

Abstract
Filamentous actinomycetes are commercially widely used as producers of natural products. However, the mycelial lifestyle of actinomycetes has been a major bottleneck in their commercialization, and screening is difficult due to their poor growth on microtiter plates. We previously demonstrated that the enhanced expression of the cell division activator protein SsgA results in the fragmented growth of streptomycetes, with enhanced growth rates and improved product formation. We here describe a novel and efficient method to create, maintain, and screen mutant libraries in streptomycetes and the application of this method for the functional analysis of Streptomyces coelicolor ssgA. The variants were amplified directly from deep-frozen biomass suspensions. Around 800 ssgA variants, including single-amino-acid-substitution mutants corresponding to more than half of all SsgA residues, were analyzed for their abilities to restore sporulation to an ssgA mutant. The essential residues were clustered in three main sections, and hardly any were in the carboxy-terminal third of the protein. The majority of the crucial residues were conserved among all SsgA-like proteins (SALPs). However, the essential residues L29, D58, and S89 were conserved only in SsgA orthologues and not in other SALPs, suggesting an SsgA-specific function.
Filamentous microorganisms are widely used as industrial producers of products such as antibiotics, anticancer agents, antifungicides, and enzymes (1, 6, 9). These organisms include the eukaryotic filamentous fungi (in particular, ascomycetes) and the prokaryotic actinomycetes (e.g., Amycolatopsis, Nocardia, Rhodococcus, and Streptomyces species). The market capitalization for antibiotics and enzymes totals around 28 and 2 billion dollars per year, respectively. While the industrial potential of actinomycetes is widely recognized, the filamentous lifestyle makes them less favorable for growth in submerged cultures, where they produce dense clumps or pellets (3, 31). Other host-specific disadvantages include slow growth rates and high viscosity of the culture broth. The latter necessitates high stirrer speeds, resulting in uncontrolled fragmentation and lysis of the mycelium. Hence, morphological engineering of streptomycetes has become increasingly important for the optimization of fermentation processes and the improvement of productivity. For these objectives, understanding the factors that control morphogenesis in submerged cultures is of the utmost importance. On solid-grown cultures, the high-G+C-content, gram-positive streptomycetes have an unusually complex life cycle, with mycelial growth and reproduction through spores (4, 5, 8). After spore germination, a large vegetative mycelium of branching hyphae is produced. Upon starvation, sporogenic aerial hyphae are produced on top of the vegetative mycelial mat, culminating in the simultaneous formation of many sporulation septa and eventually producing chains of mature, uninucleoid spores. In submerged cultures, most streptomycetes grow only by hyphal extension, although some species produce submerged spores (14).
The SsgA-like proteins (SALPs) form a family of developmental regulators occurring exclusively in sporulating actinomycetes. The genome of Streptomyces coelicolor (2) encodes seven SsgA-like proteins (SALPs), with specific tasks in the control of the sporulation process, each protein playing distinct and important roles (13, 20, 29). SsgA plays an important role in the control of morphogenesis in both liquid- and solid-grown cultures and is a known activator of sporulation-specific cell division (11, 28). The overproduction of SsgA in S. griseus and S. coelicolor results in mycelial fragmentation (12, 28). The industrial importance of ssgA for a number of Streptomyces species was recently highlighted, and increased expression of ssgA results in fragmented growth of S. coelicolor, “S. lividans,” and S. roseosporus and enhanced growth rates in batch fermentations, with strongly improved enzyme production by S. lividans (27). The SALPs do not share significant homology to other proteins and are one of the few protein families that lack any clear protein motif, and so far the three-dimensional structure is unknown. A mutational analysis of the SsgA protein would provide more insight into the functional importance of individual residues. However, screening a library of perhaps thousands of transformants is feasible only when microtiter plate (MTP)-based screening and colony PCR are an option. While the growth of streptomycetes on microtiter plates has been reported previously (19), PCR-based screening is difficult and often requires mycelium pretreatment (lysis) and DNA isolation steps, and the direct screening of transformants has been reported only for transformants carrying high-copy-number plasmids (23, 26).
In this work, we describe an efficient method to create, propagate, and screen a large strain collection of random ssgA mutants and present a way to amplify and sequence the individual mutant clones directly from deep-frozen samples stored on microtiter plates. For each of the amino acid residues of the SsgA protein, the importance for in vivo function of the protein was analyzed.
MATERIALS AND METHODS
Bacterial strains and culturing conditions.
The bacterial strains used in this work are listed in Table 1. Escherichia coli K-12 strains JM109 (22) and ET12567 (18) were used for the propagation of plasmids and were grown and transformed using standard procedures (22). S. coelicolor M145 was obtained from the John Innes Centre strain collection. S. coelicolor GSA3 (van Wezel 2000) is an ssgA null mutant of M145, created by the insertion of the spectinomycin and streptomycin resistance cassette aadA (21). GSA3 produces aerial hyphae but produces very few spores and only on mannitol-containing media. The introduction of a plasmid carrying ssgA and its promoter region fully restores sporulation (25). Preparation of media, protoplast preparation, and transformations were done according to methods described in reference 15. Soya flour agar medium (SFM) (15) was used to make spore suspensions and to check for the complementation of the GSA3 phenotype by ssgA mutant-containing plasmids. R2YE agar plates were used for regenerating protoplasts and selecting transformants.
TABLE 1.
Bacterial strains, plasmids, and libraries
| Bacterial strain, plasmid, or construct | Relevant genotype or description | Reference |
|---|---|---|
| Strains | ||
| E. coli JM109 | F′ traD36 proA+ proB+ lacIq Δ(lacZ)M15 Δ(lac-proAB) glnV44 gyrA96 recA1 relA1 endA1 thi hsdR17 | 22 |
| E. coli ET12567 | dam dcm hsdM hsdS hsdR cat tet | 18 |
| S. coelicolor M145 | Wild type (SCP1− SCP2−) | 15 |
| S. coelicolor GSA3 | M145 ΔssgA(::aadA) | 28 |
| Plasmids and constructs | ||
| pIJ2925 | Derivative of pUC19 with BglII sites flanking the multiple cloning site | 10 |
| pHJL401 | Shuttle vector with ori for maintenance in E. coli (pUC) and ori for replication in Streptomyces (SCP2*) | 16 |
| pGWS32 | pHJL401 with a 2-kb fragment harboring the ssgR and ssgA gene cluster (including promoter regions) | This study |
| pGWS278 (library) | pIJ2925 with the random mutagenic PCR product of ssgA (reaction with additional dGTP) | This study |
| pGWS279 (library) | pIJ2925 with the random mutagenic PCR product of ssgA (reaction with additional dTTP) | This study |
| pGWS280 (library) | pGWS32 with the wild-type ssgA sequence from the BamHI site replaced with inserts from pGWS278 and pGWS279 | This study |
Creating a library of random mutants.
PCRs were performed in an iCycler thermal cycler (Bio-Rad). Oligonucleotides used in this research are listed in Table 2. PCR-mediated random mutagenesis was performed using Taq polymerase in the buffer supplied, with 5% dimethyl sulfoxide, 200 μM deoxynucleoside triphosphates, and 100 pmol (each) of appropriate oligonucleotides and in the presence of 4.5 mM MgCl2, 0.1 or 0.5 mM MnCl2, and an additional 3.2 mM dATP, dCTP, dGTP, or dTTP to allow occasional errors. A fragment of approximately 540 bp containing the complete ssgA sequence of S. coelicolor (nucleotide positions +1 to +540 relative to the translational start site) was amplified by PCR with primers Q5 and Q6 (Table 2) and Taq DNA polymerase. A large excess of pGWS32 template (around 50 ng) was used to ensure enough yield within 15 cycles. A control PCR was performed (without additional agents) to make a preliminary judgment on the extent of mutations. Samples were separated on a 1% agarose gel in Tris-acetate-EDTA buffer, correct products were gel purified, and the yield was optimized by an additional PCR amplification using proofreading Pfu polymerase (Stratagene, La Jolla, CA) in the buffer supplied.
TABLE 2.
Oligonucleotides used in this studya
| Primer | DNA sequence (5′ to 3′) | Nucleotide location of 5′ end |
|---|---|---|
| Q1 | CTGGAATTCTAGCATCGAGGGCAGGACATCAGA | −1450 |
| Q5 | CTGGAATTCATATGAGCTTTCTCGTGTCCGAGG | +1 |
| Q6 | CTGAAGCTTCACCGCTGCCTTGCTGGCCGGGTC | +540 |
| ssgA W41A | CCGAAGGCCGCGGTCACCGGCGCGTC | +105 |
| ssgA S89A | CGCGGCGGAGGCACGGAACAGCG | +254 |
| ssgA L94A | GAAGGCCACCGCGGGCGCGGCG | +270 |
| ssgA P106A | CCTGCCCCAGCGCCACCAGCTTG | +308 |
Restriction sites used for cloning are presented in bold. Restriction sites: GAATTC, EcoRI; AAGCTT, HindIII; CATATG, NdeI. Underlined triplets are present to introduce codons encoding alanine. The locations of the 5′ ends of oligonucleotides are given relative to the start of ssgA.
Site-directed mutagenesis.
PCR-based site-directed mutagenesis was performed using Q5 as a forward primer and reverse primers containing specific mutations (Table 2) so as to create fragments of ssgA with a specific codon replaced by either GCG or GCC, the most frequently occurring Ala codons in Streptomyces. Subsequently, a second PCR was done with the mutated PCR products as forward primers and primer Q6 as a reverse primer, resulting in a full-length ssgA sequence specifying an SsgA mutant with a single amino acid substitution. By using a strategy similar to that described in “General cloning vectors” below, these mutants with site-directed ssgA mutations were then cloned as BamHI-HindIII-digested fragments into pGWS32 (Table 1) so as to replace the wild-type copy of ssgA. The clones were verified by DNA sequencing.
Plasmids and constructs. (i) General cloning vectors.
pIJ2925 (10) is a pUC19-derived plasmid used for routine subcloning. For cloning in Streptomyces, we used shuttle vector pHJL401 (16), containing an E. coli pUC19 origin of replication (ori) and a Streptomyces SCP2* ori (17) (approximately five copies per chromosome). pIJ2925 and pHJL401 contain an ampicillin resistance marker for selection in E. coli. For Streptomyces transformants, thiostrepton was used for the selection of pHJL401. Plasmid DNA was isolated from ET12567 (Table 1) prior to the transformation of Streptomyces.
(ii) Construction of ssgA clone pGWS32.
For the construction of pGWS32, a DNA fragment of approximately 2 kb was PCR amplified from genomic DNA of S. coelicolor M145 by using primers Q1 to Q6 and was inserted as an EcoRI-HindIII fragment into pHJL401 digested with the same enzymes. The introduction of this plasmid into the ssgA null mutant GSA3 fully restored sporulation (see Results). pGWS32 was used as a basis for the cloning of the plasmid library of ssgA variants.
(iii) Construction of mutant libraries.
Mutations in ssgA of S. coelicolor were introduced by random mutagenic PCR. PCR-generated mutant libraries were cloned as EcoRI-HindIII-digested fragments into EcoRI-HindIII-digested pIJ2925 (Table 1). Subsequently, E. coli TG1 cells were transformed by electroporation. Colonies were grown separately in 96-well MTPs in 200 μl of Luria-Bertani medium overnight. All cultures reached similar optical densities (fully grown) and were therefore expected to contribute approximately equal amounts of plasmid DNA. After replication of the cultures on fresh MTPs containing 200 μl of Luria-Bertani medium with 10% glycerol for storage, the biomasses were pooled and DNA purified in batches. ssgA variants were inserted as BamHI-HindIII fragments into pGWS32 (Table 1) by using the BamHI restriction site present approximately 60 bp downstream of the translational start site of ssgA and the HindIII restriction site from the multiple cloning site. After the transformation of E. coli TG1 cells, colonies were grown separately on 96-well MTPs in 200 μl of Luria-Bertani medium overnight. Cultures were replicated for storage before being pooled and DNA purified in batches. This process resulted in plasmid library pGWS280 (Table 1). Prior to the transformation of Streptomyces, E. coli ET12567 was transformed with the library DNA by electroporation and plated onto 12-cm-square petri dishes, giving around 2 × 104 colonies per transformation, after which cells were harvested and DNA was isolated. GSA3 was transformed with this DNA (Table 1) and plated onto R2YE agar plates; transformants were selected by screening for thiostrepton resistance.
Recovering ssgA variants and DNA sequencing.
To sequence the individual clones of the mutant library, the ssgA gene was amplified by PCR in 96-well PCR plates. As a template, we used 3 μl of frozen mycelial or spore stock, replicated from deep-frozen 96-deep-well plates by using a metal pin replicator (Enzyscreen, Leiden, The Netherlands). Pretreatment of the mycelium and/or spores was not required. All DNA sequencing was performed directly with PCR-amplified DNA fragments by BaseClear BV (Leiden, The Netherlands).
Statistics to determine the cutoff for the analysis of multiple mutants.
Of the 790 sequenced clones, 348 ssgA mutant clones (approximately 44%) failed to complement the sporulation-deficient phenotype of the ssgA mutant. This figure therefore illustrates the chance that any clone in the library would be noncomplementing. This figure was used to get a statistically relevant indication of the number of independent clones required to judge the importance of a certain residue for protein function. If a particular residue was mutated in n independent clones, the approximate probability of this being coincidental becomes P = (0.44)n. To obtain a reliability score above 0.95, a minimum of four independent clones was required.
Computing.
Alignments were done using ClustalW software (24). Figure S1 in the supplemental material was generated using Boxshade software (http://www.ch.embnet.org/software/BOX_form.html). Sequences of SsgA variants were aligned with the wild-type sequence of SsgA by the commercial SeqScape package at BaseClear, allowing the rapid identification of amino acid changes. The data were then introduced into Excel (Microsoft).
RESULTS
Alignment of all known SALPs from streptomycetes.
As of this moment, 24 sequences of SALPs from different streptomycetes are available in the databases. These include seven different SALPs corresponding to the fully sequenced genome of S. coelicolor (designated SsgA to SsgG), seven from S. scabies, and six from S. avermitilis. Additionally, four orthologues of SsgA have been annotated, namely, proteins from S. albus, “S. goldeniensis,” S. griseus, and S. netropsis. Hybridization data suggest that orthologues of SsgA, SsgB, SsgD, SsgE, and SsgG (S. coelicolor nomenclature) occur in most if not all streptomycetes (our unpublished data). Furthermore, several homologues were identified by analyzing the genomes of other sporulating actinomycetes, namely, Thermobifida fusca, Kineococcus radiotolerans, Nocardioides, Acidothermus cellulolyticus, Salinispora tropica, and several Frankia species. Without exception, these non-Streptomyces orthologues share highest sequence homology with members of the SsgB-SsgG branch of the phylogenetic tree (20). Due to the low sequence homology to SsgA, we have not considered these orthologues in this work. An alignment of all Streptomyces SALPs (Fig. S1 in the supplemental material) highlighted several conserved areas, with 16 residues conserved in all 24 sequences (Fig. S1A in the supplemental material).
Creation of a library of ssgA variants.
The strict alignment (100% conservation) (Fig. S1A in the supplemental material) highlighted a number of residues that are conserved among all SALPs. To further address the question of which residues are crucial for SsgA function, we set out to create a mutant library of SsgA.
In order to create mutant variants of S. coelicolor ssgA as a basis for a mutant library, we performed mutagenic PCR in four reactions with mixtures containing an excess of either dATP, dCTP, dGTP, or dTTP and in the presence of 0.1 mM or 0.5 mM MnCl2 (inducing amplification errors). Reaction mixtures containing additional dGTP and dTTP were much more efficient than those with dCTP or dATP (not shown). The PCR products were cloned as EcoR-HindIII-digested fragments into pIJ2925 (Table 1) digested with the same enzymes, and E. coli JM109 was transformed by electroporation. As a test of each transformation, five individual clones were sequenced; this sequencing revealed the desired two to seven nucleotide changes in the region of interest when 0.1 mM MnCl2 was used in the initial PCRs and 15 to 20 nucleotide changes when 0.5 mM MnCl2 was used. The products of the latter reaction were discarded. A total of 1,056 transformants (528 transformants derived from the excess-dGTP PCR and another 528 from the excess-dTTP PCR) representing the maximum number of independent mutants in our library were grown separately in 96-well MTPs overnight. To ensure satisfactory recovery of the different mutants, a large excess (at least fivefold) of this number of colonies was grown in the subsequent cloning steps (Fig. 1). DNA was purified in batches, resulting in plasmid libraries pGWS278 (generated with an excess of dGTP) and pGWS279 (generated with an excess of dTTP) (Table 1).
FIG. 1.

A schematic overview of cloning and propagation in E. coli of the plasmid library of ssgA variants.
To transfer the ssgA variants from pIJ2925 to a vector that allows for expression in Streptomyces, pGWS278 and pGWS279 were digested with BamHI-HindIII by using the BamHI restriction site naturally occurring in ssgA and the HindIII restriction site from the multiple cloning site, generating ssgA fragments corresponding to all residues from amino acid position 25 onwards (amino acids 25 to 135). The fragments were then inserted into pGWS32, a low-copy-number E. coli-Streptomyces shuttle vector that harbors the entire ssgRA gene cluster and its natural promoters (Table 1). Using the BamHI restriction site for cloning replaced the nucleotides encoding amino acids 25 to 135 with the PCR-generated ssgA variants, while the nucleotides encoding the first 24 amino acids remained unchanged (i.e., wild type). After the transformation of E. coli JM109, colonies were again grown separately in 96-well MTPs and DNA was purified in batches, resulting in library pGWS280 (Table 1). A schematic representation of the different cloning steps leading to pGWS280 is given in Fig. 1.
Protoplasts of the ssgA null mutant GSA3 (Table 1) were transformed with pGWS280 and plated on R2YE agar plates. After approximately 3 days, thiostrepton-resistant colonies were individually transferred to solid SFM cultures on 24-well plates. After 7 days, biomass samples (mycelia and, where relevant, spores) from each of the GSA3 transformants were individually prepared. Due to the high number of transformants (around 1,500), the standard procedures to harvest mycelia and spores were not feasible and a new and much more efficient method was required, which was developed based on the following procedure.
A novel method to prepare and maintain Streptomyces libraries.
Spores and/or mycelia were harvested with sterile cotton swabs from the wells of 24-well tissue culture plates in glycerol (20%, wt/vol), and large debris was removed by taking up the suspensions with a syringe through the cotton of the swab. Suspensions were stored separately at −80°C in 96-deep-well MTPs. In this manner, 1,440 samples were produced. By using a spring-loaded replicator (19), individual transformants from deep-frozen MTPs were replicated onto squared SFM plates (12 by 12 cm) for phenotypic screening. When a functional ssgA clone was present in GSA3, the colonies had a typical gray, sporulation phenotype, while colonies of GSA3 with an inactive copy of ssgA remained white (no gray spores); occasionally, an intermediate (light gray) phenotype was observed. In this way, the abilities of individual clones to restore sporulation to the ssgA mutant could be assessed. A typical example is shown in Fig. 2A. The replicator was used to transfer approximately 3 μl from the deep-frozen suspensions directly onto 96-well PCR plates for colony PCR (for details, see “Recovering ssgA variants and DNA sequencing” in Materials and Methods). To optimize the yield, all PCR products were amplified once more using proofreading Pfu DNA polymerase with 2 μl of the initial reaction mixtures as templates. This led to saturated amounts of product for all reactions as confirmed by gel electrophoresis (Fig. 2B). While successful PCRs with Streptomyces biomass have been described previously (26), the authors of the previous report used colonies harboring high-copy-number plasmids (based on pIJ486 [30]). Finally, 790 PCR products were sequenced using the same primers used for amplifications and linked to the phenotypes of their respective transformants.
FIG. 2.
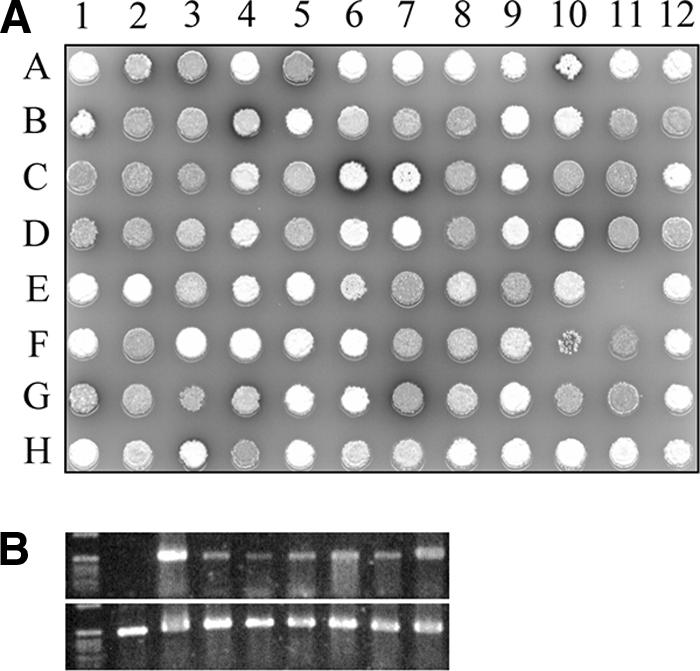
(A) Typical example of an SFM plate containing a replicate of transformants from deep-frozen 96-deep-well MTPs. The colony at position A1 is the control (GSA3 containing pHJL401 without the insert), displaying the white, nonsporulating phenotype characteristic of the ssgA null mutant. All other colonies represent different GSA3 transformants of the library. Several phenotypes are observed: nonsporulating phenotypes (no complementation) such as those in colonies in positions A4 and E1, sporulating (full complementation) phenotypes such as those in colonies in positions A5 and B2, and intermediate phenotypes (partial complementation) such as those in colonies in positions B6 and H2. (B) PCR products for DNA sequencing. An initial PCR was done using a few microliters of deep-frozen suspensions of individual mutants as the template. The amounts of product formed in different reactions were highly variable (upper panel). A second PCR was performed using products of the first PCR as the DNA template, resulting in saturated amounts for all samples (lower panel).
Functional analysis of the ssgA mutants.
Three hundred forty-eight ssgA mutant clones (approximately 44%) failed to complement the sporulation-deficient phenotype, while the remaining 442 clones (approximately 56%) restored sporulation either partially or completely. Of these active clones, 145 clones encoded wild-type SsgA, while 297 encoded a functional mutant SsgA. No wild-type sequences were found among the noncomplementing clones. This is an important observation, as it indicates that invariably the constructs properly expressed SsgA.
A collection of SsgA mutants with single amino acid substitutions.
Out of the 790 analyzed ssgA variants, 180 specified a single amino acid substitution. For 62 out of the 111 amino acids that were encoded by the part of ssgA that was subjected to mutagenesis (residues 25 to 135), we obtained at least one single-amino-acid-substitution mutant (Table 3). For 13 residues we obtained two different substitutions, and for residue 36 we obtained three different ones (D36G, D36N, and D36V). Additionally, we replaced residues W41, S89, L94, and P106 with an alanine residue by site-directed mutagenesis (see Materials and Methods). Of the 66 mutated residues, a total of 22 resulted in an inactive SsgA protein. These loss-of-function mutations (i.e., present in noncomplementing clones) among the single-substitution mutants were mostly found in the central part of the protein (covering residues W41 to L98), with small clusters from residues W41 to G51, residues V74 to L78, and residues A85 to F87. Another small group of loss-of-function mutations was found from residues Y25 to L29. Few loss-of-function mutations were found in the C-terminal part of the protein, with only two substitutions between residues D99 and G135 resulting in nonfunctional SsgA (Table 3; Fig. 3).
TABLE 3.
Single-amino-acid mutations in a collection of clones obtained for the library expressing mutant SsgAa
| Mutation | Sporulation | Mutation | Sporulation | |
|---|---|---|---|---|
| Y25C | Not restored | Q79R | Restored | |
| V27L | Not restored | S82C | Restored | |
| R28C | Restored | A85P | Not restored | |
| L29P | Not restored | L86P | Not restored | |
| L29Q | Not restored | F87S | Not restored | |
| T30A | Restored | S89A* | Restored | |
| T30S | Restored | S90P | Restored | |
| F31L | Restored | A92V | Restored | |
| F31S | Restored | L94A* | Not restored | |
| L33P | Restored | V95A | Restored | |
| D36G | Restored | L98H | Not restored | |
| D36N | Restored | D99E | Restored | |
| D36V | Restored | D99G | Restored | |
| P38L | Restored | T101A | Restored | |
| T40A | Restored | D102N | Restored | |
| W41A* | Not restored | K103E | Restored | |
| G44D | Restored | K103M | Restored | |
| R45L | Not restored | L104P | Not restored | |
| E46G | Restored | L104V | Restored | |
| L47Q | Not restored | V105M | Restored | |
| L48P | Not restored | P106A* | Restored | |
| D50V | Restored | L107P | Restored | |
| G51R | Not restored | G111D | Restored | |
| V52A | Restored | G111S | Restored | |
| G53S | Restored | L113F | Restored | |
| R54Q | Restored | L113P | Restored | |
| C56R | Restored | A114T | Restored | |
| G57S | Not restored | F116L | Restored | |
| R62G | Restored | F116S | Restored | |
| I63T | Not restored | S118G | Restored | |
| P65R | Restored | H119P | Restored | |
| P70Q | Not restored | D121V | Restored | |
| P70L | Restored | L124P | Not restored | |
| L71P | Restored | D125V | Restored | |
| V74A | Not restored | I127T | Restored | |
| V74E | Not restored | L128P | Restored | |
| L75P | Not restored | S133C | Restored | |
| I76T | Not restored | A134E | Restored | |
| R77G | Restored | G135D | Restored | |
| R77Q | Restored | G135S | Restored | |
| L78P | Not restored |
Asterisks indicate point mutations made by site-directed mutagenesis.
FIG. 3.

Amino acid sequence of SsgA. The theoretical secondary structure is shown; barrels indicate predicted α-helices, and arrows indicate predicted β-sheets. Residues not subjected to mutagenesis in this study (and hence invariably wild type) are shown in lower case. Residues 100% conserved (identical or similar) among all 24 known Streptomyces SALPs (see the text for further explanation) are underlined. Identical amino acids are further highlighted in bold. Bullets above the sequence indicate single amino acid substitutions causing loss of function (Table 3). Asterisks above the sequence indicate amino acids which obtained an importance score of 80% or higher from this study (see Results and Fig. 4). Amino acid numbering is shown below the primary sequence (numbers correspond to the amino acid above the first digit in each number).
Analysis of the multiple mutants.
The majority of the SsgA variants specified multiple mutations. In this manner, we obtained mutants corresponding to mutations in nearly all the residues subjected to mutagenesis, albeit in combination with one or more other mutations. To allow us to also distill information from the multiple mutants on the importance of individual amino acids, we used the following approach. The deduced amino acid sequences of all 790 clones were aligned, and the total number of times a certain residue was modified, alone or in combination with other mutations, was counted. For each residue, the fraction that occurred in clones unable to restore sporulation was determined, resulting in a so-called importance score (Fig. 4). As an example, residue D83 was replaced in 12 clones, five of which failed to complement GSA3; hence the importance score was 42%. The residues with the highest importance scores (80% or higher) are highlighted in Fig. 3. To ensure an accuracy of 95% or more (P > 0.95), residues that were found to be mutated in fewer than four clones were considered below statistical reliability (see “Statistics to determine the cutoff for the analysis of multiple mutants” in Materials and Methods). While the initial cutoff was set at four mutations, of all the residues with a high importance score (>80%), none were found in four independent clones; two were found to be mutated in five different clones (P of 0.99) and all others were found to be mutated in six or more independent clones.
FIG. 4.

Graph showing the importance scores (gray bars) and specificity scores (black bars) in percentages on the y axis. The amino acid sequence of SsgA is shown along the x axis. The SsgA importance score represents the frequency at which a certain mutation occurs in a nonfunctional ssgA clone, where 100% would indicate that an amino acid is essential for SsgA function. By dividing the importance score by the conservation value, amino acids are identified as primarily important for the function of SsgA and less important for the other SALPs. A score of 100% would indicate that an amino acid is essential for SsgA function and unique for the SsgA protein. Data sets below the statistical threshold (specific residues mutated in fewer than four clones) were not included in the analysis.
Many of the important residues were clustered. The two most noticeable clusters were residues 39 to 49 (VTWAFGRELL) and residues 55 to 61 (PCGDGDV). Another relatively clustered group of important residues covered a somewhat larger section, namely, residues V74 to L98. Outside of these three regions, the effect of amino acid substitutions was significantly less obvious (Fig. 3). These regions all showed high conservation among all SALPs and therefore most likely relate to a general function, such as target binding. The overall amino acid identity of the SALPs from S. coelicolor is between 30 and 50% (7).
An important question is, “Can we identify residues that are important for SsgA but not conserved among the other SALPs?” Such residues may be involved in a unique aspect of SsgA function. For this purpose, we calculated a specificity score (Fig. 4). We divided the importance score obtained from our library by the degree of conservation (Fig. 4), expressed as the occurrence of a particular residue in SsgA in all other SALPs of S. coelicolor, with a conservation value of 1 if a residue was unique for SsgA and a value of 7 if a residue occurred in all seven S. coelicolor SALPs. In the example described above, we calculated an importance score of 42% for residue D83. This residue occurred in two SALPs, namely, SsgA and SsgF, and hence the specificity score was 21%. Among the 111 possible candidates, residues L29, D58, and S89 obtained high specificity scores, i.e., they were found to be important for SsgA function, but they were not significantly conserved among the other SALPs. Expectedly, L29 and D58 were well conserved among all SsgA orthologues (L29 was replaced with methionine in S. griseus and S. netropsis). However, S89 was less well conserved (it was replaced with valine in S. albus, S. goldeniensis, and S. scabies and alanine in S. griseus and S. netropsis).
DISCUSSION
So far, little structural information on the SsgA protein and its relatives (SALPs) is available. The SALPs do not share significant sequence homology to any known protein family, and even structural characteristics (protein motifs) could not be identified. However, the proteins play a major role in the control of development (20) and have been used to efficiently improve growth and product formation (27). Learning more about the intrinsic properties of the proteins is the next logical step in understanding the way the proteins function. For this purpose, we designed a new mutational analysis strategy to identify the residues that are most important for the function of SsgA. One of the main bottlenecks of working with streptomycetes is the lack of a good system for batch handling of many individual Streptomyces colonies, mainly due to the fact that cultivation in microtiter plates and cloning of DNA directly from colonies are very difficult when working with filamentous, obligate-anaerobe bacteria.
Producing a mutant library in streptomycetes with high efficiency.
In this work, we detail the creation of a plasmid library of random S. coelicolor ssgA mutants and discuss a new method to efficiently maintain the colonies and screen the library. The PCR-based random mutant library resulted in a maximum of more than 1,000 different E. coli clones, a figure that represents the maximum number of different mutant clones. Since the standard procedure of harvesting mycelia and spores is time-consuming (and thus costly), the daunting task of producing a library of thousands of transformants required a novel approach. We were able to produce 1,440 individual transformants, stored as spore and/or mycelium suspensions in 96-deep-well MTPs at −80°C, in a short time. By using a spring-loaded replicator (19), these suspensions were efficiently replicated onto SFM plates to analyze the phenotypes (sporulating or nonsporulating) of transformants of the ssgA null mutant GSA3 harboring ssgA mutant clones (Table 1). The same replicator was used to transfer a few microliters of deep-frozen samples into PCR mixtures. In this way, a total of 790 ssgA variants were amplified and sequenced while steps involving DNA purification or pretreatment of the mycelium were eliminated.
Amino acid clusters important for SsgA function.
A comparison of the data obtained from the single-substitution mutants and the data obtained from the complete library shows that similar results were found. However, while mutants with the single-amino-acid mutations Y25C, G51R, I63T, P70Q, L75P, A85P, and L104P were all inactive, the respective residues were not highlighted by the analysis of multiple mutants. This apparent discrepancy is at least in part explained by the lack of sufficient data for some residues. For instance, the single mutation G51R failed to complement the ssgA null mutant but since G51 was mutated in fewer than four clones it was excluded from the analysis. Alternatively, the differences can be explained by the nature of particular amino acid changes; SsgA with P70Q was inactive, while SsgA with P70V was active and the corresponding clone restored sporulation to an ssgA mutant.
We identified several residues that are required for proper SsgA function, or at least for its ability to activate sporulation-specific cell division. A combination of all data highlights three particularly interesting regions, namely, clusters of residues V39 to G51 and residues P55 to I63 and a larger region covering residues V74 to L98. These regions are of great interest as they possibly represent one or more motifs essential for the function of SsgA. The penalty for mutations in the C-terminal third of the protein is much lower, highlighted by the fact that only two of the single mutations in this region inactivated SsgA, both of which involved a leucine residue changed into a much more bulky proline residue (L104P and L124P).
Most of the important residues considered show high levels of conservation among all SALPs. Therefore, we compared SsgA to the other six SALPs of S. coelicolor to try and identify residues that are important for the function of SsgA but not conserved among the other SALPs. Thorough analysis revealed that particularly residues L29, D58, and S89 stood out as candidates. Of these, L29 and D58 were particularly highly conserved among the SsgA orthologues of different organisms, while S89 was less well conserved. For residue L29, both the L29P and L29Q single mutations failed to restore sporulation to GSA3. For residues D58 and S89, no single mutants were obtained, but an S89A substitution introduced by site-directed mutagenesis did result in a functional protein, restoring sporulation to GSA3. Hence, out of all the residues identified as crucial for SsgA function in sporulation, only two residues are of particular importance for the function of only SsgA itself.
In conclusion, we designed a new method to efficiently create, maintain, and screen mutant libraries for the industrially important streptomycetes. The technology was successfully applied to create a diverse mutant library of SsgA, with half of all SsgA residues mutated in single mutants, and a large collection of multiple mutants. Obviously, the full potential of this library will be unlocked once the three-dimensional structure of the SsgA protein is available, which is a prerequisite to learning more on the structure-function relationship. This insight will better our understanding of the role of SsgA in the control of the growth and development of streptomycetes and will provide new means to exploit SsgA in strain development to further improve streptomycetes as industrial production hosts.
Supplementary Material
Acknowledgments
We are grateful to Wouter Duetz and to Bas Reichert for discussions.
This work was supported by a grant from the Royal Academy for Arts and Sciences to G.P.V.W.
Footnotes
Published ahead of print on 9 February 2007.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Bennett, J. W. 1998. Mycotechnology: the role of fungi in biotechnology. J. Biotechnol. 66:101-107. [DOI] [PubMed] [Google Scholar]
- 2.Bentley, S. D., K. F. Chater, A. M. Cerdeno-Tarraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C. H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M. A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill, and D. A. Hopwood. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141-147. [DOI] [PubMed] [Google Scholar]
- 3.Bushell, M. E. 1988. Actinomycetes in biotechnology, p. 185-217. Academic Press, London, United Kingdom.
- 4.Chater, K. F. 2001. Regulation of sporulation in Streptomyces coelicolor A3(2): a checkpoint multiplex? Curr. Opin. Microbiol. 4:667-673. [DOI] [PubMed] [Google Scholar]
- 5.Chater, K. F. 1998. Taking a genetic scalpel to the streptomyces colony. Microbiology 144:1465-1478. [DOI] [PubMed] [Google Scholar]
- 6.Demain, A. L. 1991. Production of beta-lactam antibiotics and its regulation. Proc. Natl. Sci. Counc. Repub. China B 15:251-265. [PubMed] [Google Scholar]
- 7.Flärdh, K., and G. P. van Wezel. 2003. Cell division during growth and development of Streptomyces, p. 71-90. In S. G. Pandalai (ed.), Recent developments in bacteriology. Transworld Research Network, Trivandrum, India.
- 8.Hopwood, D. A. 1999. Forty years of genetics with Streptomyces: from in vivo through in vitro to in silico. Microbiology 145:2183-2202. [DOI] [PubMed] [Google Scholar]
- 9.Hopwood, D. A., K. F. Chater, and M. J. Bibb. 1995. Genetics of antibiotic production in Streptomyces coelicolor A3(2), a model streptomycete. Biotechnology 28:65-102. [DOI] [PubMed] [Google Scholar]
- 10.Janssen, G. R., and M. J. Bibb. 1993. Derivatives of pUC18 that have BglII sites flanking a modified multiple cloning site and that retain the ability to identify recombinant clones by visual screening of Escherichia coli colonies. Gene 124:133-134. [DOI] [PubMed] [Google Scholar]
- 11.Jiang, H., and K. E. Kendrick. 2000. Characterization of ssfR and ssgA, two genes involved in sporulation of Streptomyces griseus. J. Bacteriol. 182:5521-5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawamoto, S., H. Watanabe, A. Hesketh, J. C. Ensign, and K. Ochi. 1997. Expression analysis of the ssgA gene product, associated with sporulation and cell division in Streptomyces griseus. Microbiology 143:1077-1086. [DOI] [PubMed] [Google Scholar]
- 13.Keijser, B. J., E. E. Noens, B. Kraal, H. K. Koerten, and G. P. van Wezel. 2003. The Streptomyces coelicolor ssgB gene is required for early stages of sporulation. FEMS Microbiol. Lett. 225:59-67. [DOI] [PubMed] [Google Scholar]
- 14.Kendrick, K. E., and J. C. Ensign. 1983. Sporulation of Streptomyces griseus in submerged culture. J. Bacteriol. 155:357-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kieser, T., M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom.
- 16.Larson, J. L., and C. L. Hershberger. 1986. The minimal replicon of a streptomycete plasmid produces an ultrahigh level of plasmid DNA. Plasmid 15:199-209. [DOI] [PubMed] [Google Scholar]
- 17.Lydiate, D. J., F. Malpartida, and D. A. Hopwood. 1985. The Streptomyces plasmid SCP2*: its functional analysis and development into useful cloning vectors. Gene 35:223-235. [DOI] [PubMed] [Google Scholar]
- 18.MacNeil, D. J., K. M. Gewain, C. L. Ruby, G. Dezeny, P. H. Gibbons, and T. MacNeil. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61-68. [DOI] [PubMed] [Google Scholar]
- 19.Minas, W., J. E. Bailey, and W. Duetz. 2000. Streptomycetes in micro-cultures: growth, production of secondary metabolites, and storage and retrieval in the 96-well format. Antonie Leeuwenhoek 78:297-305. [DOI] [PubMed] [Google Scholar]
- 20.Noens, E. E., V. Mersinias, B. A. Traag, C. P. Smith, H. K. Koerten, and G. P. van Wezel. 2005. SsgA-like proteins determine the fate of peptidoglycan during sporulation of Streptomyces coelicolor. Mol. Microbiol. 58:929-944. [DOI] [PubMed] [Google Scholar]
- 21.Prentki, P., and H. M. Krisch. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303-313. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Soliveri, J., A. K. Scheu, A. Hernandez, J. L. Copa-Patino, and K. F. Chater. 1999. Faster recombinant DNA procedures for Streptomyces. Bio Techniques 26:394-396. [DOI] [PubMed] [Google Scholar]
- 24.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Traag, B. A., G. H. Kelemen, and G. P. Van Wezel. 2004. Transcription of the sporulation gene ssgA is activated by the IclR-type regulator SsgR in a whi-independent manner in Streptomyces coelicolor A3(2). Mol. Microbiol. 53:985-1000. [DOI] [PubMed] [Google Scholar]
- 26.Van Dessel, W., L. Van Mellaert, N. Geukens, and J. Anne. 2003. Improved PCR-based method for the direct screening of Streptomyces transformants. J. Microbiol. Methods 53:401-403. [DOI] [PubMed] [Google Scholar]
- 27.van Wezel, G. P., P. Krabben, B. A. Traag, B. J. Keijser, R. Kerste, E. Vijgenboom, J. J. Heijnen, and B. Kraal. 2006. Unlocking Streptomyces spp. for use as sustainable industrial production platforms by morphological engineering. Appl. Environ. Microbiol. 72:5283-5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Wezel, G. P., J. van der Meulen, S. Kawamoto, R. G. M. Luiten, H. K. Koerten, and B. Kraal. 2000. ssgA is essential for sporulation of Streptomyces coelicolor A3(2) and affects hyphal development by stimulating septum formation. J. Bacteriol. 182:5653-5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Wezel, G. P., and E. Vijgenboom. 2004. Novel aspects of signalling in Streptomyces development. Adv. Appl. Microbiol. 56:65-88. [DOI] [PubMed] [Google Scholar]
- 30.Ward, J. M., G. R. Janssen, T. Kieser, M. J. Bibb, and M. J. Buttner. 1986. Construction and characterisation of a series of multicopy promoter-probe plasmid vectors for Streptomyces using the aminoglycoside phosphotransferase gene from Tn5 as indicator. Mol. Gen. Genet 203:468-478. [DOI] [PubMed] [Google Scholar]
- 31.Wardell, J. N., S. M. Stocks, C. R. Thomas, and M. E. Bushell. 2002. Decreasing the hyphal branching rate of Saccharopolyspora erythraea NRRL 2338 leads to increased resistance to breakage and increased antibiotic production. Biotechnol. Bioeng. 78:141-146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.