52. In contrast, short treatments with APH (less than 1 hour) are thought to inhibit replication fork progression without induction of DSBs 53; 54. The data reported here reveal that a short (10 minutes) treatment with APH rapidly activated transient γ-H2AX foci, which mark DSBs, in an ATR-dependent manner. This surge of γ-H2AX foci could occur in cells that had been treated with a Chk1 inhibitor, suggesting that γ-H2AX foci formation did not depend on the activation of the S-phase checkpoint through the Chk1-mediated signaling cascade. In cells with normal DNA-PK, γ-H2AX foci induced by low level of APH rapidly disappeared, indicating that the initial group of DSBs was repaired without inducing a cell cycle checkpoint and that the subsequent low progression of replication forks did not lead to further induction of DSBs. Our data further indicate that DSBs were recognized by the Ku protein and that the reduction of γ-H2AX levels after exposure to a low dose of APH required DNA-PK activity. These observations suggest that an activity of DNA-PK, most likely the activation of the NHEJ pathway, rapidly repaired APH-induced DNA DSBs when damage was low. It is likely that the induction of a cell cycle checkpoint by ATR only occurred when DNA-PK activity was absent or insufficient to tackle the damage.
The data reported here suggest that the ATR kinase is primarily responsible for the surge or DSBs and the inhibition of DNA replication after exposure to low levels of APH. By contrast, the cellular response to other drugs that inhibit DNA replication such as CPT are mediated primarily by ATM, suggesting that the two responses are activated by different lesions. Exposure to CPT in actively replicating cells directly forms DSBs that trigger the ATM-mediated damage response and can be prevented by APH, which inhibits replication and prevents the formation of replication-mediated DNA breaks 53. As shown here, cells with functional DNA-PK do not exhibit DNA breaks in the presence of mild doses of APH. In those cells, replication is inhibited by APH and no further DSBs are formed after the repair of the first surge of DSBs. This repair process, coupled with APH-induced inhibition of DNA replication, facilitates prevention of replication-dependent DNA breaks after exposure to CPT.
Cells deficient in DNA-PK were not deficient in the activation of the S-phase checkpoint, which suppresses origin firing and stalls replication forks. However, these cells exhibited an increased sensitivity to low doses of APH and a lower threshold (0.1 μg/ml) beyond which the ATR-Chk1-mediated S-phase checkpoint was activated. These studies are consistent with the observation that DNA-PK inhibition strongly activates Chk1 and Chk2 after ionizing radiation that leads to sustained G2 arrest after DNA damage 55. Our studies are also consistent with the observed phosphorylation of DNA-PK, which correlates with its activation 38. DNA-PK deficient cells were reported to be highly sensitive to low doses of ionizing radiation 56; 57 and were also sensitive to the replication inhibitor hydroxyurea 7; 19. Our data demonstrate that DNA-PKcs-deficient cells were hypersensitive to APH but only at low doses (below 1 μg/ml). We further demonstrated that exposure to APH recruits the regulatory subunit of DNA-PK, Ku, to chromatin regardless of DNA-PKcs status. In cells with an active DNA-PK, exposure to APH triggered the phosphorylation of the catalytic subunit of DNA-PK. Activation of DNA-PK occurred before activation of Chk1 and was not inhibited by the Chk1 inhibitor, UCN-01. It is plausible that at low levels of APH, DNA-PK repaired DSBs by NHEJ before the activation of Chk1-mediated S-phase checkpoint. In DNA-PKcs-deficient cells, damage sensors detected persistent DSBs and triggered the S-phase checkpoint, possibly because the NHEJ pathway was not activated. Consistent with this, replication elongation continued slowly after exposure to low levels of APH in cells with an active DNA-PK but forks completely stalled when cells were exposed to 10 μg/ml APH, suggesting that high levels of DNA damage that were not repaired by NHEJ triggered the Chk1-mediated S-phase checkpoint. These data are in concert with the observation that the yeast S-phase tolerates a low level of damage-like structures and requires a threshold level of DNA damage to activate the checkpoint response that prevents late replication 58.
Mammalian cells have two distinct DNA repair pathways for DSBs, NHEJ and HR. Our data are consistent with the suggestion that the DNA-PK-mediated NHEJ pathway recognizes DSBs faster than the HR pathway and acts before the activation of the DNA damage S-phase checkpoint 7. The activation of NHEJ by DNA-PK and XRCC4 before recruitment of Rad51 to sites of replication inhibition might underlie the roles of DNA-PK and XRCC4 in suppressing spontaneous HR 59 when spontaneous HR is caused by stalled or collapsed replication forks 15,60. Our DNA fiber analysis suggests that when DNA-PK is active, replication can proceed slowly even in the presence of low levels of DNA polymerase inhibition. Presumably, under these conditions, low levels of DNA breaks are repaired via the NHEJ pathway. Although we cannot formally rule out that DNA-PK is involved in another repair pathway unrelated to NHEJ, our data are consistent with the suggestion that the ATR-Chk1-mediated S-phase checkpoint is activated in the absence or insufficiency of NHEJ and that the active S-phase checkpoint serves to stabilize replication forks to facilitate the alternative HR repair pathway.
Taken together, the observations reported here have elucidated the relative roles of DNA-PK and the S-phase checkpoint in response to replication inhibition. Perturbations of DNA replication produce a surge of DSBs that is initially repaired by DNA-PK. In this way, DNA-PK prevents the activation of the Chk1-mediated S-phase checkpoint. ATR predominantly induces this S-phase checkpoint in response to replication perturbation in the absence or the insufficiency of the initial response catalyzed by DNA-PK. These data suggest how DNA-PK activity might affect the sensitivity of cells to drugs that perturb DNA replication.
Materials and Methods
Cells and culture conditions
The M059K and M059J human glioma-derived cell lines 29, the human fibroblast cell lines GM00637 (normal ATM) and GM05849 (deficient in the ATM kinase) and the Chinese hamster XD-17 cell line were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum and L-glutamine (Gibco-BRL). M059J/Fus1 and M059J/Fus9 cells 56 were grown in DMEM supplemented with 10% fetal bovine serum 250 μg/ml G418 (Invitrogen). SV40 transformed GM847 fibroblasts harboring ATRkd 44 (ATRkd cells) were grown in DMEM supplemented with 10% heat-inactivated fetal calf serum, L-glutamine and 400 μg/ml G418.
Drugs
UCN-01 provided by the Drug Synthesis Chemistry Branch, Division of Cancer Treatment, National Cancer Institute, was dissolved at a final concentration of 100 mM in Me2SO and stored at −20°C. APH was purchased from Wako, U.S.A. Caffeine was purchased from Sigma. DNA-PK inhibitor 2 (NU7026) was purchased from Calbiochem. APH was dissolved in Me2SO (1 mg/ml) and stored at −20°C. Caffeine was dissolved in DMEM (30 mM) and stored at 4°C. DNA-PK inhibitor 2 was dissolved in Me2SO (10 mM) and stored at −20°C.
DNA fiber analysis
DNA fiber analysis was performed as described previously 48. Cells were labeled with 20 μM IdU for 10 min and then labeled with 20 μM CldU for 20 min. Cells were trypsinized and resuspended in PBS at 1 x 106 cells/ml. The cell suspension (2.5 μl) was mixed with 7.5 μl lysis buffer (0.5% SDS in 200 mM Tris-HCl, pH 7.4, 50 mM EDTA) on an uncoated glass slide (Daigger). After 8 min, DNA spreads were fixed in 3:1 methanol:acetic acid for 5 minutes and stored in 70% ethanol at 4°C. Double immunostaining of CldU and IdU was performed according to Dimitrova and Gilbert 61. The slides were incubated in 100% methanol at room temperature for 5 minutes and rehydrated with PBS. DNA was denatured with 2.5 N HCl at 37°C for 30 minutes, then washed and incubated with primary antibodies. The anti-CldU (Accurate Chemical and Scientific Corporation) and anti-IdU (Becton Dickinson) antibodies were diluted in PBS with 0.5% bovine serum albumin (BSA). Cells were incubated with the antibodies for 1 hour at 37°C. The slides were then washed 3 times with 0.1% Triton X-100 in PBS and incubated for 1 hour at 37°C with secondary antibody conjugated with Alexa 488 (Molecular Probes for rat immunoglobin G) and Cy-3 (Jackson Immuno Research Laboratories, Inc. for mouse immunoglobin). The slides were washed 3 times with 0.1% Triton X-100 in PBS and counterstained for DNA with 4 μg/ml 4′-6-diamino-2-phenylindole in aqueous mounting medium (Biomeda Corp.). Images of DNA fibers were captured by epifluorescence microscopy using 100X objective lens.
FACS analysis
Cells were labeled with 20 μM BrdU and 0.25 μM fluorodeoxyuridine (FdU, Fluka), washed with PBS, and fixed in 70% ethanol overnight. DNA was denatured with 1 M HCl-0.1% Triton X-100 on ice for 10 minutes followed by boiling for 10 minutes. Cells were incubated with fluorescein isothiocyanate-conjugated anti-BrdU antibody (Becton Dickinson) for 1 hour, and DNA was stained with propidium iodide in the presence of RNase. BrdU-positive cells were detected and quantified by FACScan (Becton Dickinson).
Immunofluorescence
Cells were grown on 18mm x 18mm x 1mm coverslips (Fisher 12-548-A). After treatment with APH, cells were washed with PBS, treated with a hypotonic lysis solution (10 mM Tris-HCl pH 7.4, 2.5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, and 0.5% Nonidet P-40) for 8 minutes on ice. Cells were fixed in 4% paraformaldehyde in PBS for 10 minutes, washed in PBS, made permeable in 100% methanol at −20°C for 15 minutes, and then washed and blocked with PBS containing 1% BSA and 0.1% Triton X-100 for 30 minutes. Cells were incubated with anti-PCNA (Santa Cruz), anti-γ-H2AX (Upstate), anti-phospho-DNA-PKcs-threonine-2609 (Abcam, ab18356), anti-phospho-Chk1-Serine-317 (Cell Signaling), or anti-Ku70 (Santa Cruz, sc-1486) antibodies. Antibodies were diluted in PBS with 0.5% BSA for 1 hour at 37°C. Slides were then washed 3 times with 0.1% Triton X-100 in PBS and incubated for 1 hour at 37°C with secondary antibody conjugated with Alexa 488 (Molecular Probes) or Cy-3 (Jackson Immuno Research Laboratories). Slides were washed 3 times with 0.1% Triton X-100 in PBS and counterstained for DNA with 4′-6-diamino-2-phenylindol. Images were captured by confocal microscopy (Nikon; PCM 2000) using 100X objective lens.
Neutral Comet Assay
Neutral comet assay was performed using the CometAssay Kit (Trevigen) following the manufacture’s protocol. Cells were treated with APH for indicated times. Cells were collected and suspended in low melting point agarose. The agarose was applied to CometSlidesTM and allowed to set at 4°C in the dark. After lysis of the agarose-embedded cells in lysis solution (2.5 M NaCl, 100 mM EDTA, pH 10, 10 mM Tris base, 1% sodium lauryl sarcosinate, 0.01% Triton X-100), the slides were electrophoresed in TBE, pH 8 (0.089 M Tris, 0.089 M boric acid, 0.003 M EDTA). The samples were then fixed in 70% ethanol and dried overnight before staining with SyBr® Green (Molecular Probes, Eugene, OR) to visualize cellular DNA. Images of nuclei were captured by CCD camera (Roter Scientific; Cool SNAP FX) with epifluorescence microscopy (Olympus; IX70) using a 20X objective lens. For each sample, 50 cells were scored for the length of tail. Tail length was manually measured using the IPLab software. Two independent experiments were performed for each data set.
Western Blot Analyses
Cells were treated with 1 ug/ul (final concentration) APH for the indicated times. Cells were lysed at room temperature in 200 μl sucrose buffer (0.32 m sucrose, 10mM 1 M Tris-HCL pH=7.4, 3mM CaCl2, 2mM 1 M magnesium acetate, 0.1 mM 0.5M EDTA, 0.5%NP-40, 1 mM DTT, and 0.5 PMSF), centrifuged at 500g for 5 min and the nuclear extract pellet was washed with sucrose buffer solution without NP-40. Nuclei were sequentially washed with low salt buffer (20 mM Hepes, 1.5 mM MgCl2, 20 mM KCL, 0.2 mM 0.5 EDTA, and 25% glycerol) and high salt buffer (20 mM Hepes, 1.5 mM MgCl2, 20 mM KCL, 0.2 mM 0.5 EDTA, 25% glycerol, and 1% NP-40). Proteins were recovered through centrifugation at 14000 rpm for 15 min. Proteins were separated by SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked with 5% non-fat milk for 1 hour and incubated with either mouse anti-phospho-Histone H2AX (Ser 139) clone JBW 103 (Upstate), rabbit anti-phopho-Chk1 (Ser317) (Cell Signaling) or mouse [10B1] phospho DNA PKcs (AbCam) overnight at 4°C. To verify that we can detect DNA-PK in M059K but not in M059K, we used mouse [7A4] DNA PKcs (data not shown). Secondary incubation with peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody (Santa Cruz) was performed for 1 hour and detection was achieved with SuperSignal west pico chemiluminescent substrates (Pierce).
Acknowledgments
We thank Drs. Yves G. Pommier, William M. Bonner, and Kurt W. Kohn for their critical reading of the manuscript and for numerous helpful suggestions. We are grateful to Chii-Mei Lin, Lixin Wang, Haiqing Fu, Elsa Bronze Da Rocha, Ashutosh Rao, Chiara Conti, and Asako Nakamura for helpful suggestions. This study was supported by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute; T.S. was supported by fellowships from the Uehara Memorial Foundation and the Japan Society for the Promotion of Science.
Abbreviations
- (APH)
Aphidicolin
- (BSA)
bovine serum albumin
- (ATM)
ataxia telangiectasia mutated
- (ATR)
ATM- and Rad3-related
- (ATRkd)
ATR kinase dead
- (BrdU)
bromodeoxyuridine
- (CldU)
5-chloro-2′-deoxyuridine
- (DMEM)
Dulbecco’s modified Eagle’s medium
- (DNA-PK)
DNA-dependent protein kinase
- (DNA-PKcs)
catalytic subunit of DNA-PK
- (DSBs)
double-strand breaks
- (FACS)
fluorescence-activated cell sorting
- (FdU)
fluorodeoxyuridine
- (HR)
homologous recombination
- (HU)
hydroxyurea
- (IdU)
5-Iodo-2′-deoxyuridine
- (NHEJ)
nonhomologous end-joining
- (PBS)
phosphate-buffered saline
- (pol α)
polymerase α
- (PCNA)
proliferating cell nuclear antigen
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang J, Yu Y, Hamrick HE, Duerksen-Hughes PJ. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis. 2003;24:1571–80. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- 2.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13:225–31. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 3.Gottifredi V, Prives C. The S phase checkpoint: when the crowd meets at the fork. Semin Cell Dev Biol. 2005;16:355–68. doi: 10.1016/j.semcdb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Osborn AJ, Elledge SJ, Zou L. Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol. 2002;12:509–16. doi: 10.1016/s0962-8924(02)02380-2. [DOI] [PubMed] [Google Scholar]
- 5.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 6.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949–61. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 7.Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol. 2002;22:5869–78. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev. 1999;13:916–34. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- 9.Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909–18. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 10.Pastwa E, Blasiak J. Non-homologous DNA end joining. Acta Biochim Pol. 2003;50:891–908. [PubMed] [Google Scholar]
- 11.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 12.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 13.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–45. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 16.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 17.Kuzminov A. DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc Natl Acad Sci U S A. 2001;98:8461–8. doi: 10.1073/pnas.151260698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–7. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J. 2001;20:3861–70. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikegami S, Taguchi T, Ohashi M, Oguro M, Nagano H, Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–60. doi: 10.1038/275458a0. [DOI] [PubMed] [Google Scholar]
- 21.Pedrali-Noy G, Belvedere M, Crepaldi T, Focher F, Spadari S. Inhibition of DNA replication and growth of several human and murine neoplastic cells by aphidicolin without detectable effect upon synthesis of immunoglobulins and HLA antigens. Cancer Res. 1982;42:3810–3. [PubMed] [Google Scholar]
- 22.Pedrali-Noy G, Spadari S, Miller-Faures A, Miller AO, Kruppa J, Koch G. Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin. Nucleic Acids Res. 1980;8:377–87. doi: 10.1093/nar/8.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheaff R, Ilsley D, Kuchta R. Mechanism of DNA polymerase alpha inhibition by aphidicolin. Biochemistry. 1991;30:8590–7. doi: 10.1021/bi00099a014. [DOI] [PubMed] [Google Scholar]
- 24.Decker RS, Yamaguchi M, Possenti R, DePamphilis ML. Initiation of simian virus 40 DNA replication in vitro: aphidicolin causes accumulation of early-replicating intermediates and allows determination of the initial direction of DNA synthesis. Mol Cell Biol. 1986;6:3815–25. doi: 10.1128/mcb.6.11.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levenson V, Hamlin JL. A general protocol for evaluating the specific effects of DNA replication inhibitors. Nucleic Acids Res. 1993;21:3997–4004. doi: 10.1093/nar/21.17.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oguro M, Suzuki-Hori C, Nagano H, Mano Y, Ikegami S. The mode of inhibitory action by aphidicolin on eukaryotic DNA polymerase alpha. Eur J Biochem. 1979;97:603–7. doi: 10.1111/j.1432-1033.1979.tb13149.x. [DOI] [PubMed] [Google Scholar]
- 27.Spadari S, Sala F, Pedrali-Noy G. Aphidicolin and eukaryotic DNA synthesis. Adv Exp Med Biol. 1984;179:169–81. doi: 10.1007/978-1-4684-8730-5_17. [DOI] [PubMed] [Google Scholar]
- 28.Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–23. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS, 3rd, Barron GM, Allalunis-Turner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–5. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 30.Peng Y, Woods RG, Beamish H, Ye R, Lees-Miller SP, Lavin MF, Bedford JS. Deficiency in the catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer Res. 2005;65:1670–7. doi: 10.1158/0008-5472.CAN-04-3451. [DOI] [PubMed] [Google Scholar]
- 31.Gately DP, Hittle JC, Chan GK, Yen TJ. Characterization of ATM expression, localization, and associated DNA-dependent protein kinase activity. Mol Biol Cell. 1998;9:2361–74. doi: 10.1091/mbc.9.9.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003;63:6008–15. [PubMed] [Google Scholar]
- 33.Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol. 2003;23:5836–48. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees-Miller SP. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J. 2002;368:243–51. doi: 10.1042/BJ20020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, Chen DJ. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–8. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yajima H, Lee KJ, Chen BP. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol. 2006;26:7520–8. doi: 10.1128/MCB.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodarzi AA, Block WD, Lees-Miller SP. The role of ATM and ATR in DNA damage-induced cell cycle control. Prog Cell Cycle Res. 2003;5:393–411. [PubMed] [Google Scholar]
- 38.Drouet J, Delteil C, Lefrancois J, Concannon P, Salles B, Calsou P. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem. 2005;280:7060–9. doi: 10.1074/jbc.M410746200. [DOI] [PubMed] [Google Scholar]
- 39.Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol. 2003;81:123–9. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- 40.Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol Ther. 2003;2:233–5. doi: 10.4161/cbt.2.3.373. [DOI] [PubMed] [Google Scholar]
- 41.Crumpton MJ, Collins AR. Are environmental electromagnetic fields genotoxic? DNA Repair (Amst) 2004;3:1385–7. doi: 10.1016/j.dnarep.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 42.Delacote F, Han M, Stamato TD, Jasin M, Lopez BS. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002;30:3454–63. doi: 10.1093/nar/gkf452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryans M, Valenzano MC, Stamato TD. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat Res. 1999;433:53–8. doi: 10.1016/s0921-8777(98)00063-9. [DOI] [PubMed] [Google Scholar]
- 44.Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. Embo J. 1998;17:159–69. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 46.Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709–15. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- 47.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merrick CJ, Jackson D, Diffley JF. Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem. 2004;279:20067–75. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 49.Szuts D, Krude T. Cell cycle arrest at the initiation step of human chromosomal DNA replication causes DNA damage. J Cell Sci. 2004;117:4897–908. doi: 10.1242/jcs.01374. [DOI] [PubMed] [Google Scholar]
- 50.Hammond EM, Green SL, Giaccia AJ. Comparison of hypoxia-induced replication arrest with hydroxyurea and aphidicolin-induced arrest. Mutat Res. 2003;532:205–13. doi: 10.1016/j.mrfmmm.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 51.Musio A, Montagna C, Mariani T, Tilenni M, Focarelli ML, Brait L, Indino E, Benedetti PA, Chessa L, Albertini A, Ried T, Vezzoni P. SMC1 involvement in fragile site expression. Hum Mol Genet. 2005;14:525–33. doi: 10.1093/hmg/ddi049. [DOI] [PubMed] [Google Scholar]
- 52.Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. 1984;67:136–42. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- 53.Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–12. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 54.Liu JS, Kuo SR, Melendy T. Comparison of checkpoint responses triggered by DNA polymerase inhibition versus DNA damaging agents. Mutat Res. 2003;532:215–26. doi: 10.1016/j.mrfmmm.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 55.Sturgeon CM, Knight ZA, Shokat KM, Roberge M. Effect of combined DNA repair inhibition and G2 checkpoint inhibition on cell cycle progression after DNA damage. Mol Cancer Ther. 2006;5:885–92. doi: 10.1158/1535-7163.MCT-05-0358. [DOI] [PubMed] [Google Scholar]
- 56.Hoppe BS, Jensen RB, Kirchgessner CU. Complementation of the radiosensitive M059J cell line. Radiat Res. 2000;153:125–30. doi: 10.1667/0033-7587(2000)153[0125:cotrmc]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 57.Daido S, Yamamoto A, Fujiwara K, Sawaya R, Kondo S, Kondo Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005;65:4368–75. doi: 10.1158/0008-5472.CAN-04-4202. [DOI] [PubMed] [Google Scholar]
- 58.Shimada K, Pasero P, Gasser SM. ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev. 2002;16:3236–52. doi: 10.1101/gad.239802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Allen C, Kurimasa A, Brenneman MA, Chen DJ, Nickoloff JA. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc Natl Acad Sci U S A. 2002;99:3758–63. doi: 10.1073/pnas.052545899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25:7158–69. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell. 1999;4:983–93. doi: 10.1016/s1097-2765(00)80227-0. [DOI] [PubMed] [Google Scholar]