

Abstract
The gene Rv0813c from Mycobacterium tuberculosis, which codes for a hypothetical protein of unknown function, is conserved within the order Actinomycetales but absent elsewhere. The crystal structure of Rv0813c reveals a new family of proteins that resemble the fatty acid-binding proteins (FABPs) found in eukaryotes. Rv0813c adopts the 10-stranded β-barrel fold typical of FABPs but lacks the double-helix insert that covers the entry to the binding site in the eukaryotic proteins. The barrel encloses a deep cavity, at the bottom of which a small cyclic ligand was found to bind to the hydroxyl group of Tyr192. This residue is part of a conserved Arg-X-Tyr motif much like the triad that binds the carboxylate group of fatty acids in FABPs. Most of the residues forming the internal surface of the cavity are conserved in homologous protein sequences found in CG-rich prokaryotes, strongly suggesting that Rv0813c is a member of a new family of bacterial FABP-like proteins that may have roles in the recognition, transport, and/or storage of small molecules in the bacterial cytosol.
Tuberculosis (TB) remains a major health problem in the world, and new antituberculosis drugs are urgently needed to shorten the time for chemotherapy, to combat the spread of drug-resistant TB, and to treat the latent form of Mycobacterium tuberculosis infection. The availability of several mycobacterial genome sequences (5, 6, 9) has opened unparalleled opportunities for the discovery of new potential targets for therapeutic intervention. Thus, comparative mycobacterial genomics has already revealed a number of genetic loci important for virulence, persistence, and survival (4), and genome-based approaches are providing new insights into the essential genes required for mycobacterial growth and pathogenicity (24). Several of these conserved genes, mostly annotated as hypothetical proteins of unknown function, are confined to mycobacteria or actinomycetales and might represent potential candidates for antituberculosis drug targets (4).
The product of the M. tuberculosis gene Rv0813c is one of these hypothetical proteins confined to the order Actinomycetales. The inspection of mycobacterial genomes reveals a conserved arrangement of the genetic locus around Rv0813c, but this conservation provides no further functional hints, since neighboring genes also lack reliable functional annotation. In an attempt to provide new elements for function discovery, we have undertaken the structural characterization of Rv0813c. Here, we show that Rv0813c is a representative of a new family of bacterial proteins strongly resembling the eukaryotic fatty acid-binding proteins, and we discuss the structural and functional implications of these observations.
MATERIALS AND METHODS
The gene Rv0813c was cloned into the Gateway (Invitrogen) expression vector pDEST17 and used to transform Escherichia coli BL21(DE3)pLysS cells. The cells were grown in 500 ml of LB medium containing 100 μg/ml ampicillin and 25 μg/ml chloramphenicol for 3.5 h at 30°C before induction with 1 mM isopropopyl β-d-thiogalactoside (IPTG). After 1.5 h, the cells were harvested by centrifugation, resuspended, and frozen in 50 mM Tris-HCl buffer containing 150 mM NaCl and 10 mM imidazole, pH 8.0. The cellular suspension was thawed, and the cells were lysed in a French press at 14,000 lb/in2. After centrifugation, the recombinant protein was purified by metal affinity (Ni-nitrilotriacetic acid) and gel filtration (Superdex 75) chromatography. The peak fractions were pooled and concentrated to 6 to 7 mg/ml for crystallization. Since Rv0813c contains only a single Met residue at the N terminus, a double mutant (Ile94Met-Ile115Met) was produced for phasing purposes using the Quickchange multi site-directed mutagenesis kit (Stratagene, La Jolla, CA). The selenomethionine (SeMet)-labeled mutant was produced in BL21(DE3) E. coli cells as described previously (28) and purified as described above.
Initial crystallization screenings were carried out on sitting drops (200 nl of protein, 5.5 mg/ml, plus 200 nl of the crystallization solution equilibrated against 150 μl of the well solution) using a Cartesian Technologies nanoliter dispenser workstation. Initial hits were manually reproduced and optimized. The best crystals were grown in 2 M NaCl, 5% (vol/vol) dioxane, and 100 mM HEPES, pH 6.5, at 18°C and reached a size of 200 by 200 by 300 μm (50 by 50 by 100 μm for the SeMet-labeled protein) in 3 days.
X-ray diffraction data were collected on beamline ID29 at the European Synchrotron Anomalous Diffraction (Grenoble, France) from frozen crystals (100 K) in mother liquor plus 25% (vol/vol) glycerol. A 1.7-Å-resolution data set was collected from a single native crystal, and complete SAD data were collected from a SeMet-labeled crystal for the X-ray wavelength corresponding to the peak of the selenium K edge. Data reduction was carried out with the programs MOSFLM and SCALA from the CCP4 software package (3). The two selenium sites were readily located with the programs SHELXD (25) and SOLVE (26). SAD phases were calculated to 2.6-Å resolution and extended to 1.7 Å using the program RESOLVE. The resulting electron density map (Fig. 1A) allowed the automatic tracing of over 80% of the polypeptide chain with the program ARP/WARP (20). Refinement of the native structure was carried out with REFMAC (21) alternated with rounds of validation and rebuilding with O (17). The final model included protein residues 27 to 226 and three molecules of dioxane, which was present in the crystallization solution.
FIG. 1.

(A) Experimental electron density map showing the triad Arg-X-Tyr and the bound dioxane molecule. (B) Overall structure of Rv0813c, color coded from the N terminus (blue) to the C terminus (red). Charged residues in the protein loops that define the entry to the cavity are labeled. (C) Molecular surface of the internal binding cavity, color coded according to electrostatic potential, showing the bound dioxane molecule and ordered solvent molecules. The entrance of the cavity (top) is open to the solvent.
Sequence database searches were performed with BLAST (1) at NCBI. Multiple sequence alignments were carried out with Muscle (8), and manually adjusted structural comparisons against databases were done using DALI (16).
Protein structure accession numbers.
The experimental structure factors and the refined coordinates for the final model (Table 1) have been deposited in the Protein Data Bank with accession code 2FWV.
TABLE 1.
Crystallographic data for Rv0813 crystals
| Parameter | Value
|
|
|---|---|---|
| SeMet labeled | Native | |
| Data collection | ||
| X-ray wavelength (Å) | 0.97910 | 0.97895 |
| Cell dimensions (Å) | a = b = 65.92 | A = b = 65.84 |
| c = 240.38 | C = 239.71 | |
| Space group | I4122 | I4122 |
| Resolution rangea | 46.61-2.70 (2.85-2.70) | 43.44-1.70 (1.74-1.70) |
| No. of unique reflections | 7,723 | 29,663 |
| Rsyma,b | 0.055 (0.197) | 0.070 (0.316) |
| Ranoma | 0.062 (0.164) | |
| Multiplicitya | 6.8 (6.7) | 6.60 (6.4) |
| <I/σ>a | 25.7 (10.3) | 19.6 (5.2) |
| Completeness (%)a | 99.9 (99.9) | 100.0 (100.0) |
| Phasing statistics | ||
| Figure of merit (SOLVE) | 0.26 (2.96 Å) | |
| Z score (SOLVE) | 7.05 (2 Se) | |
| Figure of merit (RESOLVE) | 0.54 (2.70 Å) | |
| Refinement | ||
| Unique reflections (total/test) | 29,635 (1,503) | |
| Rfactorc | 0.181 | |
| Rfreec | 0.223 | |
| No. of protein atoms | 1,497 | |
| No. of solvent molecules | 355 | |
| Average B value (Å2) | 20.17 | |
| Bond length (Å) | 0.014 | |
| Bond angle (degrees) | 1.533 | |
Values in parentheses apply to the high-resolution shell.
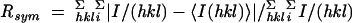 .
.
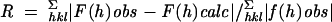 . Rfactor and Rfree were calculated from the working and test reflection sets, respectively.
. Rfactor and Rfree were calculated from the working and test reflection sets, respectively.
RESULTS AND DISCUSSION
The crystal structure of Rv0813c was determined by single-wavelength anomalous diffraction methods (Fig. 1A) and refined at 1.7-Å resolution (Table 1). The protein folds into a 10-stranded β-barrel (residues 71 to 226) enclosing a deep internal cavity, reminiscent of eukaryotic fatty acid binding proteins (FABPs), and displays an additional N-terminal domain (residues 27 to 70) that lies juxtaposed to the “bottom” of the barrel (Fig. 1B). This small domain is well conserved in homologous proteins from mycobacteria and corynebacteria (see below) and comprises a long amphipathic α-helix that extends into the solvent and is increasingly disordered toward its N terminus (no density is observed for the first 26 residues in the crystal structure). This α-helix is followed by a largely exposed proline-rich loop (residues 40 to 50) that also contains several hydrophobic side chains and is engaged in crystallographic contacts, raising the question of whether this domain could function as a docking site for protein-protein interactions. The 10-stranded β-barrel is well defined in density, except for the solvent-exposed loop 132 to 144, which is presumably disordered in the crystal structure. A short 310-helix seals the “bottom” end of the barrel, whereas the “top” end comprises five protein loops that protrude into the solvent in an open configuration, allowing access of a putative ligand to the central cavity.
The internal surface of the central cavity (Fig. 1C) is defined by an array of hydrogen-bonded polar side chains. Thus, the guanidinium group of Arg190 forms a salt bridge with Glu126 (Fig. 1A) and is also engaged in H bonds to Tyr192 and Glu20. On the other side of the pocket, Gln92 makes H-bonding interactions with the hydroxyl groups of Tyr202 and Ser108. Other H-bonding interactions connect Glu126 to Trp106 and Glu204 to Arg206. At the bottom of the cavity, the phenol group of Tyr192 binds a small ligand, which displays the chair conformation of a six-membered nonplanar ring in the experimental electron density map (Fig. 1A) and was modeled as dioxane, an additive included in the crystallization buffer. An extended array of well-ordered solvent molecules fills the remaining space of the cage (Fig. 1C), further stabilized by hydrogen-bonding interactions to several polar side chains (Arg206, Glu157, Tyr155, His152, His116, Tyr88, and Arg112).
Four of the five loops that define the entry to the cavity at the top of the β-barrel (Fig. 1B) extend into the solvent and contain two or more charged side chains each. Thus, loop 178 to 186 contains two basic residues (Arg178 and Arg180), loop 207 to 215 contains two acidic residues (Asp208 and Asp210), loop 82 to 87 contains three charged groups (Arg82, Asp85, and Asp87), and loop 112 to 121 contains another three (Arg112, Asp118, and Glu121). However, many of these charged side chains point toward the interior of the protein, making it unlikely that these charged residues could fulfill a role similar to those in FABPs, which mediate interactions of the protein with the lipid bilayer to facilitate the transfer of small hydrophobic ligands, such as fatty acids (7, 15, 23).
Rv0813c belongs to a new bacterial family of FABP-like proteins.
A search for amino acid sequence similarities in public databases (as of December 2005) revealed that Rv0813c homologs (Fig. 2) are found in mycobacteria (amino acid sequence identities of >80% to Rv0813c), nocardia (65%), and corynebacteria (40 to 50%). More distantly related proteins retain the 10-stranded β-barrel and the short 310-helix that seals the bottom of the β-barrel but lack the N-terminal α-helical region of Rv0813c. This larger group includes several proteins of bacterial and eukaryotic origin, with sequence identities of 28 to 35% to Rv0813c. Indeed the three-dimensional (3D) structures of two of these proteins, Arabidopsis thaliana At1g79260 and M. tuberculosis Rv2717c (Protein Data Bank codes 2A13 and 2FR2) (Fig. 3A), have been recently determined within the context of structural-genomics programs. Although these proteins lack the N-terminal domain of Rv0813c, their 10-stranded β-barrels are very similar (root mean square deviations of 1.2 to 1.4 Å for 133 matched Cα positions). The above observations suggest that the 10-stranded beta barrel may be a structurally conserved domain seen in many other proteins among prokaryotes and eukaryotes.
FIG. 2.

Multiple alignment of Rv0813c homologs. Secondary-structure elements are indicated for Rv0813c (above) and Atlg79260 (below). Invariant residues are in white (red background), and those conserved in at least 70% of the sequences are in red (boxed). Genes and species abbreviations: Rv0813, M. tuberculosis Rv0813c; Nf, Nocardia farcinica; Ml, Mycobacterium leprae; Ma, Mycobacterium avium subsp. paratuberculosis; Cj, Corynebacterium jeikeium; Cd, Corynebacterium diphtheriae; Ce, Corynebacterium efficiens; Cg, Corynebacterium glutamicum; Os, Oryza sativa; Pa, Propionibacterium acnes; Nsp, Nocardioides sp.; Tf, Thermobifida fusca; Sa, Streptomyces avermitilis; Sc, Streptomyces coelicolor; Fsp, Frankia sp.; Kr, Kineococcus radiotolerans; Asp, Arthrobacter sp.; Bl, Brevibacterium linens; Lx, Leifsonia xyli; Mtb, M. tuberculosis Rv2717c; At, Arabidopsis thaliana Atlg79260.
FIG. 3.

(A) Structural superposition of Rv0813c (red) with At1g79260 (2A13; green) and Rv2717c (2FR2; light cyan). (B) Structural superposition (93 core Ca positions) of Rv0813c (red) with human muscle FABP (1HMT; blue).
Despite the absence of sequence similarities, detailed topological comparisons with known protein structures identify Rv0813c as a member of the calycin superfamily, which includes lipocalins, cellular retinol binding proteins (CRBPs), cellular retinoic acid binding proteins (CRABPs), and FABPs. Lipocalins are functionally diverse eight-stranded β-barrel proteins that bind small hydrophobic molecules and are found mainly, but not exclusively, in eukaryotes. On the other hand, CRBPs, CRABPs, and FABPs (here referred to as the FABP family) are 10-stranded β-barrel proteins having a double α-helical insertion between the first and second β-strands that covers the entrance to the internal binding site. So far, proteins belonging to the FABP family have been found only in eukaryotes, where they are thought to function in the uptake, transport, and storage of retinoids and fatty acids, as well as in signaling, although their precise cellular functions remain unclear (11, 12, 14, 18, 19, 30). The 10-stranded antiparallel β-barrel of Rv0813 superposes well (root mean square deviation, 2.3 to 2.5 Å) with those of CRBPs, CRABPs, and FABPs, as shown in Fig. 3B for human muscle FABP (29), and also shares the short irregular 310-helix (Rv0813c residues 63 to 71) immediately preceding the first β-strand. Sequence comparisons suggest that this helix may represent a general motif that primes the folding of the 10-stranded β-barrel and may serve as a signature for identifying additional Rv0813c-like proteins. On the other hand, Rv0813c differs from eukaryotic FABPs in some features. For instance, the mycobacterial protein lacks the gap observed between the fourth and fifth β-strands of the FABP β-barrel, which was proposed to serve as an entrance to the fatty acid-binding site. In Rv0813c (and in other bacterial homologs), these two strands are occluded within the dimer interface (see below) and are thus not accessible to a putative ligand. Instead, the top of the barrel (opposite the conserved 310-helix) is open to the solvent in Rv0813c, which lacks the double α-helix insertion that caps the entrance to the ligand-binding site in FABPs.
Extensive crystallographic and nuclear magnetic resonance studies of FABPs show that these proteins usually bind a single fatty acid molecule with its hydrophilic group deep within the cavity, although in some cases, two ligand molecules are found to occupy the internal cavity (22, 27). Bound fatty acids are completely enclosed in the interior of the β-barrel with their carboxyl groups forming direct or water-mediated H bonds to three polar amino acids, which always include at least one arginine (13). Two of these three binding residues are usually found within an Arg-X-Tyr motif. A similar motif, Arg190-Leu191-Tyr192, is also found at the bottom of the β-barrel cavity in Rv0813c, where the phenol group of Tyr192 is engaged in H-bonding interactions with a dioxane molecule (Fig. 1A). There are important differences, however, since the side chain of Arg190 might be unavailable for ligand binding, as it is locked into a conserved salt bridge with Glu126. Also, the Arg-X-Tyr motif in Rv0813c is located on the 8th strand of the β-barrel, rather than in the 10th, as seen in FABPs, and the Rv0813c internal cavity is more negatively charged than those in FABPs, resembling more closely those of the CRBPs.
Several glycine residues are strictly conserved in the β-strands of Rv0813c homologs (Fig. 2), probably because of structural constraints required to allow bending of the corresponding β-strands. These include three glycine residues in the largely conserved motif GXWXGXG (residues 73 to 79 in β-strand 1), Gly91 in strand 2, Gly128 in β-strand 4, Gly161 in β-strand 6, and Gly193 in β-strand 8. Interestingly, only Gly73 and Trp75 of the GXWXGXG motif correspond in part to the PROSITE signature patterns of FABPs and lipocalins (PROSITE entries PS00214 and PS00213, respectively) (10). A hydrophobic pattern, HHXHXXHH (where H stands for a hydrophobic residue, usually Leu), is also conserved in the 310 helix preceding the first β-strand. More importantly, several residues defining the internal cavity are also largely conserved (Fig. 2), suggesting that these proteins might bind similar ligands. They include the salt bridge between Glu126 and Arg190, as well as the residues in contact with the dioxane molecule in the Rv0813c structure (Tyr192, Tyr202, and Gln92) and others located close to the putative binding site (Trp106, Ser 108, Glu157, Ala219, and Leu221). In one homolog (P. acnes), residues Gln92 and Ser108, which are H bonded to each other in Rv0813c, are both replaced by Cys, suggesting the formation of a disulfide bridge. Unlike the FABP family, however, neither the long loops above the β-barrel that protrude into the solvent nor the residues defining the entry to the binding site contain any conserved patch of residues.
A dimer occluding ∼2,100 Å2 from each monomer at the interface is observed in the Rv0813c crystal structure (Fig. 4A), and the residues defining this interface are well conserved in bacterial Rv0813c homologs (Fig. 4B). This dimeric arrangement is compatible with a putative ligand-binding function of the protein, since the entrance to the cavity in each monomer remains fully accessible from the solvent. To investigate the oligomeric state of Rv0813c in solution, we carried out dynamic light-scattering and gel filtration studies of the recombinant protein. As shown in Fig. 4, the results clearly indicated that Rv0813c behaves as a dimer in solution. Since the homologs At1g79260 and Rv2717c also crystallize as similar dimers, the above-mentioned results strongly suggest that, as in eukaryotic FABPs (12), the formation of homodimers might be a common feature of this bacterial protein family.
FIG. 4.

(A) The crystallographic Rv0813c dimer. (B) The monomer-monomer interface, shown in red, for one monomer. The labeled residues correspond to highly conserved amino acid positions in the bacterial-protein family, which account for over 70% of the dimer interface. (C) Monomodal size distribution dynamic light scattering profile of recombinant Rv0813c. The mean value of the molecular radius (3.2 nm) is consistent with a dimeric form (∼50 kDa) of the protein in solution. (D) Gel filtration chromatography of Rv0813c. The chromatograph was obtained in a SMART system (Amersham Pharmacia) with a Superdex 200 column at a flow rate of 40 ml/min. The retention time (39.7 min) is between those of bovine serum albumin (67 kDa; 36.5 min) and Chymotrypsin A (25 kDa; 44.3 min), consistent with a dimeric form of Rv0813c in solution.
Concluding remarks.
In conclusion, the 3D structure determination of Rv0813c has led to the identification of a new bacterial family of proteins, which resembles the eukaryotic FABPs, CRABPs, and CRBPs. These bacterial proteins may fulfill similar molecular functions in the transport and storage of small hydrophobic ligands, since organisms such as M. tuberculosis produce a vast repertoire of lipophilic molecules (lipids, glycolipids, and glycopeptidolipids), most of which are found in the cell envelope. Bacterial FABP-like proteins might thus serve to shuttle these molecules from their sites of synthesis near the plasma membrane to the cell envelope. The known binding promiscuity of FABPs makes the identification of the putative physiological ligand(s) of Rv0813c an arduous task. Inspection of mass spectra of the supernatant after denaturing recombinant Rv013c expressed in E. coli identified a small molecule with a molecular mass of 207 Da (data not shown), but this putative ligand was not observed in the crystal structure. Furthermore, crystals of Rv0813c were soaked in the crystallization solution containing either retinol or M. tuberculosis lipids dissolved in ethanol, but the 3D structures (determined to 1.8 Å and 2.3 Å, respectively) showed only a network of H-bonded solvent molecules in each case (data not shown). Further experimental work on the native proteins, such as the mass spectrometry screenings for ligands carried out for FABPs (2), are clearly required to gain insight into the ligand binding specificity of the Rv0813c family of proteins.
Acknowledgments
We acknowledge V. Bondet, J. Bellalou, and S. Petres for help in protein production.
This work was partially supported by grants from the Institut Pasteur (GPH-Tuberculose), the National Genopole Network (contract RG-2002-08), and the European Commission (SPINE, contract no. QLG2-CT-2002-00988, and X-TB, contract no. QLK2-CT-2001-02018).
Footnotes
Published ahead of print on 15 December 2006.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benkestock, K., C. K. Van Pelt, T. Akerud, A. Sterling, P. O. Edlund, and J. Roeraade. 2003. Automated nano-electrospray mass spectrometry for protein-ligand screening by noncovalent interaction applied to human H-FABP and A-FABP. J. Biomol. Screen 8:247-256. [DOI] [PubMed] [Google Scholar]
- 3.CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 4.Cole, S. T. 2002. M comparative mycobacterial genomics as a tool for drug target and antigen discovery. Eur. Respir. J. Suppl. 36:78s-86s. [DOI] [PubMed] [Google Scholar]
- 5.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 6.Cole, S. T., K. Eiglmeier, J. Parkhill, K. D. James, N. R. Thomson, P. R. Wheeler, N. Honore, T. Garnier, C. Churcher, D. Harris, K. Mungall, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. M. Davies, K. Devlin, S. Duthoy, T. Feltwell, A. Fraser, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, C. Lacroix, J. Maclean, S. Moule, L. Murphy, K. Oliver, M. A. Quail, M. A. Rajandream, K. M. Rutherford, S. Rutter, K. Seeger, S. Simon, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, K. Taylor, S. Whitehead, J. R. Woodward, and B. G. Barrell. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007-1011. [DOI] [PubMed] [Google Scholar]
- 7.Corsico, B., H. L. Liou, and J. Storch. 2004. The alpha-helical domain of liver fatty acid binding protein is responsible for the diffusion-mediated transfer of fatty acids to phospholipid membranes. Biochemistry 43:3600-3607. [DOI] [PubMed] [Google Scholar]
- 8.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garnier, T., K. Eiglmeier, J. C. Camus, N. Medina, H. Mansoor, M. Pryor, S. Duthoy, S. Grondin, C. Lacroix, C. Monsempe, S. Simon, B. Harris, R. Atkin, J. Doggett, R. Mayes, L. Keating, P. R. Wheeler, J. Parkhill, B. G. Barrell, S. T. Cole, S. V. Gordon, and R. G. Hewinson. 2003. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 100:7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gattiker, A., E. Gasteiger, and A. Bairoch. 2002. ScanProsite: a reference implementation of a PROSITE scanning tool. Appl. Bioinformatics 1:107-108. [PubMed] [Google Scholar]
- 11.Glatz, J. F., and J. Storch. 2001. Unravelling the significance of cellular fatty acid-binding proteins. Curr. Opin. Lipidol. 12:267-274. [DOI] [PubMed] [Google Scholar]
- 12.Glatz, J. F., and G. J. van der Vusse. 1996. Cellular fatty acid-binding proteins: their function and physiological significance. Prog. Lipid Res. 35:243-282. [DOI] [PubMed] [Google Scholar]
- 13.Hamilton, J. A. 2004. Fatty acid interactions with proteins: what X-ray crystal and NMR solution structures tell us. Prog. Lipid Res. 43:177-199. [DOI] [PubMed] [Google Scholar]
- 14.Haunerland, N. H., and F. Spener. 2004. Fatty acid-binding proteins—insights from genetic manipulations. Prog. Lipid Res. 43:328-349. [DOI] [PubMed] [Google Scholar]
- 15.Herr, F. M., J. Aronson, and J. Storch. 1996. Role of portal region lysine residues in electrostatic interactions between heart fatty acid binding protein and phospholipid membranes. Biochemistry 35:1296-1303. [DOI] [PubMed] [Google Scholar]
- 16.Holm, L., and C. Sander. 1998. Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 26:316-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 18.Kaikaus, R. M., N. M. Bass, and R. K. Ockner. 1990. Functions of fatty acid binding proteins. Experientia 46:617-630. [DOI] [PubMed] [Google Scholar]
- 19.Li, E., and A. W. Norris. 1996. Structure/function of cytoplasmic vitamin A-binding proteins. Annu. Rev. Nutr. 16:205-234. [DOI] [PubMed] [Google Scholar]
- 20.Morris, R. J., A. Perrakis, and V. S. Lamzin. 2003. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 374:229-244. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53:240-255. [DOI] [PubMed] [Google Scholar]
- 22.Nichesola, D., M. Perduca, S. Capaldi, M. E. Carrizo, P. G. Righetti, and H. L. Monaco. 2004. Crystal structure of chicken liver basic fatty acid-binding protein complexed with cholic acid. Biochemistry 43:14072-14079. [DOI] [PubMed] [Google Scholar]
- 23.Nolan, V., M. Perduca, H. L. Monaco, B. Maggio, and G. G. Montich. 2003. Interactions of chicken liver basic fatty acid-binding protein with lipid membranes. Biochim. Biophys. Acta 1611:98-106. [DOI] [PubMed] [Google Scholar]
- 24.Sassetti, C. M., D. H. Boyd, and E. J. Rubin. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77-84. [DOI] [PubMed] [Google Scholar]
- 25.Schneider, T. R., and G. M. Sheldrick. 2002. Substructure solution with SHELXD. Acta Crystallogr. D 58:1772-1779. [DOI] [PubMed] [Google Scholar]
- 26.Terwilliger, T. C. 2003. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 374:22-37. [DOI] [PubMed] [Google Scholar]
- 27.Thompson, J., N. Winter, D. Terwey, J. Bratt, and L. Banaszak. 1997. The crystal structure of the liver fatty acid-binding protein. A complex with two bound oleates. J. Biol. Chem. 272:7140-7150. [DOI] [PubMed] [Google Scholar]
- 28.Wingfield, P. 2000. Production of recombinant proteins, p. 5.3.9-5.3.14. In J. Coligan, B. Dunn, H. Ploegh, D. Speicher, and P. Wingfield (ed.), Current protocols in protein science, vol. 1. Wiley, New York, NY. [Google Scholar]
- 29.Young, A. C., G. Scapin, A. Kromminga, S. B. Patel, J. H. Veerkamp, and J. C. Sacchettini. 1994. Structural studies on human muscle fatty acid binding protein at 1.4 Å resolution: binding interactions with three C18 fatty acids. Structure 2:523-534. [DOI] [PubMed] [Google Scholar]
- 30.Zimmerman, A. W., and J. H. Veerkamp. 2002. New insights into the structure and function of fatty acid-binding proteins. Cell Mol. Life Sci. 59:1096-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]