

Abstract
Competition between pioneer colonizing bacteria may determine polymicrobial succession during dental plaque development, but the ecological constraints are poorly understood. For example, more Streptococcus sanguinis than Streptococcus gordonii organisms are consistently isolated from the same intraoral sites, yet S. gordonii fails to be excluded and survives as a species over time. To explain this observation, we hypothesized that S. gordonii could compete with S. sanguinis to adhere to saliva-coated hydroxyapatite (sHA), an in vitro model of the tooth surface. Both species bound similarly to sHA, yet 10- to 50-fold excess S. gordonii DL1 reduced binding of S. sanguinis SK36 by 85 to >95%. S. sanguinis, by contrast, did not significantly compete with S. gordonii to adhere. S. gordonii competed with S. sanguinis more effectively than other species of oral streptococci and depended upon the salivary film on HA. Next, putative S. gordonii adhesins were analyzed for contributions to interspecies competitive binding. Like wild-type S. gordonii, isogenic mutants with mutations in antigen I/II polypeptides (sspAB), amylase-binding proteins (abpAB), and Csh adhesins (cshAB) competed effectively against S. sanguinis. By contrast, an hsa-deficient mutant of S. gordonii showed significantly reduced binding and competitive capabilities, while these properties were restored in an hsa-complemented strain. Thus, Hsa confers a selective advantage to S. gordonii over S. sanguinis in competitive binding to sHA. Hsa expression may, therefore, serve as an environmental constraint against S. sanguinis, enabling S. gordonii to persist within the oral cavity, despite the greater natural prevalence of S. sanguinis in plaque and saliva.
Within the oral cavity there are hundreds of species with the potential to colonize the tooth surface. Thus, microbes that have a competitive binding advantage are likely to be successful. Competition occurs both between and within species and is strongest for those microbes that target a similar pool of receptors. Furthermore, the outcome of competitive events for one set of bacteria can be expected to influence that of another. This is particularly pertinent for pioneer colonizers, since initial adhesion to the tooth surface by oral microbes is essential for the development and composition of dental plaque. Understanding the factors that influence initial colonization could, therefore, contribute to our understanding of the ecological constraints that govern this polymicrobial community and may suggest novel preventive and control measures for dental plaque.
The pioneer colonizers Streptococcus sanguinis and Streptococcus gordonii are readily able to attach to the tooth surface (37). The promiscuous adhesive capabilities of these bacteria are facilitated, in part, by specific cell surface adhesins (16, 43, 59). S. sanguinis and S. gordonii have a high level of 16S rRNA sequence homology and, until recently, were classified as the same species (19). In addition, these two streptococcal species are isolated from the same intraoral sites. Since these streptococci express similar surface proteins, it would be predicted that these two species compete for binding to the same array of available host receptors.
That oral streptococci will compete to adhere to saliva-coated teeth has been suggested by in vitro studies (1, 23), but limited classification of the species and limited characterization of streptococcal surface adhesins meant definitive conclusions were not possible. While interspecies competition was sometimes observed, no explanation of the underlying mechanisms could be provided. Many streptococcal adhesins have now been characterized (16). This study, therefore, aimed to revisit competitive interspecies binding between S. sanguinis and S. gordonii and establish a molecular basis. Adhesion of a radiolabeled reference species to saliva-coated hydroxyapatite (sHA), an in vitro model of the tooth surface in the mouth, was measured in the presence of an unlabeled competing species to test the hypothesis that S. gordonii and S. sanguinis compete for binding to salivary receptors. S. gordonii was shown to be a strong competitor of S. sanguinis adhesion, while S. sanguinis was not competitive with S. gordonii. A panel of isogenic mutants abrogated in the expression of surface-expressed proteins associated with streptococcal adhesion and colonization, including SspAB, CshAB, ScaA, AbpAB, and Hsa, was screened. From these and subsequent complementation studies, the sialic acid-binding protein Hsa was shown to confer the competitive binding advantages to S. gordonii.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Streptococci (listed in Table 1) were routinely grown in chemically defined synthetic medium (FMC) (56, 57) at 37°C in 5% CO2. Escherichia coli DH5α cells were grown aerobically at 37°C in Luria-Bertani (LB) medium. When required, antibiotics were added to the medium at the following concentrations: erythromycin, 1 μg ml−1 (S. gordonii OB390) or 5 μg ml−1 (S. gordonii OB277, abpAB hsa srtA mutants); kanamycin, 50 μg ml−1 (E. coli), 200 μg ml−1 (S. gordonii OB390), or 250 μg ml−1 (S. gordonii abpAB mutant); tetracycline, 10 μg ml−1 (S. gordonii); spectinomycin, 100 μg ml−1 (S. gordonii); ampicillin, 100 μg ml−1 (E. coli); and chloramphenicol, 25 μg ml−1 (E. coli).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ−thi-1 gyrA96 relA1 | Invitrogen |
| S. gordonii strains | ||
| DL1 (Challis) | Wild type | 33 |
| V288 | Wild type | G. Dunny, University of Minnesota |
| Blackburn | Wild type | J. Rudney, University of Minnesota |
| OB277a | cshA3::cat cshB3::ermAM | 30 |
| OB390a | sspAB::ermAM cshA::aphA3 cshB::cat | H. Jenkinson, University of Bristol |
| OB470a | scaA::tet | 14 |
| UB1360a | Δ(sspA sspB)::aad9 | 11 |
| srtA mutanta | Δ(srtA)::ermAM | This study |
| srtA complementeda | Δ(srtA)::ermAM(pDL276 srtA+) | This study |
| hsa mutanta | Δ(hsa)::ermAM | This study |
| hsa complementeda | Δ(hsa)::ermAM(pAS8741 hsa+) | This study |
| abpAB mutanta | Δ(abpA)::ermAM Δ(abpB)::aphA3 | This study |
| S. sanguinis strains | ||
| SK36 | Wild type | 18 |
| 133-79 | Wild type | 12 |
| S. cristatus CC5A | Wild type | J. Rudney, University of Minnesota |
| S. mitis SK306 | Wild type | J. Rudney, University of Minnesota |
| S. oralis SK100 | Wild type | J. Rudney, University of Minnesota |
| S. parasanguinis 15912 | Wild type | J. Rudney, University of Minnesota |
| Plasmids | ||
| pGEM-T Easy | 3.0 kb; Apr; ColE1ori | Promega |
| pGEM-abpA::ermAM | pGEM-T Easy derived; containing ermAM cassette within flanking sequences of abpA gene | This study |
| pGEM-abpB::aphA3 | pGEM-T Easy derived; containing aphA3 cassette within flanking sequences of abpB gene | This study |
| pVA891 | 5.4 kb; Emr, Cmr; pACYCori; E. coli-streptococcal shuttle vector | 27 |
| pSF151 | 3.5 kb; Kmr; ColE1ori; E. coli-streptococcal-integration shuttle vector | 53 |
| pDL276 | 6.9 kb; Kmr, ColE1ori, E. coli-streptococcal-integration shuttle vector | 53 |
| pAS8741 | pAS40S carrying hsa within a 7.4-kb HindIII-SphI cloned fragment of S. gordonii DL1 genomic DNA; Spr | 48 |
Derived from DL1 (Challis).
sHA adhesion assay.
The sHA adhesion assay employed was originally described by Liljemark et al. (25) and Tellefson and Germaine (55) and modified by Gong and Herzberg (6). In brief, human whole saliva was collected from five adult volunteers (as approved by the Committee on the Use of Human Subjects in Research, University of Minnesota) into a chilled tube on ice, pooled, and clarified by centrifugation (2,000 × g, 20 min, 4°C). Whole salivary supernatant (1 ml) was then incubated at ambient temperature for 1 h with 20 mg ceramic HA (ICN Biomedicals, Inc.) that had been equilibrated with modified Gibbons' buffer (1 mM KH2PO4-K2HPO4 buffer [pH 6.8] with 50 mM KCl, 1 mM CaCl2, and 0.1 mM MgCl2). The sHA was then washed three times with Gibbons' buffer and transferred to a fresh tube. Streptococci were grown in FMC for 16 to 20 h in the presence of 10 μCi ml−1 [methyl-3H]thymidine to a specific activity of between 2 × 10−4 and 1.2 × 10−5 cpm cell−1, centrifuged, and adjusted with Gibbons' buffer to give a suspension of 2.5 × 1010 cells ml−1. Cells (ranging from 107 to 1010) were incubated with 20 mg of sHA (1-ml final volume) for 1 h at ambient temperature with continuous inversion on a rototorque. Unattached cells were aspirated, and cells loosely associated with the sHA were removed by washing a further three times. The radioactivity associated with sHA was monitored by liquid scintillation counting. Bacterial suspensions of known concentration were used to calculate the number of cells per radioactive count per minute. From this, numbers of adherent or sHA-associated bacteria were determined. At the lowest input of 107 cells, radioactivity values ranged from 50 to 100 cpm. Background values were ≤15 cpm.
sHA interspecies competition assay.
Competing species were grown in the absence of [methyl-3H]thymidine, harvested, and adjusted to a 2.5 × 1010 cells ml−1 suspension in Gibbons' buffer. Noncompeting, radiolabeled cells (5 × 108) were prepared as described above for the sHA adhesion assay and then incubated with 20 mg sHA using continuous inversion for 1 h at ambient temperature in the presence of a 10- or 50-fold excess of unlabeled, competing cells (1-ml final volume). The sHA was then washed three times in Gibbons' buffer, and numbers of labeled cells bound to the sHA were determined by liquid scintillation counting, as described above for the sHA adhesion assay.
Genetic manipulations.
Standard recombinant DNA techniques were employed, as described by Sambrook et al. (41). Plasmids (listed in Table 1) were purified from E. coli cells by using the QIAquick Spin Miniprep Purification kit (QIAGEN). Oligonucleotides (listed in Table 2) were synthesized by Integrated DNA Technologies, Inc. Chromosomal DNA was prepared from mutanolysin-treated streptococcal cells by using the QIAGEN 100/G Genomic Tip system. PCR products were purified using the High Pure PCR Product Purification kit (Roche). DNA restriction and modification enzymes were used under the conditions specified by the manufacturer (Promega).
TABLE 2.
Primers used in this study
| Primer | Sequencea | Source or reference |
|---|---|---|
| abpA.F1 | TGATACTGCAGGAACCATC | This study |
| abpA.F2 | CCATCGATGGTCGTAACTCGGAGTTACTACC | This study |
| abpA.R1 | CCATCGATGGAATATCACTGGGCCATTG | This study |
| abpA.R2 | CATGACCAAAGAGCCAAG | This study |
| abpB.F1 | ACCAAGTCCAGAGGAAGC | This study |
| abpB.R1 | CCATCGATGGTCTGTTGAGAAAGCCAGTC | This study |
| abpB.F2 | CCATCGATGGTTTGAAAGTCCTAGACTCCC | This study |
| abpB.R2 | CGGTGATTCTACCAGTTTC | This study |
| ermAM.F | CCATCGATGGCCATATCATAAAAATCGAAACAGC | This study |
| ermAM.R | CCATCGATGGTAGGGACCTCTTTAGCTCC | This study |
| aphA3.F | CCATCGATGGGGCTCCGTCGATACTATG | This study |
| aphA3.R | CCATCGATGGCCGATACAAATTCCTCGTAG | This study |
| srtA.F1 | CGGGGTACCCATGGCCTGTAGCTCAATC | This study |
| srtA.R1 | GGATCGATGGAAGGAAGCATAAGTTTAATGC | This study |
| srtA.F2 | CCATCGATCCTTCTCGTCTTGCAACTC | This study |
| srtA.R2 | CGCGAGCTCACCTAAGAGACGGTGACCAG | This study |
| hsaL3BamHI | CGCGGATCCGGATAAGATTTATCAAGACGATCACA | This study |
| hsaR3SacI | CGCGAGCTCTGTGGCAGACGATGGACTTA | This study |
| hsaL5KpnI | CGGGGTACCCAAACATAGTTTAAATGCTTTGGA | This study |
| hsaR5EcoRI | CCGGAATTCCCCCTCTACTTAATTTAATATCCCAAAA | This study |
| RealsrtA.F | ATGGAAAGCACAGCAACTTC | This study |
| RealsrtA.R | CTGTAATCCCGAAAACATGG | This study |
| hsaL1 | CAGAGCTGCAAATCCAAACA | 65 |
| hsaR1 | GCCGAGATACTTGCGCTTAC | 65 |
Underlined letters indicate restriction enzyme sites.
The abpA and abpB genes of S. gordonii DL1 were inactivated by allelic exchange with the erythromycin resistance determinant ermAM (amplified from pVA891) or the kanamycin resistance determinant aphA3 (amplified from pSF151), respectively. PCR amplification with primers abpA.F1/abpA.R1 and abpA.F2/abpA.R2 of S. gordonii DL1 DNA template generated two fragments comprising the flanking sequences of the abpA gene (468 bp and 512 bp) with a unique ClaI site at their 3′ and 5′ ends, respectively. These were ligated via the ClaI site and cloned into pGEM-T Easy. A DNA fragment (1,023 bp) containing the ermAM gene was PCR amplified from plasmid pVA891 by using primers ermAM.F/ermAM.R, which incorporated ClaI restriction sites. The PCR product was digested with ClaI and ligated into the unique site within the combined abpA fragments, generating plasmid pGEM-abpA::ermAM. This process was then repeated to generate plasmid pGEM-abpB::aphA3, using the primers listed in Table 2. The insert DNA from these two plasmids was PCR amplified using primers abpA.F1/abpA.R2 or primers abpB.F1/abpB.R2, purified, and transformed into S. gordonii DL1, thus generating the double mutant. Confirmation of predicted insertions was obtained by PCR amplification and sequencing; loss of function was demonstrated by amylase blot overlay (36).
The srtA gene of S. gordonii DL1 was inactivated by allelic exchange with the erythromycin resistance determinant ermAM as described above, using primers srtA.F1/srtA.R1 and srtA.F2/srtA.R2.
To complement the srtA deletion mutant, a DNA fragment (1,484 bp) incorporating the entire srtA gene was PCR amplified from S. gordonii DL1 chromosomal DNA by using primers srtA.F1/srtA.R2. This was cloned into E. coli-streptococcal shuttle vector pDL276, generating plasmid pDL276-srtA. This construct was purified and used to transform the S. gordonii srtA mutant. Confirmation of predicted insertions was obtained by PCR amplification and sequencing. Complementation was confirmed by detection of the srtA RNA transcript by using primers RealsrtA.F/RealsrtA.R. RNA extraction and cDNA synthesis were performed as described below.
The hsa gene of S. gordonii DL1 was inactivated by allelic exchange with the erythromycin resistance determinant ermAM, using primers hsaL3BamH1/hsaR3SacI and hsaL5KpnI/hsaR5EcoR1, as described above.
To complement the hsa deletion mutant, the S. gordonii hsa mutant was transformed with plasmid pAS8741 (a gift of Y. Takahashi, Nippon Dental University, Tokyo, Japan), which carries hsa within a 7.4-kb HindIII-SphI cloned fragment of S. gordonii DL1 genomic DNA (48). Complementation was confirmed by detection of the hsa RNA transcript with primers hsaL1/hsaR1 (65) and by Western immunoblotting with a goat anti-Hsa antiserum (a gift of B. Bensing, University of California, San Francisco). RNA extraction and cDNA synthesis were performed as described below.
RNA extraction and cDNA synthesis.
Bacterial cultures (2 ml) were harvested (3,000 × g, 20 min, 4°C) and resuspended in a lysing reagent containing 222 μl H2O and 778 μl Buffer RLT (RNeasy Mini Kit; QIAGEN). Suspensions were transferred to FastPrep Blue tubes (QBIOgene, Morgan Irvine, CA) and processed in a FastPrep FP120 vibrator (Bio101) at a speed rating of 6 for 2 min. Cell debris was removed by centrifugation (12,000 × g, 10 min), the supernatant was collected, and RNA was prepared using an RNeasy Mini Kit (QIAGEN) according to manufacturer's instructions. The integrity of the RNA was confirmed by gel electrophoresis, and it was then treated with DNase I (Promega) for 2 h at 37°C. The concentration of RNA was determined by measuring the absorbance at 260 nm in a spectrophotometer. RNA (2 μg) was reverse transcribed into cDNA with random hexamer primers as described previously (64).
Data analysis.
Analysis of adhesion data and determination of significance were performed using Student's t test. Data are presented based on at least two independent experiments performed in duplicate. Differences were considered significant when a P value of <0.05 was obtained.
RESULTS
sHA adhesion by S. gordonii and S. sanguinis.
As S. gordonii and S. sanguinis are pioneer colonizers, their ability to adhere to sHA models adhesion to the tooth. Given the array of surface adhesins that could potentially mediate binding to sHA, well-characterized laboratory strains were chosen to represent both species. Using a range of input bacteria, S. gordonii (strain DL1 or V288) and S. sanguinis SK36 were shown to adhere directly to sHA with similar isotherms (Fig. 1A). In each case, the binding curve appeared to approach saturation at approximately 109 bound cells per 20 mg of sHA. A comparable binding profile was also seen for S. sanguinis 133-79 (data not shown). In the absence of saliva, an input of 109 cells resulted in ≤5% adhesion for each strain (data not shown), indicating that these streptococcal strains interact primarily with a salivary component(s) in the saliva coating film or pellicle.
FIG. 1.

Adhesion and competition between S. gordonii and S. sanguinis for sHA. (A) Symbols: ▪, S. gordonii DL1; ▴, S. gordonii V288; ○, S. sanguinis SK36. Radioactively labeled bacteria were incubated with sHA for 1 h, and numbers of attached cells were determined as described in Materials and Methods. Data are presented as the mean ± standard deviation. (B and C) Radioactively labeled S. sanguinis SK36 cells (B) or S. gordonii V288 cells (C) were incubated with sHA in the presence of a 10-fold (□) or 50-fold (□) excess of unlabeled competing cells (S. gordonii strain DL1 or V288 [B] or S. sanguinis SK36 [C]). Numbers of attached reference (labeled) cells were then determined as described in Materials and Methods. As a control, reference cells were incubated with sHA in the absence of any competing cells (▪), and these adhesion levels were set to 100%. Data are shown as the relative percentage of cells bound ± standard deviation. **, P < 0.01 compared to controls in the absence of competition.
Competitive binding between S. gordonii and S. sanguinis.
As S. gordonii and S. sanguinis bound sHA similarly, we hypothesized that these two species may compete when present together. This hypothesis was tested using an adapted sHA adhesion assay in which one (noncompeting) species is radioactively labeled, while a second (competing) species is unlabeled. Based upon the sHA binding profiles of S. gordonii and S. sanguinis (Fig. 1A), an input of 5 × 108 noncompeting cells was selected as producing half-maximal binding to sites on sHA. The adhesion of 5 × 108 noncompeting radiolabeled cells was arbitrarily set as 100%. Competition would, therefore, reduce the percentage bound; cooperativity would increase the percentage as maximal binding was approached. Noncompeting radiolabeled cells were then incubated with sHA in the presence of a 10- or 50-fold excess of competing cells, and numbers of bound noncompeting cells were determined by liquid scintillation counting. When present at a 10-fold excess, S. gordonii DL1 or V288 reduced binding levels of S. sanguinis SK36 by >80%, which increased to >95% at a 50-fold excess (Fig. 1B). A similar reduction in adhesion was also found with S. sanguinis 133-79 (data not shown). By contrast, S. sanguinis did not significantly impair the ability of S. gordonii DL1 to adhere to sHA at any concentration tested (data not shown) and actually promoted adhesion by S. gordonii V288 up to 50% (Fig. 1C), suggesting interspecies cooperativity in binding. The ability of S. gordonii to resist competition by S. sanguinis was dependent upon the presence of saliva and was not seen using HA alone (data not shown). In self-inhibition studies, in which unlabeled and labeled cells of the same strain were combined, the presence of a 10-fold excess of competing cells resulted in 65 to 75% inhibition (data not shown).
Competitive binding by other oral streptococci.
To determine if the one-way competition was species specific, the abilities of S. gordonii and S. sanguinis to resist competition by other oral streptococci were examined. It was first confirmed that each of the species tested bound similarly to sHA. Competition with S. sanguinis SK36 was then investigated. Of the six competing strains tested, S. gordonii strains DL1 and Blackburn reduced adhesion levels of S. sanguinis SK36 by 85% and 75%, respectively (Fig. 2A). This level of inhibition was significantly higher than that seen with the other species tested. S. mitis SK306 reduced S. sanguinis binding by only 29%, while S. oralis SK100, S. cristatus CC5A, or S. parasanguinis 15912 had no significant effects (Fig. 2A). Competition for adhesion to sHA by S. gordonii DL1 was then tested. S. gordonii Blackburn was found to be the strongest competitor, reducing strain DL1 adhesion by 62%, followed by S. oralis SK100, which inhibited by 53% (Fig. 2B). A 20 to 30% reduction in DL1 adhesion levels was found in the presence of excess S. mitis SK306 or S. parasanguinis 15912 (Fig. 2B). Neither S. sanguinis SK36 nor S. cristatus CC5A had any significant effects on binding to sHA by S. gordonii DL1 (Fig. 2B). Taken collectively, these data suggest that competitive binding by oral streptococci is species dependent. S. gordonii was consistently shown to be a strong competitor of S. sanguinis. Unlike S. sanguinis, other species of streptococci could inhibit binding of S. gordonii to sHA.
FIG. 2.

Adhesion of S. sanguinis SK36 or S. gordonii DL1 to sHA in the presence of competing oral streptococci. Radioactively labeled S. sanguinis SK36 cells (A) or S. gordonii DL1 cells (B) were incubated with sHA in the presence of a 10-fold excess of unlabeled competing streptococcal cells. Numbers of attached reference (labeled) cells were then determined as described in Materials and Methods. As a control, reference cells were incubated with sHA in the absence of any competing cells, and these adhesion levels were set to 100%. Data are shown as the relative percentage of cells bound ± standard deviation. *, P < 0.05; **, P < 0.01 (compared to controls in the absence of competition).
LPXTG-containing adhesins, notably Hsa, mediate the competitive capabilities of S. gordonii.
To explain the one-way competitive binding, putative S. gordonii surface adhesins were considered for their potential to confer a selective binding advantage over S. sanguinis cells. A series of S. gordonii isogenic mutants and parent strains (Table 1) were tested in sHA adhesion and competition assays. Potential candidates for the observed competition included mutants with deletions in the following genes: srtA, as a general screen for the involvement of LPXTG motif-containing adhesins; hsa, encoding an adhesin known to bind sialylated substrates, as are found in the sHA model; and abpAB, encoding amylase-binding proteins that are present in S. gordonii but not S. sanguinis. Mutants lacking proteins SspA and -B (multifunctional polypeptides), CshA and -B (associated with fibronectin binding and hydrophobicity), and ScaA (virulence-associated lipoprotein) were also tested; these are all adhesins previously reported to promote streptococcal adhesion and colonization.
Binding of the mutants and binding of wild-type S. gordonii to sHA were compared at a range of input bacteria (107 to 1010 cells per 20 mg of sHA). The maximal binding capacity of the sHA was confirmed to be approximately 109 bound cells. Using 109 cells as the input dose, strains abrogated in the production of ScaA, AbpAB, SspAB, and CshAB bound to sHA at levels comparable to that of the wild-type strain (Fig. 3). By contrast, srtA and hsa mutants at this input dose bound sHA 15- to 20-fold less effectively than the parent strain (Fig. 3). To rule out that potential indirect effects of the mutations were responsible for this loss of binding ability, complementation of the srtA and hsa mutants was performed. Reverse transcription-PCR and Western immunoblotting were first used to confirm restoration of protein expression (Fig. 4), and sHA adhesion assays were then performed. Adhesion levels for srtA and hsa complementation mutants were restored to 70% and 100% of those of the wild-type strain, respectively (Fig. 3).
FIG. 3.

Adhesion of isogenic mutants of S. gordonii DL1 to sHA. Radioactively labeled bacteria (input of 109) were incubated with sHA for 1 h, and numbers of attached cells were determined as described in Materials and Methods. Data are presented as the mean ± standard deviation. **, P < 0.01 compared to the parent strain.
FIG. 4.
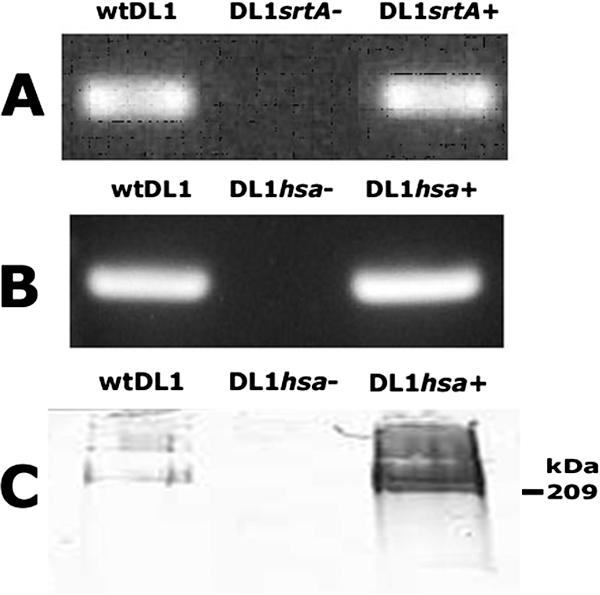
Confirmation of the genetic manipulations of S. gordonii DL1 srtA and hsa genes. (A) RNA was extracted from wild-type S. gordonii (wtDL1) and the srtA deletion (DL1srtA−) and complemented (DL1srtA+) mutants and reverse transcribed into cDNA, and the srtA transcript was detected by PCR. (B) RNA was extracted from wild-type S. gordonii and the hsa deletion (DL1hsa−) and complemented (DL1hsa+) mutants and reverse transcribed into cDNA, and the hsa transcript was detected by PCR. (C) Cell wall protein fractions were collected by mutanolysin digest and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and Hsa was detected by Western immunoblotting.
Isogenic mutants were then tested in sHA competition assays. The wild-type and isogenic mutants of S. gordonii were compared initially for competition with S. sanguinis SK36 for binding to sHA. Like the wild-type DL1, mutants with mutations in scaA, abpAB, sspAB, and cshAB reduced adhesion of S. sanguinis to sHA by 80 to more than 95% when present at 10- and 50-fold excesses, respectively (Table 3). Compared to the wild type, however, the srtA and hsa mutants used at identical input doses were unable to compete with S. sanguinis (Table 3). Indeed, S. sanguinis binding may be slightly increased in the presence of 10- and 50-fold excesses of the srtA mutant. The srtA-complemented mutant, however, regained the ability to compete, inhibiting by 73% and 85%, respectively (Table 3). At a 10-fold excess, the hsa mutant reduced S. sanguinis binding by only 13%, which increased to 39% at a 50-fold excess. Levels of inhibition similar to those of wild-type S. gordonii, however, were shown upon complementation of the hsa mutation. When present at 10- and 50-fold excesses, the hsa-complemented mutant reduced S. sanguinis adhesion by 60% and 94%, respectively (Table 3).
TABLE 3.
Adhesion of S. sanguinis SK36 to sHA in the presence of excess competing species
| Competing S. gordonii strain | % of S. sanguinis cells bound relative to control (mean ± SD) witha:
|
|
|---|---|---|
| 10-fold excess competitor | 50-fold excess competitor | |
| Control (no competitor) | 100.0 ± 11.3 | 100.0 ± 11.3 |
| DL1 (Challis) | 13.3 ± 2.6 | 3.4 ± 0.4 |
| scaA mutant (OB470) | 10.7 ± 1.0 | 3.7 ± 1.4 |
| abpAB mutant | 12.4 ± 1.3 | 4.9 ± 0.2 |
| sspAB mutant (UB1360) | 8.3 ± 1.3 | 4.4 ± 1.9 |
| cshAB mutant (OB277) | 7.5 ± 1.4 | 2.4 ± 0.2 |
| sspAB cshAB mutant (OB390) | 17.4 ± 7.8 | 4.1 ± 1.8 |
| srtA mutant | 116.0 ± 12.1** | 126.7 ± 30.8** |
| srtA-complemented strain | 26.8 ± 9.8 | 15.0 ± 5.9 |
| hsa mutant | 86.8 ± 16.7** | 61.1 ± 8.5** |
| hsa-complemented strain | 39.8 ± 6.8** | 5.5 ± 0.9 |
Radioactively labeled S. sanguinis SK36 cells were incubated with sHA in the presence of a 10- or 50-fold excess of unlabeled competing streptococcal cells. Numbers of attached S. sanguinis cells were then determined as described in Materials and Methods. As a control, S. sanguinis was incubated with sHA in the absence of any competing cells, and these adhesion levels were set to 100%.
, value is significantly higher (P < 0.01) than that for wild-type DL1.
Each mutant was then compared for adhesion in the presence of excess S. sanguinis. In the presence of a 10-fold excess of S. sanguinis cells, binding by wild-type S. gordonii DL1 or abpAB or cshAB mutants was unaffected. In the presence of S. sanguinis, adhesion of scaA, sspAB, and sspABcshAB mutants tended to increase by 15 to 20%; the increase was statistically significant (P < 0.05) for the scaA mutant (Table 4). Adhesion of wild-type S. gordonii and the scaA mutant was unaffected by 50-fold excess S. sanguinis cells, while binding by abpAB, sspAB, cshAB, or sspAB cshAB mutants was reduced by just 10 to 25% (Table 4). Once again, the srtA and hsa mutants exhibited a different profile. In the presence of 10-fold excess S. sanguinis cells, adhesion by the srtA mutant was unaffected, but at 50-fold excess, its binding was inhibited by 70% (Table 4). Similar inhibition was seen with the hsa mutant, the binding of which was also reduced by 40% at a 10-fold excess of S. sanguinis cells (Table 4). Like wild-type cells, the srtA and hsa-complemented mutants showed no significant reduction in adhesion, even at a 50-fold excess of S. sanguinis cells (Table 4).
TABLE 4.
Adhesion of wild-type S. gordonii DL1 and isogenic mutants to sHA in the presence of excess competing S. sanguinis cells
| S. gordonii strain | % of S. gordonii cells bound relative to control (mean ± SD) witha:
|
|
|---|---|---|
| 10-fold excess SK36 cells | 50-fold excess SK36 cells | |
| Control (no competitor) | 100.0 | 100.0 |
| DL1 (Challis) | 111.1 ± 6.3 | 89.8 ± 4.8 |
| scaA mutant (OB470) | 118.8 ± 14.1 | 90.4 ± 12.0 |
| abpAB mutant | 105.9 ± 5.6 | 77.6 ± 11.2** |
| sspAB mutant (UB1360) | 117.6 ± 13.6 | 74.6 ± 9.0** |
| cshAB mutant (OB277) | 102.8 ± 8.7 | 88.3 ± 2.9 |
| sspAB cshAB mutant (OB390) | 121.9 ± 34.4 | 87.4 ± 1.7** |
| srtA mutant | 102.6 ± 11.5 | 30.5 ± 7.9** |
| srtA-complemented strain | 105.0 ± 6.6 | 79.0 ± 15.0 |
| hsa mutant | 58.9 ± 8.2** | 29.5 ± 5.5** |
| hsa-complemented strain | 93.4 ± 13.9 | 94.7 ± 9.8 |
Radioactively labeled S. gordonii cells were incubated with sHA in the presence of a 10- or 50-fold excess of unlabeled S. sanguinis cells. Numbers of attached S. gordonii cells were then determined as described in Materials and Methods. As a control, S. gordonii strains were incubated with sHA in the absence of any competing cells, and these adhesion levels were set to 100%.
, value is significantly lower (P < 0.01) than that for controls in absence of competition.
DISCUSSION
Dental plaque is a dynamic environment in which microbes must overcome constant challenges to survive. With more than 600 species occupying the oral cavity at any one time, bacteria relentlessly compete or cooperate with other microflora for available receptors, while contending with a multitude of host defenses. In addition, bacteria must colonize in continuously changing environmental conditions generated by salivary flow, vacillating temperature and humidity, and dietary intake. The success with which each species can utilize available resources and tolerate adverse conditions determines its ability to thrive and thereby establish its own unique environmental niche. Collectively, these niches then specify the polymicrobial community that constitutes dental plaque.
Employing one or more adhesins, bacteria such as S. gordonii and S. sanguinis interact with a wide range of potential host receptors and other oral microbes in the vicinity to facilitate colonization of the oral cavity. Salivary pellicle constituents identified as putative receptors for S. sanguinis can also be utilized by S. gordonii and vice versa. Such receptors include proline-rich proteins (5, 40), glycoproteins (31, 40, 49), salivary agglutinin (3, 9), secretory immunoglobulin A (sIgA) (24, 39), lysozyme (38, 55), and lactoferrin (38). An ability to bind directly to α-amylase has been shown for S. gordonii and is lacking for S. sanguinis (45), while S. sanguinis has been shown to utilize a binding site formed by a complex of α-amylase and the light chain of sIgA (7). Given the genetic similarities between these two species (17), it is perhaps unsurprising that they showed similar isotherms for binding to sHA. Despite this similarity in binding, however, S. gordonii and S. sanguinis differed significantly in their abilities to compete with one another to adhere.
Remarkably, the numbers of S. gordonii cells bound to sHA were unaffected by the presence of excess S. sanguinis cells. Conversely, excess S. gordonii cells significantly reduced S. sanguinis adhesion levels, an example of interspecies antagonism for binding sites. The principle that colonization of the tooth surface by one bacterium could impair attachment by a second species has been fundamental to the models of plaque development proposed over the years. This study provides, however, the first proof of principle of a specific surface-expressed adhesin playing a role in the mechanism of interspecies competition for adhesion to the tooth surface.
Other forms of interspecies antagonism among oral bacteria are often associated with the production of bacteriocins or toxic metabolic by-products (8, 13, 21). S. gordonii DL1 (Challis) produces both bacteriocins (58) and hydrogen peroxide (2), which impair bacterial replication. In our experimental conditions, however, cells neither synthesize new proteins (M. C. Herzberg and H. F. Jenkinson, unpublished observations) nor replicate during the short period of incubation with sHA. Furthermore, extensive washing of the cells prior to incubation with sHA would effectively remove any products potentially released during the labeling phase. Thus, since such extracellular antagonisms are unlikely to be operative, the competition with S. gordonii appears to reflect the specific inability of S. sanguinis to bind to sHA in the presence of the competitor.
To characterize the basis of its competitive advantage, a series of isogenic S. gordonii adhesin mutants were screened. Mutants with mutations in antigen I/II polypeptides (3), Csh adhesins (30), and amylase-binding protein (Abp) (42, 44, 45) were selected, since there is evidence that these enable colonization. In addition, Hamada et al. (9) recently found that synthetic SspB peptides of S. gordonii 10558 could inhibit the binding of S. sanguinis 10556 to salivary components, as detected using a BIAcore system. Abp expression by S. gordonii is also a taxonomic distinction from S. sanguinis, which lacks the abpAB genes (20). The sialic acid-binding protein Hsa (48) was studied since abundant sialylated proteins in saliva could form potential binding sites in the salivary pellicle (60). ScaA was tested since it forms part of a compensatory adhesin mechanism in S. gordonii utilized during sHA biofilm formation (65), while a sortase (SrtA) mutant allowed a general screen for the role of LPXTG-containing adhesins. Despite their putative roles in facilitating colonization of the oral cavity, loss of the antigen I/II polypeptides or the Csh or Abp adhesins had no significant effects on the ability of S. gordonii to bind to sHA or to effectively compete with S. sanguinis for adhesion. The functions of such adhesins may be compensated for by altered expression of alternative adhesins, a system that has recently been demonstrated during S. gordonii biofilm formation in vitro (65). By contrast, mutations in both the srtA and hsa genes strongly impaired S. gordonii binding to sHA. In addition, these mutants were less effective competitors than the parent strain, and excess S. sanguinis cells inhibited their adhesion to sHA. This implied that LPXTG-containing adhesins, notably Hsa, are required for maximal binding to sHA by S. gordonii and for competition resistance against S. sanguinis. This hypothesis was then further confirmed, with restoration of wild-type S. gordonii competitive capabilities upon complementation of the srtA or hsa mutations.
Hsa is a member of a family of serine-rich repeat polypeptides common among oral streptococci. Hsa or its homologue, GspB, have been found in all strains of S. gordonii tested to date. These proteins, originally identified in S. gordonii strains DL1 and M99, respectively, are nearly identical, and both have been shown to promote attachment to platelets (48, 49, 51, 63), epithelial cells (15), and the α2-3-linked sialoglycoconjugates of salivary mucins and sIgA (26, 52). An Hsa-like protein, designated SrpA, was recently identified in S. sanguinis strains SK36 and 133-79 (34, 47) and S. cristatus CC5A (10). SrpA also mediates adhesion to platelets, but expression appears to be strain dependent (34, 47). A serine-rich repeat protein (Fap1) similar to Hsa is expressed by S. parasanguinis FW213 (61, 62) and appears to promote adhesion to sHA (46).
The GspB homologues can be split into two groups based on the isoelectric point (pI) of the intervening region between the two sets of serine-rich repeats (51). Hsa and GspB have an intervening region with a basic pI (termed the basic region [BR]), and SrpA and Fap1 have region with an acidic pI (termed the acidic region [AR]). The BR mediates adhesion of both S. gordonii strains DL1 and M99 to carbohydrate moieties on platelets, but variations in primary sequence generate, at least in part, differences in sialylated glycoprotein specificities and affinities between these strains (51). By contrast, the AR-containing GspB homologues of Streptococcus agalactiae and S. parasanguinis bind poorly to platelets (34, 51). SrpA of S. sanguinis does mediate adhesion to platelet receptor GPIbα in a sialic acid-dependent manner (34). However, unlike the AR of S. agalactiae, the SrpA AR is somewhat similar to that of Hsa, sharing 48% sequence identity (51). Thus, although the carbohydrate structures recognized by SrpA have yet to be determined, it might be predicted that they will prove to be similar to that of S. gordonii. Nevertheless, given the variation in primary sequence and the evidence that other domains within the protein and glycosylation levels may influence adhesion (49, 50), it seems likely that differences in specific binding properties could exist between these proteins. In this study, all Hsa-carrying strains were strong competitors against S. sanguinis, whereas species with an acidic product of the srpA gene competed poorly. We speculate, therefore, that the presence of an Hsa-like adhesin and the structure of the intervening BR specify the competitive capabilities of a strain. S. gordonii srtA and hsa mutants do, however, partially bind sHA and compete with S. sanguinis. Hence, other, non-LPXTG-containing adhesins may also facilitate these interactions, albeit it to a lesser extent. Thus, for some streptococcal species and strains, adhesins other than Hsa may play major roles in sHA adhesion. Nonetheless, for S. gordonii, Hsa is essential for efficient binding.
Competitive binding of S. gordonii Hsa is strongly suggested to occur through interactions with a sialylated pellicle constituent(s), maintaining adhesion to sHA even in the presence of competing cells. Likely pellicle receptors for Hsa include gp340, mucin MG2, and the sIgA heavy chain (15, 52). By elucidating the salivary receptor(s) in this competition phenomenon, we will determine whether Hsa-mediated attachment of S. gordonii directly blocks the same receptor utilized by S. sanguinis or prevents access of S. sanguinis to its own pellicle receptor(s). For example, both S. gordonii and S. sanguinis have been shown to bind salivary mucin MG2 (35, 52). Alternatively, salivary pellicle binding sites identified for S. sanguinis include a complex of α-amylase and the light chain of sIgA (7). One could speculate, therefore, that S. gordonii Hsa may bind to the sialylated residues of sIgA and sterically interfere with attachment of S. sanguis to the α-amylase-sIgA complex. Another mechanism that cannot be ruled out is that Hsa facilitates binding of S. gordonii to surface components of S. sanguinis and in this way physically blocks attachment of S. sanguinis to the salivary pellicle. It should be noted, however, that no aggregates of cells could be visualized during these studies, and the one-sided nature of this competition phenomenon makes this unlikely. It is also possible that the range of binding specificities of Hsa allow S. gordonii to target a larger number of salivary pellicle receptors in comparison to S. sanguinis, enabling S. gordonii to attach more effectively. Future work will now aim to elucidate the precise mechanism(s) underlying this competitive binding phenomenon in greater detail.
In vivo, oral streptococci represent approximately 20% of the total bacteria present in saliva (29) and can constitute up to 80% of early plaque (37). Regardless of the detection technique employed, S. sanguinis is consistently found in excess relative to S. gordonii in saliva (22, 54), dental plaque (4, 28, 32), and oral soft tissue surfaces (28). Nevertheless, S. gordonii seems to defy the evolutionary odds and persists over time in comparatively low numbers within the oral cavity, even when outnumbered by one of its major competitors, S. sanguinis. Thus, in facilitating adhesion to the salivary pellicle, S. gordonii Hsa also appears to serve as an environmental constraint, limiting the binding of S. sanguinis to the same intraoral sites. Consequently, S. gordonii is able to establish an environmental niche in the presence of S. sanguinis, which allows these two genetically similar species to coexist. S. gordonii can, however, be outcompeted by other oral streptococci, namely, S. oralis, which can also be isolated from the same oral sites. This may therefore explain why, despite its ability to bind to the salivary pellicle, S. gordonii is not present in abundance within the oral cavity but rather consistently maintains a low-level presence. Since the early-colonizer population influences the overall composition of dental plaque, such mechanisms have important implications for our understanding of the microbial ecology of dental plaque and for the development of novel preventive and control regimens.
Acknowledgments
This work was supported by NIH grant R01 DE08590.
Footnotes
Published ahead of print on 2 February 2007.
REFERENCES
- 1.Appelbaum, B., E. Golub, S. C. Holt, and B. Rosan. 1979. In vitro studies of dental plaque formation: adsorption of oral streptococci to hydroxyapatite. Infect. Immun. 25:717-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnard, J. P., and M. W. Stinson. 1996. The alpha-hemolysin of Streptococcus gordonii is hydrogen peroxide. Infect. Immun. 64:3853-3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demuth, D. R., Y. Duan, W. Brooks, A. R. Holmes, R. McNab, and H. F. Jenkinson. 1996. Tandem genes encode cell-surface polypeptides SspA and SspB which mediate adhesion of the oral bacterium Streptococcus gordonii to human and bacterial receptors. Mol. Microbiol. 20:403-413. [DOI] [PubMed] [Google Scholar]
- 4.Frandsen, E. V., V. Pedrazzoli, and M. Kilian. 1991. Ecology of viridans streptococci in the oral cavity and pharynx. Oral Microbiol. Immunol. 6:129-133. [DOI] [PubMed] [Google Scholar]
- 5.Gibbons, R. J., D. I. Hay, and D. H. Schlesinger. 1991. Delineation of a segment of adsorbed salivary acidic proline-rich proteins which promotes adhesion of Streptococcus gordonii to apatitic surfaces. Infect. Immun. 59:2948-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gong, K., and M. C. Herzberg. 1997. Streptococcus sanguis expresses a 150-kilodalton two-domain adhesin: characterization of several independent adhesin epitopes. Infect. Immun. 65:3815-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong, K., L. Mailloux, and M. C. Herzberg. 2000. Salivary film expresses a complex, macromolecular binding site for Streptococcus sanguis. J. Biol. Chem. 275:8970-8974. [DOI] [PubMed] [Google Scholar]
- 8.Grenier, D. 1996. Antagonistic effect of oral bacteria towards Treponema denticola. J. Clin. Microbiol. 34:1249-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamada, T., M. Kawashima, H. Watanabe, J. Tagami, and H. Senpuku. 2004. Molecular interactions of surface protein peptides of Streptococcus gordonii with human salivary components. Infect. Immun. 72:4819-4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handley, P. S., F. F. Correia, K. Russell, B. Rosan, and J. M. DiRienzo. 2005. Association of a novel high molecular weight, serine-rich protein (SrpA) with fibril-mediated adhesion of the oral biofilm bacterium Streptococcus cristatus. Oral Microbiol. Immunol. 20:131-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heddle, C., A. H. Nobbs, N. S. Jakubovics, M. Gal, J. P. Mansell, D. Dymock, and H. F. Jenkinson. 2003. Host collagen signal induces antigen I/II adhesin and invasin gene expression in oral Streptococcus gordonii. Mol. Microbiol. 50:597-607. [DOI] [PubMed] [Google Scholar]
- 12.Herzberg, M. C., K. Gong, G. D. MacFarlane, P. R. Erickson, A. H. Soberay, P. H. Krebsbach, G. Manjula, K. Schilling, and W. H. Bowen. 1990. Phenotypic characterization of Streptococcus sanguis virulence factors associated with bacterial endocarditis. Infect. Immun. 58:515-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hillman, J. D., S. S. Socransky, and M. Shivers. 1985. The relationships between streptococcal species and periodontopathic bacteria in human dental plaque. Arch. Oral Biol. 30:791-795. [DOI] [PubMed] [Google Scholar]
- 14.Holmes, A. R., R. McNab, and H. F. Jenkinson. 1996. Candida albicans binding to the oral bacterium Streptococcus gordonii involves multiple adhesin-receptor interactions. Infect. Immun. 64:4680-4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakubovics, N. S., S. W. Kerrigan, A. H. Nobbs, N. Strömberg, C. J. van Dolleweerd, D. M. Cox, C. G. Kelly, and H. F. Jenkinson. 2005. Functions of cell surface-anchored antigen I/II family and Hsa polypeptides in interactions of Streptococcus gordonii with host receptors. Infect. Immun. 73:6629-6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkinson, H. F., and R. J. Lamont. 1997. Streptococcal adhesion and colonization. Crit. Rev. Oral Biol. Med. 8:175-200. [DOI] [PubMed] [Google Scholar]
- 17.Kawamura, Y., X. G. Hou, F. Sultana, H. Miura, and T. Ezaki. 1995. Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int. J. Syst. Bacteriol. 45:406-408. [DOI] [PubMed] [Google Scholar]
- 18.Kilian, M., and K. Holmgren. 1981. Ecology and nature of immunoglobulin A1 protease-producing streptococci in the human oral cavity and pharynx. Infect. Immun. 31:868-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kilian, M., L. Mikkelsen, and J. Henrichsen. 1989. Taxonomic study of viridans streptococci: description of Streptococcus gordonii sp. nov. and emended descriptions of Streptococcus sanguis (White and Niven 1946), Streptococcus oralis (Bridge and Sneath 1982), and Streptococcus mitis (Andrewes and Horder 1906). Int. J. Syst. Bacteriol. 39:471-484. [Google Scholar]
- 20.Kilian, M., and B. Nyvad. 1990. Ability to bind salivary alpha-amylase discriminates certain viridans group streptococcal species. J. Clin. Microbiol. 28:2576-2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreth, J., J. Merritt, W. Shi, and F. Qi. 2005. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J. Bacteriol. 187:7193-7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, J., E. J. Helmerhorst, C. W. Leone, R. F. Troxler, T. Yaskell, A. D. Haffajee, S. S. Socransky, and F. G. Oppenheim. 2004. Identification of early microbial colonizers in human dental biofilm. J. Appl. Microbiol. 97:1311-1318. [DOI] [PubMed] [Google Scholar]
- 23.Liljemark, W. F., and S. V. Schauer. 1977. Competitive binding among oral streptococci to hydroxyapatite. J. Dent. Res. 56:157-165. [DOI] [PubMed] [Google Scholar]
- 24.Liljemark, W. F., C. G. Bloomquist, and J. C. Ofstehage. 1979. Aggregation and adherence of Streptococcus sanguis: role of human salivary immunoglobulin A. Infect. Immun. 26:1104-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liljemark, W. F., C. G. Bloomquist, and G. R. Germaine. 1981. Effect of bacterial aggregation on the adherence of oral streptococci to hydroxyapatite. Infect. Immun. 31:935-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loimaranta, V., N. S. Jakubovics, J. Hytönen, J. Finne, H. F. Jenkinson, and N. Strömberg. 2005. Fluid- or surface-phase human salivary scavenger protein gp340 exposes different bacterial recognition properties. Infect. Immun. 73:2245-2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macrina, F. L., R. P. Evans, J. A. Tobian, D. L. Hartley, D. B. Clewell, and K. R. Jones. 1983. Novel shuttle plasmid vehicles for Escherichia-Streptococcus transgeneric cloning. Gene 25:145-150. [DOI] [PubMed] [Google Scholar]
- 28.Mager, D. L., L. A. Ximenez-Fyvie, A. D. Haffajee, and S. S. Socransky. 2003. Distribution of selected bacterial species on intraoral surfaces. J. Clin. Periodontol. 30:644-654. [DOI] [PubMed] [Google Scholar]
- 29.Marsh, P. D. 1999. Microbiologic aspects of dental plaque and dental caries. Dent. Clin. N. Am. 43:599-614. [PubMed] [Google Scholar]
- 30.McNab, R., H. F. Jenkinson, D. M. Loach, and G. W. Tannock. 1994. Cell-surface-associated polypeptides CshA and CshB of high molecular mass are colonization determinants in the oral bacterium Streptococcus gordonii. Mol. Microbiol. 14:743-754. [DOI] [PubMed] [Google Scholar]
- 31.Murray, P. A., A. Prakobphol, T. Lee, C. I. Hoover, and S. J. Fisher. 1992. Adherence of oral streptococci to salivary glycoproteins. Infect. Immun. 60:31-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nyvad, B., and M. Kilian. 1990. Comparison of the initial streptococcal microflora on dental enamel in caries-active and in caries-inactive individuals. Caries Res. 24:267-272. [DOI] [PubMed] [Google Scholar]
- 33.Pakula, R., and W. Walczak. 1963. On the nature of competence of transformable streptococci. J. Gen. Microbiol. 31:125-133. [DOI] [PubMed] [Google Scholar]
- 34.Plummer, C., H. Wu, S. W. Kerrigan, G. Meade, D. Cox, and C. W. I. Douglas. 2005. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 129:101-109. [DOI] [PubMed] [Google Scholar]
- 35.Plummer, C., and C. W. Douglas. 2006. Relationship between the ability of oral streptococci to interact with platelet glycoprotein Ibalpha and with the salivary low-molecular-weight mucin, MG2. FEMS Immunol. Med. Microbiol. 48:390-399. [DOI] [PubMed] [Google Scholar]
- 36.Rogers, J. D., E. M. Haase, A. E. Brown, C. W. I. Douglas, J. P. Gwynn, and F. A. Scannapieco. 1998. Identification and analysis of a gene (abpA) encoding a major amylase-binding protein in Streptococcus gordonii. Microbiology 144:1223-1233. [DOI] [PubMed] [Google Scholar]
- 37.Rosan, B., and R. J. Lamont. 2000. Dental plaque formation. Microbes Infect. 2:1599-1607. [DOI] [PubMed] [Google Scholar]
- 38.Rudney, J. D., Z. Ji, C. J. Larson, W. F. Liljemark, and K. L. Hickey. 1995. Saliva protein binding to layers of oral streptococci in vitro and in vivo. J. Dent. Res. 74:1280-1288. [DOI] [PubMed] [Google Scholar]
- 39.Ruhl, S., A. L. Sandberg, M. F. Cole, and J. O. Cisar. 1996. Recognition of immunoglobulin A1 by oral actinomyces and streptococcal lectins. Infect. Immun. 64:5421-5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruhl, S., A. L. Sandberg, and J. O. Cisar. 2004. Salivary receptors for the proline-rich protein-binding and lectin-like adhesins of oral actinomyces and streptococci. J. Dent. Res. 83:505-510. [DOI] [PubMed] [Google Scholar]
- 41.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 42.Scannapieco, F. A., G. Torres, and M. J. Levine. 1993. Salivary alpha-amylase: role in dental plaque and caries formation. Crit. Rev. Oral Biol. Med. 4:301-307. [DOI] [PubMed] [Google Scholar]
- 43.Scannapieco, F. A. 1994. Saliva-bacterium interactions in oral microbial ecology. Crit. Rev. Oral Biol. Med. 5:203-248. [DOI] [PubMed] [Google Scholar]
- 44.Scannapieco, F. A., L. Solomon, and R. O. Wadenya. 1994. Emergence in human dental plaque and host distribution of amylase-binding streptococci. J. Dent. Res. 73:1627-1635. [DOI] [PubMed] [Google Scholar]
- 45.Scannapieco, F. A., G. I. Torres, and M. J. Levine. 1995. Salivary amylase promotes adhesion of oral streptococci to hydroxyapatite. J. Dent. Res. 74:1360-1366. [DOI] [PubMed] [Google Scholar]
- 46.Stephenson, A. E., H. Wu, J. Novak, M. Tomana, K. Mintz, and P. Fives-Taylor. 2002. The Fap1 fimbrial adhesin is a glycoprotein: antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol. Microbiol. 43:147-157. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi, Y., A. L. Sandberg, S. Ruhl, J. Muller, and J. O. Cisar. 1997. A specific cell surface antigen of Streptococcus gordonii is associated with bacterial hemagglutination and adhesion to alpha2-3-linked sialic acid-containing receptors. Infect. Immun. 65:5042-5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi, Y., K. Konishi, J. O. Cisar, and M. Yoshikawa. 2002. Identification and characterization of hsa, the gene encoding the sialic acid-binding adhesin of Streptococcus gordonii DL1. Infect. Immun. 70:1209-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi, Y., A. Yajima, J. O. Cisar, and K. Konishi. 2004. Functional analysis of the Streptococcus gordonii DL1 sialic acid-binding adhesin and its essential role in bacterial binding to platelets. Infect. Immun. 72:3876-3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takamatsu, D., B. A. Bensing, and P. M. Sullam. 2004. Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol. Microbiol. 52:189-203. [DOI] [PubMed] [Google Scholar]
- 51.Takamatsu, D., B. A. Bensing, H. Cheng, G. A. Jarvis, I. R. Siboo, J. A. López, J. M. Griffiss, and P. M. Sullam. 2005. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibα. Mol. Microbiol. 58:380-392. [DOI] [PubMed] [Google Scholar]
- 52.Takamatsu, D., B. A. Bensing, A. Prakobphol, S. J. Fisher, and P. M. Sullam. 2006. Binding of the streptococcal surface glycoproteins GspB and Hsa to human salivary proteins. Infect. Immun. 74:1933-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao, L., D. J. LeBlanc, and J. J. Ferretti. 1992. Novel streptococcal-integration shuttle vectors for gene cloning and inactivation. Gene 120:105-110. [DOI] [PubMed] [Google Scholar]
- 54.Tappuni, A. R., and S. J. Challacombe. 1993. Distribution and isolation frequency of eight streptococcal species in saliva from predentate and dentate children and adults. J. Dent. Res. 72:31-36. [DOI] [PubMed] [Google Scholar]
- 55.Tellefson, L. M., and G. R. Germaine. 1986. Adherence of Streptococcus sanguis to hydroxyapatite coated with lysozyme and lysozyme-supplemented saliva. Infect. Immun. 51:750-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Terleckyj, B., and G. D. Shockman. 1975. Amino acid requirements of Streptococcus mutans and other oral streptococci. Infect. Immun. 11:656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terleckyj, B., N. P. Willett, and G. D. Shockman. 1975. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect. Immun. 11:649-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tompkins, G. R., M. A. Peavey, K. R. Birchmeier, and J. R. Tagg. 1997. Bacteriocin production and sensitivity among coaggregating and noncoaggregating oral streptococci. Oral Microbiol. Immunol. 12:98-105. [DOI] [PubMed] [Google Scholar]
- 59.Whittaker, C. J., C. M. Klier, and P. E. Kolenbrander. 1996. Mechanisms of adhesion by oral bacteria. Annu. Rev. Microbiol. 50:513-552. [DOI] [PubMed] [Google Scholar]
- 60.Wu, A. M., G. Csako, and A. Herp. 1994. Structure, biosynthesis and function of salivary mucins. Mol. Cell Biochem. 137:39-55. [DOI] [PubMed] [Google Scholar]
- 61.Wu, H., K. P. Mintz, M. Ladha, and P. M. Fives-Taylor. 1998. Isolation and characterization of Fap1, a fimbriae-associated adhesin of Streptococcus parasanguis FW213. Mol. Microbiol. 28:487-500. [DOI] [PubMed] [Google Scholar]
- 62.Wu, H., and P. M. Fives-Taylor. 1999. Identification of dipeptide repeats and a cell wall sorting signal in the fimbriae-associated adhesin, Fap1 of Streptococcus parasanguis. Mol. Microbiol. 34:1070-1081. [DOI] [PubMed] [Google Scholar]
- 63.Yajima, A., Y. Takahashi, and K. Konishi. 2005. Identification of platelet receptors for the Streptococcus gordonii DL1 sialic acid-binding adhesin. Microbiol. Immunol. 49:795-800. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, Y., Y. Lei, A. Khammanivong, and M. C. Herzberg. 2004. Identification of a novel two-component system in Streptococcus gordonii V288 involved in biofilm formation. Infect. Immun. 72:3489-3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang, Y., Y. Lei, A. Nobbs, A. Khammanivong, and M. C. Herzberg. 2005. Inactivation of S. gordonii SspAB alters expression of multiple adhesin genes. Infect. Immun. 73:3351-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]