

Abstract
Saccharomyces cerevisiae L2612 transformed with genes for xylose reductase and xylitol dehydrogenase (XYL1 and XYL2) grows well on glucose but very poorly on d-xylose. When a gene for d-xylulokinase (XYL3 or XKS1) is overexpressed, growth on glucose is unaffected, but growth on xylose is blocked. Spontaneous or chemically induced mutants of this engineered yeast that would grow on xylose could, however, be obtained. We therefore used insertional transposon mutagenesis to identify two loci that can relieve this xylose-specific growth inhibition. One is within the open reading frame (ORF) of PHO13, and the other is approximately 500 bp upstream from the TAL1 ORF. Deletion of PHO13 or overexpression of TAL1 resulted in a phenotype similar to the insertional mutation events. Quantitative PCR showed that deletion of PHO13 increased transcripts for TAL1, indicating that the growth inhibition imposed by the overexpression of XYL3 on xylose can be relieved by an overexpression of transcripts for downstream enzymes. These results may be useful in constructing better xylose-fermenting S. cerevisiae strains.
Saccharomyces cerevisiae affords several advantages over the use of other yeasts for ethanol fermentations. It is ethanol tolerant, knowledge of its genetics and physiology is highly developed, and it is generally regarded as being safe. Native S. cerevisiae strains, however, are unable to grow on d-xylose. Attempts to develop a strain of S. cerevisiae capable of using xylose have focused on adapting the xylose metabolic pathway from xylose-utilizing yeasts such as Pichia stipitis or on the expression of xylose isomerase (16).
In P. stipitis, conversion of xylose to xylulose is catalyzed by two oxidoreductases. Xylose is reduced to xylitol by an NAD[P]H+-linked xylose reductase (XR or P. stipitis XYL1 [PsXYL1]), and xylitol is oxidized to xylulose by an NAD+-linked xylitol dehydrogenase (XD or PsXYL2). Finally, d-xylulokinase (XK or PsXYL3) phosphorylates d-xylulose to form d-xyluose-5-phosphate (11), which is metabolized further via the pentose phosphate pathway and glycolysis (9).
Native S. cerevisiae strains encode homologs of PsXYL1 and PsXYL2, but they do not express sufficient xylose reductase or xylitol dehydrogenase activity to enable significant growth on xylose. S. cerevisiae encodes a different xylulokinase (XKS1) (4, 18), which is expressed at a low level and which enables S. cerevisiae to grow on and ferment xylulose. Recombinant S. cerevisiae expressing PsXYL1 and PsXYL2 was found to grow on xylose (18). Ethanol production from xylose, however, was not significant because a substantial portion of xylose was converted to xylitol (13, 15, 20).
Early reports of xylose fermentation by S. cerevisiae indicated that the overexpression of xylulokinase (XKS1) is essential to growth and fermentation. Ho et al. previously reported that the overexpression of the endogenous S. cerevisiae xylulose kinase gene (XKS1) (ScXKS1) along with PsXYL1 and PsXYL2 increased ethanol production and decreased xylitol production from xylose (7). Recombinant S. cerevisiae cells transformed with a single copy of PsXYL1 and multiple copies of PsXYL2 accumulate xylulose (10, 11), which suggested that the native level of XK activity in S. cerevisiae limits xylose assimilation when PsXYL1 or PsXYL2 is overexpressed.
Rodriguez-Pena et al. showed, however, that the overexpression of XKS1 in S. cerevisiae inhibits growth on pure d-xylulose (18). Similarly, overexpressing PsXYL3 along with high levels of PsXYL1 and PsXYL2 in S. cerevisiae completely inhibited cell growth on xylose, whereas an S. cerevisiae transformant expressing PsXYL3 at a moderate level was able to grow on xylose (13a). Johansson et al. previously found that the overexpression of ScXKS1 reduced xylose consumption by 50 to 80% in S. cerevisiae transformants, even though it increased the yield of ethanol from xylose, and those researchers cautioned against the unmodulated overexpression of ScXKS1 (14). Other studies by Toivari et al. (21) and Richard et al. (17) did not show an inhibitory effect of xylulose kinase overexpression. Those reports seemed contradictory, but studies in our laboratory suggested that spontaneous mutations might play a role in overcoming growth inhibition on xylose. The present research sought to characterize mutational events in genes that could contribute to the growth of and fermentation by engineered strains on xylose.
MATERIALS AND METHODS
Microbial strains, primers, and plasmids.
S. cerevisiae L2612 (MATα leu2-3 leu2-112 ura3-52 trp1-298 can1 cyn1 gal+) was the parental strain. S. cerevisiae YS1020 (MATα leu2::LEU2-TDH3P-PsXYL1-TDH3T ura3::URA3-TDH3P-PsXYL2-TDH3T trp1-298 can1 cyn1 gal+) was the recipient host for control experiments and the parental strain for transposon mutagenesis. S. cerevisiae YSX3 (MATα leu2::LEU2-TDH3P-PsXYL1-TDH3T ura3::URA3-TDHP-PsXYL2-TDH3T Ty3::G418R-PsXYL3 trp1-298 can1 cyn1 gal+) was developed in a previous study (13a). Escherichia coli DH5α was used for plasmid preparations. The primers and plasmids used in this study are listed in Tables 1 and 2.
TABLE 1.
Oligonucleotides used in this study
| Oligonucleotide | Sequence | Use |
|---|---|---|
| LacZ | TGTGCTGCAAGGCGATTAAG | Sequence insertion site from mTn |
| LEU2-S20C | GAACACACATGAACAAGGAAG | PCR from LEU2 promoter to upstream |
| PHO13 Nl | GGATCCTGGTGGAAACTATTTCTCG | PCR from PHO13 promoter to downstream |
| PHO13 C2 | CCAACAAAGTGAACAGAATCC | PCR from middle of PHO13 ORF to upstream |
TABLE 2.
Plasmids used in this study
| Plasmid | Function | Expression element(s) |
|---|---|---|
| pRS314 TRPI | Centromere | None (starting vector) |
| pRS424 TRPI | 2μm origin | None (tarting vector) |
| pYPR2831 TRPI | 2μm origin | Control vector with promoter/terminator TDH3P-TDH3T |
| pYS32 TRP1 | 2μm origin | TDH3P-PsXYL3-TDH3T |
| pRS314-X123 | TRPI, centromere | TDH3P-PsXYL1-TDH3T TDH3P-PsXYL2-TDH3T TDH3P-PsXYL3-TDH3T |
| pRS314-X123-T2 | TRP1, centromere | TDH3P-PsXYL1-TDH3T TDH3P-PsXYL2-TDH3T TDH3P-PsXYL3-TDH3T TDH3P-ScTAL1-CYC1T TDH3P-ScTKLl-CYC1T |
| pRS424-X123 | TRP1, 2μm origin | TDH3P-PsXYLI-TDH3T TDH3P- |
| pYES2 | URA3, 2μm origin | PsXYL2-TDH3T TDH3P- |
| PsXYL3-TDH3T | ||
| pYES2-X123 | URA3, 2μm origin | TDH3P-PsXYL1-TDH3T TDH3P-PsXYL2-TDH3T TDH3P-PsXYL3-TDH3T |
| pRSQ2-URA3 | Integration vector | None (starting vector) |
| pRS314-312tal | TRP1, centromere | ScTAL1 with upstream insertion from Mut312 |
| pRS314-512tal | Trp1, centromere | ScTAL1 with upstream insertion from Mut512 |
| pRS314-ADHtal | TRP1, Centromere | ADH1P-ScTAL1-CYC1T |
| pBlue-DR PHO13 | pBluescript | PHO13P-LEU2-PHO13T |
Media.
Yeast strains without plasmids were grown on YPD medium (20 g/liter yeast extract, 10 g/liter peptone, and 20 g/liter glucose). Strains with plasmids carrying TRP1 and/or URA3 markers were grown on yeast synthetic medium, which was made of yeast nitrogen base (YNB) and Casamino Acids (10 g/liter), supplemented with glucose (20 g/liter) (YNBG) or xylose (20 g/liter for growth test and 40 g/liter for fermentation) (YNBX). Complete synthetic mix (CSM) without certain amino acids and nucleotides was used to select other auxotrophic markers.
Overexpression of PsXYL3 in S. cerevisiae YSX3.
pYS32 was transformed into YSX3 using the lithium acetate-heat shock method. The transformants were selected on yeast synthetic medium with dextrose (YSD).
Insertional mutagenesis.
The mTn-lacZ/LEU2-mutagenized library (3, 19) was obtained from the Yale Genome Analysis Center, New Haven, CT (http://ygac.med.yale.edu/mtn/reagent/avail_reagents/lacZ_LEU2_lib_p.stm). Plasmid DNA from the pools of the mTn inserted genomic library was digested with NotI to release the mutated insertional library of S. cerevisiae genes from the bacterial vector. The digested DNA was purified with a GENECLEAN kit from Qbiogene (Carlsbad, CA). The strain for mutagenesis, S. cerevisiae L2612(pYES2-X123), was grown on YSD with Trp and Leu to an optical density at 600 nm of between 1 and 2. Cells grew faster in this medium than in YNBG with CSM and without Ura. Either medium could prevent the plasmid with URA3 from being lost. Digested library DNA was transformed by following the lithium acetate-heat shock protocol described previously by Gietz and Woods (6) After transformation, cells were incubated in YSD with Trp for one doubling (ca. 4 h). They were then harvested, washed, and resuspended in water. A portion (1%) of the cell suspension was plated onto YNBG with CSM and without Leu and Trp to count the number of total transformants. The rest was plated onto YNBX with CSM and without Leu and Trp to screen for mutants that would grow on the xylose plates (Fig. 1).
FIG. 1.
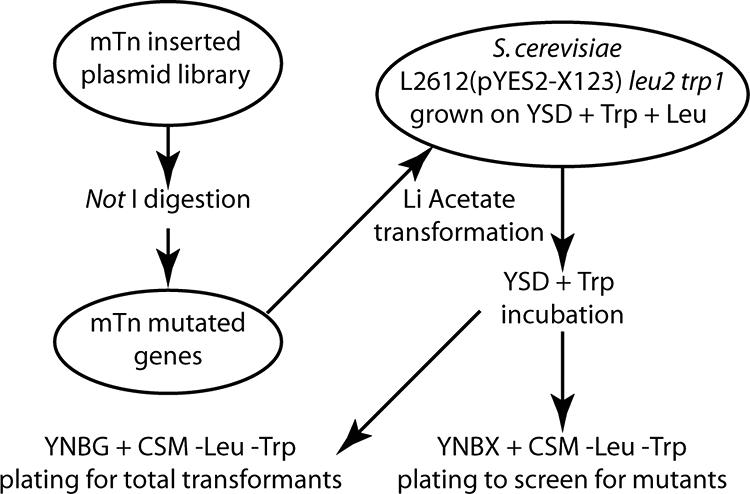
Protocol for insertional mutagenesis of S. cerevisiae bearing genes that inhibit growth on xylose.
Identification of insertion site.
Yeasts were cured of pYES2-X123 by plating the mutants onto YPD plus 0.1% 5-fluoroorotic acid. The resulting colonies were plated onto YSD with Trp to make sure that they were Ura−. Vector pRSQ2-URA3 was linearized with BamHI to leave a portion of lacZ at either end. The digested pRSQ2-URA3 was purified with the GENECLEAN kit and transformed into the URA3-cured mutants by lithium acetate-heat shock, whereupon it integrated into the transposon-borne lacZ sequence. Total DNA was recovered from the transformants using lyticase, proteinase K, sodium dodecyl sulfate, RNase A, and added salt to 0.2 mol/liter and 2 volumes of cold absolute ethanol to precipitate DNA.
For plasmid rescue, 5 μg of transformed yeast DNA was digested with a restriction enzyme that cut once in the polylinker of pRSQ2-URA3 (ClaI, EcoRI, EcoRV, HindIII, KpnI, PstI, SalI, or XhoI) and at some other point in the flanking yeast DNA. This released a linear fragment that contained the bacterial replication origin, the β-lactamase gene, a portion of lacZ, and some adjacent yeast DNA. The digestion mixture was heated to inactivate the restriction enzyme using conditions recommended in the New England Biolabs catalog. The mixture was then cooled on ice and purified by ethanol precipitation. The DNA pellet was dissolved in water and supplemented with T4 DNA ligase buffer. T4 DNA ligase (2 units/μl) was added. To favor intramolecular ligation, the final DNA concentration was kept low, in the range of 2 to 10 μg/ml. After ligation at 16°C overnight, DNA was purified by ethanol precipitation. The pellet was dissolved in 10 μl water. One-half (5 μl) was used to transform E. coli DH5α by electroporation. The transformants were selected for ampicillin resistance. DNA from plasmid minipreps was analyzed by double digestion with BamHI and the rescue enzyme, which could release a piece of pRSQ2-URA3. The plasmids that appeared to have appropriate inserts were sequenced using a primer from the lacZ sequence in pRSQ2-URA3. The sequences, each containing a part of lacZ and adjacent yeast DNA, were then obtained. If plasmids could not be properly rescued, another rescue enzyme was tried because the nearest restriction site in the yeast DNA was probably too far from the insertion site.
After obtaining the sequence, we identified it by using the BLAST algorithm available at the NCBI website (http://www.ncbi.nlm.nih.gov:80/BLAST/).
Confirmation of transposition event.
To confirm that the insertion rather than a spontaneous mutation enabled growth on xylose, we transferred the transposon insertion along with the genomic flanking regions back to the parental strain to see if it resulted in the same phenotype.
To rescue the whole insertion site from the genomic DNA preparation, a restriction enzyme that could cut neither mTn-lacZ/LEU2 nor pRSQ2-URA3 but that would cut on both sides near the insertion site in the yeast DNA was selected. The resulting large plasmid was recovered by transformation into E. coli that was then digested with this rescue enzyme and transformed into L2612. Transformants were selected on plates of YNBG with CSM and without Leu and Ura. We picked four transformants to confirm each mutation and transformed each one with pRS424-X123. The resulting strains were selected on YSD plates, and their potential for growth on YSX plates was observed.
Subcloning the mutant TAL1 into a plasmid.
For mutants Mut312 and Mut512, which contain mTn inserted upstream of TAL1, PstI was used to rescue DNA fragments containing the insertion sites. The resulting plasmids were named pRSQ312 and pRSQ512, respectively. They were each digested with DraI (cut in the amp open reading frame [ORF]) and EcoRV (cut in the pRSQ2-URA3 polylinker) to release a fragment that contained the N terminus of amp, the full LacZ ORF from pRSQ2-URA3, the promoter (after the insertion), and the ORF and terminator (400 bp to the PstI site) of TAL1. The fragment was subcloned into the SmaI site of pRS314. The resulting plasmids were named pRS314-312tal and pRS314-512tal.
pho13 knockout.
The plasmid used for the PHO13 knockout was subcloned in three steps. First, LEU2 was cut out with AccI and DraIII from pRS315 and inserted into the SmaI site of pBluescript (pBlue-LEU2). Second, a fragment containing the PHO13 ORF C-terminal 180 bp and the 250-bp terminator was inserted between the PstI and XhoI sites of pBlue-LEU2 by blunt-end ligation (pBlue-LEU2-PHO13 C). Finally, an XhoI fragment, which was obtained from positions −60 to −1680 upstream of PHO13, was inserted between SacI and SpeI sites of pBlue-LEU2-PHO13 C by blunt-end ligation (pBlue-DR PHO13). In each step, the orientation of the insert was determined by restriction analysis, and the correct orientation was selected. In pBlue-DR PHO13, PHO13 upstream, LEU2, and PHO13 C terminus and terminator were all oriented in the same direction.
pBlue-DR PHO13 was digested with HindIII and ScaI to release a fragment containing PHO13 upstream of −1220 to −60 bp, LEU, and the 180-bp C terminus of the PHO13 ORF plus 250 bp of the terminator. The digested DNA was transformed into L2612, and the transformants were selected on plates of YNBG with CSM and without Leu.
Genomic DNA was prepared from several transformants. The correct knockout mutants were confirmed with PCR using primers between the PHO13 promoter and the LEU2 promoter and primers between the PHO13 promoter and the PHO13 ORF, which gave a PCR product of the expected size and no products, respectively. The correct strain was named S. cerevisiae DR PHO13 (MATa pho13::LEU2 ura3-52 trp1-298 can1 cyn1 gal+).
qPCR.
Quantitative PCR (qPCR) was performed as previously described (12). Cells from four independent cultures were used for each condition. mRNA was extracted according to methods described previously by Holstege et al. (8). cDNA was constructed using random oligonucleotides and the Reverse Transcription System kit (Promega). Reverse transcription-PCR analyses of the samples were done with SYBR green PCR master mix (Applied Biosystems) as recommended by the manufacturer, except that 15 pmol of oligonucleotides and a final volume of 25 μl per reaction were used. Actin was used to normalize for the mRNA concentration. Except where noted, all data points were collected in triplicate. The Student t test was used to determine if differences in the values were statistically significant.
Xylose fermentation.
Yeast strains were grown in YSD medium with Ura. Cells were harvested, washed with water, and inoculated into duplicate 125-ml flasks each containing 50 ml YSX with Ura (45 g/liter xylose). The shaking speed was 200 rpm. The xylose and xylitol concentrations in fermentation samples were determined by high-performance liquid chromatography, while the ethanol concentrations were determined by gas chromatography.
Enzymes, primers, and chemicals.
Restriction enzymes, DNA-modifying enzymes, and other molecular reagents were obtained from New England Biolabs (Beverly, MA), Promega (Madison, WI), Stratagene (La Jolla, CA), or Roche Biochemical (Indianapolis, IN). Reaction conditions employed were recommended by the suppliers. All general chemicals were purchased from Sigma (St. Louis, MO). Sigma-Genosys (The Woodlands, TX) and Invitrogen (Carlsbad, CA) synthesized the primers for PCR and sequencing.
Nucleotide sequence accession numbers.
Mutant strains designated DR PHO13 [S. cerevisiae L2612(pho13Δ)], GX312, and GX512 were deposited with the Agricultural Research Service Culture Collection in Peoria, IL, on 2 September 2 2004 under the Budapest Treaty and assigned accession numbers NRRL Y-30771, NRRL Y-30770, and NRRL Y-30769, respectively.
RESULTS
Toxicity of PsXYL3 overexpression can be relieved by spontaneous mutation.
S. cerevisiae YS1020 was constructed by integrating PsXYL1 and PsXYL2 under the strong ScGAPDH promoter TDH3P. PsXYL3 with its native promoter was randomly integrated into the YS1020 chromosome. S. cerevisiae YSX3 was selected from the resulting strains for its fast growth on, and ethanol production from, xylose. Our previous study showed that the overexpression of PsXYL3 in YS1020 inhibited growth and ethanol production under these conditions (13a), a finding that was confirmed in subsequent trials. Growth was almost completely inhibited on xylose agar plates but would occur slowly in xylose broth. Because xylulokinase is an essential enzyme in xylose metabolism, we concluded that moderate xylulokinase levels made YSX3 a good xylose-fermenting strain. To test this conclusion, we overexpressed PsXYL3 by plasmid transformation in the YSX3 background. Surprisingly, the transformant, YSX3(pYS32), could grow on xylose. In fact, growth in xylose broth was somewhat better than that of YSX3 carrying the empty control vector, pYPR2831 (Fig. 2). Consistent with our previous findings, YSX3 transformed with either pYS32 or the control vector grew better than YS1020(pYS32). The latter expresses xylulokinase at a high level and barely grows on xylose plates. The YS1020(pYS32) strain was plated onto xylose, and a few colonies (ca. 1 per 106 CFU) capable of more rapid growth on xylose plates were obtained. One of these colonies, which we assumed to be a spontaneous mutant, was designated “Ef.” This mutant grew in a manner similar to that of YSX3 transformed with the control vector. Fermentation data showed that YSX3 transformed with either pYS32 or the control vector consumed xylose quickly. We did not observe significant ethanol production with YSX3(pYS32), and there was even less ethanol from xylose than the amount produced by YSX3(pYPR2831); however, YSX3(pYS32) grew slightly faster, and converted xylose into 15% more cell mass (Fig. 2) and xylitol (Fig. 3A), than did YSX3(pYPR2831).
FIG. 2.

Growth of four engineered S. cerevisiae strains on d-xylose. Cells were cultivated under aerobic conditions on 4.5% xylose in synthetic medium. ○, S. cerevisiae YSX3(pYS32); •, S. cerevisiae YSX3(pYPR2831); ▴, S. cerevisiae YS1020(pYS32); ▪, S. cerevisiae YS1020(pYS32) Ef.
FIG. 3.
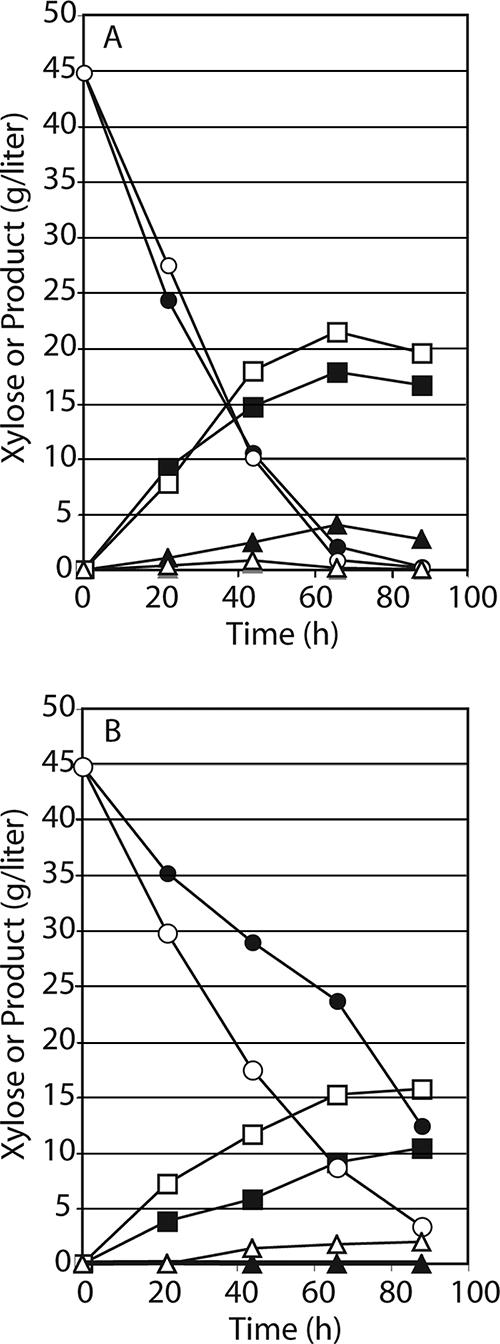
Product formation by four engineered S. cerevisiae strains on d-xylose. ○ and •, xylose; ▪ and □, xylitol; ▴ and ▵, ethanol. (A) Closed symbols, S. cerevisiae YSX3(pYPR2831); open symbols, S. cerevisiae YSX3(pYS32). (B) Closed symbols, S. cerevisiae YS1020(pYS32); open symbols, S. cerevisiae YS1020(pYS32) Ef.
Identification of genetic changes that are responsible for relieving the toxicity of XYL3.
To identify potential mutational events that could be contributing to the improved growth of engineered S. cerevisiae on xylose, S. cerevisiae L2612(pYES2-X123), which barely grows on xylose plates, was used as the parental strain for transposon mutagenesis. The parental S. cerevisiae L2612, which had never been exposed to growth on xylose, was transformed with the pYES2-X123 cassette. After transforming this host with the mTn inserted genomic library, we selected mutants that grew on xylose (Fig. 1). The insertion sites in these mutants were identified by plasmid rescue and sequencing. To confirm that it was the insertion rather than spontaneous mutations that caused the phenotype, we transferred DNA bearing the insertion back to the L2612 parental strain. If the overexpression of pYES2-X123 in the mutated L2612 strain still caused growth inhibition on xylose, the insertion site was eliminated. From an initial series of approximately 18 putative mTn insertional mutants that grew on xylose plates, we were able to identify two genes that enabled growth on xylose with high levels of xylulokinase expression. Each of these was obtained in two independent mTn transformation events. The confirmed mutants had mTn insertions either upstream of the TAL1 ORF or in the PHO13 ORF (Table 3). All but 2 of the remaining 14 unique insertion events were recovered and identified by sequencing. The effects of these mutational events could not be confirmed by the techniques used here.
TABLE 3.
mTn insertional mutants
| Mutant | Growth on xylose | Insertion site | Other experiment |
|---|---|---|---|
| 312 | Medium | TAL1 promoter −439 | Dominant |
| 512 | Medium | TAL1 promoter −515 | Dominant |
| 1101 | Very fast | PHO13 ORF 252/312 amino acids | pho13 knockout also grows on xylose |
| 1201 | Very fast | PHO13 ORF 229/312 amino acids |
mTn insertion upstream of TAL1 enhanced xylose utilization through the altered expression of TAL1.
To evaluate whether the mTn insertion upstream of TAL1 affected TAL1 expression, transformants containing two copies of TAL1 were constructed. Plasmids carrying a mutant TAL1, pRS314-312tal and pRS314-512tal, were made from Mut312 and Mut512, respectively, and transformed into L2612(pYES2-X123). The transformants contained heterodiploid TAL1, with a mutant copy on the plasmid and a wild-type copy on the chromosome. The transformants could grow on xylose, suggesting that the mutant copy of TAL1 was dominant with respect to growth on xylose. mTn insertion at TAL1 upstream might enhance TAL1 expression in the strain overexpressing pYES2-X123 when grown on xylose. To test this possibility, pRS314-ADHtal, which carries TAL1 under a constitutive strong ADH1 promoter, was transformed into L2612(pYES2-X123). As expected, the transformant could grow on xylose. Therefore, increasing TAL1 expression may relieve the growth inhibition on xylose.
Deleting PHO13 relieved growth inhibition on xylose.
Mut1101 and Mut1201 contain mTn inserted into the PHO13 ORF, and it is likely that PHO13 is defective in these strains. A large part of the PHO13 gene product could, however, still be synthesized, and this might still retain activity. To determine whether deleting the complete gene would have the same effect as the insertional mutation, we made a pho13 knockout mutation (pho13Δ) in L2612 and transformed it with pYES2-X123. This transformant grew well on xylose, which was in a manner similar to that of the mutants with mTn insertions (Fig. 4, top). Therefore, reducing PHO13 expression by mutagenesis or deletion appears to relieve growth inhibition on xylose.
FIG. 4.
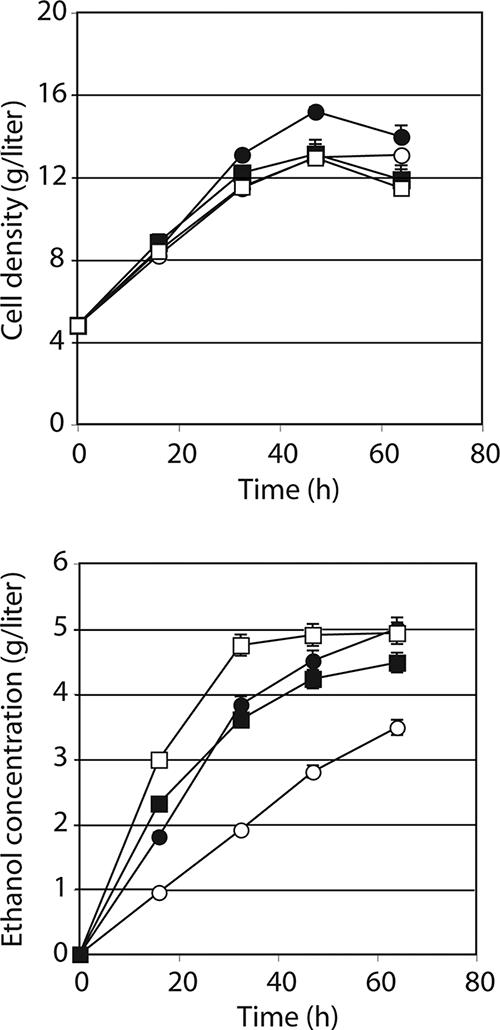
Growth and ethanol production by S. cerevisiae with native PHO13 or deleted pho13Δ plus genes for xylose metabolism. •, S. cerevisiae L2612(PHO13 pRS314-X123-T2); ○, S. cerevisiae L2612(PHO13 pRS314-X123); ▪, S. cerevisiae DR PHO13(pho13Δ pRS314-X123-T2); □, S. cerevisiae DR PHO13(pho13Δ pRS314-X123).
Improved ethanol production from xylose by recombinant strains with pho13 knockout mutation.
Fermentation was conducted under aerobic conditions in defined medium in 4% xylose using six strains made by transforming two yeasts (L2612 and a pho13Δ strain that we called DR PHO13) with three plasmids (pRS314-X123, pRS424-X123, and pRS314-X123-T2). In this latter construct, T2 consists of TAL1 and TKL1. Overall growth rates were low, and there was little difference among the four pRS314 (single-copy vector) transformants in the first 32 h (Fig. 4, top). Initial growth rates were essentially linear on a volumetric basis, which indicated that they could have been limited by the aeration rate. Growth rates declined in all strains after 48 h even though sugar analyses showed the presence of 18 to 22 g/liter of residual xylose. L2612(pRS314-X123-T2) grew to a slightly but significantly higher cell density than the other three pRS314 transformants.
Ethanol production rates were more strongly affected than growth in DR PHO13 (Fig. 4, bottom). The DR PHO13(pRS314-X123) transformant produced ethanol at almost three times the initial rate of the corresponding L2612(pRS314-X123) strain. Without TAL1 and TKL1 on the multicopy vector, the ethanol yield of the pho13Δ mutant was 92% higher than that in the PHO13 parent (0.13 versus 0.25 g/g xylose consumed), but in the presence of these two genes, the initial ethanol yield was only about 15% higher in the mutant (0.20 versus 0.23 g/g) (Table 4). Specific xylose consumption and ethanol production rates were also generally higher in the pho13Δ mutant than in the parent, but the difference was less evident when TAL1 and TKL1 were overexpressed. This indicated that most of the effect of the PHO13 deletion could be mimicked by the overexpression of TAL1 and TKL1. As in the case of cell growth, ethanol production was largely a linear function in the first 32 to 48 h, which indicated that it too could have been affected by oxygen availability.
TABLE 4.
Growth and fermentation kineticsa
| Strain | Specific growth rate, μ (h−1) | Xylose consumption (g · g−1 · h−1) | Ethanol production (g · g−1 · h−1) | Cell yield (g/g) | Ethanol yield (g/g) | Xylitol yield (g/g) |
|---|---|---|---|---|---|---|
| L2612(pRS314-X123) | 0.036 | 0.25 | 0.032 | 0.50 | 0.13 | 0.00 |
| DR PHO13(pRS314-X123) | 0.038 | 0.37 | 0.093 | 0.34 | 0.25 | 0.04 |
| L2612(pRS314 X123-T2) | 0.033 | 0.34 | 0.067 | 0.37 | 0.20 | 0.03 |
| DR PHO13(pRS314 X123-T2) | 0.035 | 0.35 | 0.080 | 0.36 | 0.23 | 0.04 |
All kinetic determinations were calculated from data in the first 16 h of cultivation. All determinations were done in triplicate.
Regardless of PHO13 status, pRS424-X123 transformants showed reduced xylose consumption, lower ethanol yields, and higher xylitol yields compared to transformants with other plasmids. Of these two strains, the DR PHO13(pRS424-X123) transformant consumed more xylose and produced more ethanol and xylitol than the L2612(pRS424-X123) transformant, which produced no ethanol (data not shown).
To further characterize the nature of the mutational events, we performed qPCR on the parental strain and three mutant strains to determine the levels of TAL1 transcript when cells were cultivated on d-xylose. Relative to L2612, preliminary experiments with the DR PHO13, Mut312, and Mut512 strains showed 1.32-, 1.53-, and 1.35-fold-higher levels of TAL1 mRNA. When these experiments were performed in replicates (n = 7) with glucose as the carbon source, the induction (n-fold) (of DR PHO13 over L2612) was 2.79 ± 0.37 for TAL1 and 1.78 ± 0.38 for TKL1.
In summary, the overexpression of pYES2-X123 (e.g., in a multiple-copy plasmid) inhibits growth on xylose in S. cerevisiae L2612 but not in a pho13 knockout strain, DR PHO13. The pho13 knockout also improved xylose fermentation when pYES2-X123 was expressed at a moderate level (in a single-copy plasmid). Increased expression of TAL1 and TKL1 (transketolase) in a pho13 knockout did not, however, exhibit a significant additive or synergistic effect.
DISCUSSION
We previously showed that the overexpression of PsXYL3 in an S. cerevisiae strain expressing PsXYL1 and PsXYL2 inhibits growth on xylose but not on glucose (13a). The present report, however, has identified spontaneous or chemically induced mutants of engineered S. cerevisiae overexpressing PsXYL3 that are not susceptible to growth inhibition on xylose. Because the overexpression of PsXYL3 in the FPL-YSX3 genetic background did not inhibit the growth of this yeast on xylose, we hypothesize that the growth advantage of YSX3 on xylose was not due solely to its moderate xylulokinase level but that it resulted at least in part from a mutation that conferred resistance to xylulokinase overexpression.
To develop and characterize mutational events that enable S. cerevisiae to grow on xylose while overexpressing PsXYL3, we carried out transposon mutagenesis on S. cerevisiae strain L2612(pYES2-X123). This parental yeast strain lacks the ability to grow on xylose. In this background, we also overexpressed PsXYL1, PsXYL2, and PsXYL3 (PsXYL123). The resulting mTn mutants were screened for the ability to grow on xylose.
Two different types of mutants capable of passing the ability to grow on xylose to their progeny were confirmed, and their respective mutations were further characterized. Two mutants with an mTn insertion at one of two sites in the transaldolase TAL1 promoter were identified. One includes an insertion at position −439 and was designated GX312; a second TAL1 promoter mutant includes an insertion at position −515 and was designated GX512. We hypothesized that a mutation in the promoter region of TAL1 results in increased expression, which protects against the inhibition of growth on xylose associated with xylulokinase overexpression. Further experiments showed that S. cerevisiae L2612(pYES2-X123) transformed with a plasmid overexpressing TAL1 is also capable of growth on xylose. Previous studies of S. cerevisiae engineered for the fermentation of xylose or arabinose showed the importance of overexpressing TAL1 (2).
A second type of transposon mutant identified as relieving xylose inhibition of S. cerevisiae overexpressing pYES2-X123 has an mTn insertion in the PHO13 ORF, which results in reduced activity. PHO13 codes for a protein with demonstrated alkaline phosphatase activity (22). Of approximately 11 substrates tested, Pho13p showed significant hydrolytic activity only against p-nitrophenylphosphate, phosphorylated histone II-A, and casein. Tuleva et al. speculated that the physiological role of the PHO13 p-nitrophenyl phosphate-specific phosphatase might involve participation in reversible protein phosphorylation (22). The possible role of phosphatases in dephosphorylating histones has been previously noted (5). These enzymes often act on a wide variety of phosphorylated proteins (1).
We noted that the growth of cells overexpressing XYL3 was almost completely inhibited on xylose agar plates but would occur slowly in xylose broth (12, 13a). While we do not fully understand this difference in response, it is possibly due to higher oxygen availability on the surface of plates than in broth. It is possible that in broth, xylose uptake is slow enough that toxicity attributed to the accumulation of sugar phosphates does not occur.
The exact function of Pho13p is unknown, but similar proteins are very widely distributed among yeasts, fungi, and other organisms. A BLAST analysis of Pho13p identified 13 closely related proteins produced by Debaryomyces hansenii, Gibberella zeae, Ustilago maydis, Schizosaccharomyces pombe, Neurospora crassa, Candida albicans, Yarrowia lipolytica, Ashbya gossypii, Kluyveromyces lactis, Candida glabrata, and Saccharomyces cerevisiae. More than 100 similar proteins from many other organisms are known, which suggests that PHO13 is highly conserved. These proteins have not yet been fully characterized with respect to their activities. It is reasonable to expect that they will be identified as orthologs having structures and functions similar to those of Pho13p and that isolates overexpressing enzymes associated with xylose fermentation and having the pho13 phenotype will exhibit improved xylose fermentation.
How PHO13 enhances growth on xylose is not entirely clear. pho13 mutants can overcome the growth inhibition caused by the overexpression of genes for xylulokinase, but simultaneous overexpression of TAL1 and TKL1 achieves some of the same effects. We do not know whether PHO13 regulates the expression of TAL1, but the qPCR results are consistent with this possibility. The different effects of TAL1 plus TKL1 overexpression versus pho13 inactivation on growth and fermentation, with the former contributing to better growth and the latter contributing to higher ethanol production on xylose, suggest that the pho13 mutation could also be enhancing fermentative activities.
Acknowledgments
This study was supported in part by U.S. Department of Agriculture NRIGCP project number 2001-35504-10695 and by National Institutes of Health Metabolic Engineering Interagency grant number 5R01GM67933-3 to T.W.J.
We acknowledge very helpful discussions with Jorge Escalante concerning mutagenic studies.
Footnotes
Published ahead of print on 2 February 2007.
REFERENCES
- 1.Barua, M., A. K. Ghosh, and G. C. Majumder. 1999. Partial purification and characterization of a phosphoprotein phosphatase from sperm plasma membrane. Reprod. Fertil. Dev. 11:379-386. [DOI] [PubMed] [Google Scholar]
- 2.Becker, J., and E. Boles. 2003. A modified Saccharomyces cerevisiae strain that consumes l-arabinose and produces ethanol. Appl. Environ. Microbiol. 69:4144-4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burns, N., B. Grimwade, P. B. Ross-Macdonald, E. Y. Choi, K. Finberg, G. S. Roeder, and M. Snyder. 1994. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes Dev. 8:1087-1105. [DOI] [PubMed] [Google Scholar]
- 4.Chang, S. F., and N. W. Ho. 1988. Cloning the yeast xylulokinase gene for the improvement of xylose fermentation. Appl. Biochem. Biotechnol. 17:313-318. [DOI] [PubMed] [Google Scholar]
- 5.Christova, N., and D. Galabova. 1998. Phosphorylase phosphatase activity in Saccharomyces cerevisiae 257. Z. Naturforsch. C 53:951-956. [DOI] [PubMed] [Google Scholar]
- 6.Gietz, R. D., and R. A. Woods. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87-96. [DOI] [PubMed] [Google Scholar]
- 7.Ho, N. W. Y., Z. D. Chen, and A. P. Brainard. 1998. Genetically engineered Saccharomyces yeast capable of effective cofermentation of glucose and xylose. Appl. Environ. Microbiol. 64:1852-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holstege, F. C., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner, M. R. Green, T. R. Golub, E. S. Lander, and R. A. Young. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95:717-728. [DOI] [PubMed] [Google Scholar]
- 9.Jeffries, T. W. 2006. Engineering yeasts for xylose metabolism. Curr. Opin. Biotechnol. 17:320-326. [DOI] [PubMed] [Google Scholar]
- 10.Jin, Y. S., and T. W. Jeffries. 2003. Changing flux of xylose metabolites by altering expression of xylose reductase and xylitol dehydrogenase in recombinant Saccharomyces cerevisiae. Appl. Biochem. Biotechnol. 105-108:277-286. [DOI] [PubMed] [Google Scholar]
- 11.Jin, Y. S., S. Jones, N. Q. Shi, and T. W. Jeffries. 2002. Molecular cloning of XYL3 (d-xylulokinase) from Pichia stipitis and characterization of its physiological function. Appl. Environ. Microbiol. 68:1232-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin, Y. S., J. M. Laplaza, and T. W. Jeffries. 2004. Saccharomyces cerevisiae engineered for xylose metabolism exhibits a respiratory response. Appl. Environ. Microbiol. 70:6816-6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin, Y. S., T. H. Lee, Y. D. Choi, Y. W. Ryu, and J. H. Seo. 2000. Conversion of xylose to ethanol by recombinant Saccharomyces cerevisiae containing genes for xylose reductase and xylitol dehydrogenase from Pichia stipitis. J. Microbiol. Biotechnol. 10:564-567. [Google Scholar]
- 13a.Jin, Y. S., H. Ni, J. M. Laplaza, and T. W. Jeffries.2003. Optimal growth and ethanol production from xylose by recombinant Saccharomyces cerevisiae require moderate d-xylulokinase activity. Appl. Environ. Microbiol. 69:495-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johansson, B., C. Christensson, T. Hobley, and B. Hahn-Hägerdal. 2001. Xylulokinase overexpression in two strains of Saccharomyces cerevisiae also expressing xylose reductase and xylitol dehydrogenase and its effect on fermentation of xylose and lignocellulosic hydrolysate. Appl. Environ. Microbiol. 67:4249-4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kötter, P., and M. Ciriacy. 1993. Xylose fermentation by Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 38:776-783. [Google Scholar]
- 16.Kuyper, M., A. A. Winkler, J. P. van Dijken, and J. T. Pronk. 2004. Minimal metabolic engineering of Saccharomyces cerevisiae for efficient anaerobic xylose fermentation: a proof of principle. FEMS Yeast Res. 4:655-664. [DOI] [PubMed] [Google Scholar]
- 17.Richard, P., M. H. Toivari, and M. Penttilä. 2000. The role of xylulokinase in Saccharomyces cerevisiae xylulose catabolism. FEMS Microbiol. Lett. 190:39-43. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Pena, J. M., V. J. Cid, J. Arroyo, and C. Nombela. 1998. The YGR194c (XKS1) gene encodes the xylulokinase from the budding yeast Saccharomyces cerevisiae. FEMS Microbiol. Lett. 162:155-160. [DOI] [PubMed] [Google Scholar]
- 19.Scholes, D. T., M. Banerjee, B. Bowen, and M. J. Curcio. 2001. Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics 159:1449-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tantirungkij, M., T. Izuishi, T. Seki, and T. Yoshida. 1994. Fed-batch fermentation of xylose by a fast-growing mutant of xylose-assimilating recombinant Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 41:8-12. [Google Scholar]
- 21.Toivari, M. H., A. Aristidou, L. Ruohonen, and M. Penttila. 2001. Conversion of xylose to ethanol by recombinant Saccharomyces cerevisiae: importance of xylulokinase (XKS1) and oxygen availability. Metab. Eng. 3:236-249. [DOI] [PubMed] [Google Scholar]
- 22.Tuleva, B., E. Vasileva-Tonkova, and D. Galabova. 1998. A specific alkaline phosphatase from Saccharomyces cerevisiae with protein phosphatase activity. FEMS Microbiol. Lett. 161:139-144. [DOI] [PubMed] [Google Scholar]