

Abstract
Pseudomonas fluorescens strain KU-7 is a prototype microorganism that metabolizes 2-nitrobenzoate (2-NBA) via the formation of 3-hydroxyanthranilate (3-HAA), a known antioxidant and reductant. The initial two steps leading to the sequential formation of 2-hydroxy/aminobenzoate and 3-HAA are catalyzed by a NADPH-dependent 2-NBA nitroreductase (NbaA) and 2-hydroxylaminobenzoate mutase (NbaB), respectively. The 216-amino-acid protein NbaA is 78% identical to a plasmid-encoded hypothetical conserved protein of Polaromonas strain JS666; structurally, it belongs to the homodimeric NADH:flavin mononucleotide (FMN) oxidoreductase-like fold family. Structural modeling of complexes with the flavin, coenzyme, and substrate suggested specific residues contributing to the NbaA catalytic activity, assuming a ping-pong reaction mechanism. Mutational analysis supports the roles of Asn40, Asp76, and Glu113, which are predicted to form the binding site for a divalent metal ion implicated in FMN binding, and a role in NADPH binding for the 10-residue insertion in the β5-α2 loop. The 181-amino-acid sequence of NbaB is 35% identical to the 4-hydroxylaminobenzoate lyases (PnbBs) of various 4-nitrobenzoate-assimilating bacteria, e.g., Pseudomonas putida strain TW3. Coexpression of nbaB with nbaA in Escherichia coli produced a small amount of 3-HAA from 2-NBA, supporting the functionality of the nbaB gene. We also showed by gene knockout and chemotaxis assays that nbaY, a chemoreceptor NahY homolog located downstream of the nbaA gene, is responsible for strain KU-7 being attracted to 2-NBA. NbaY is the first chemoreceptor in nitroaromatic metabolism to be identified, and this study completes the gene elucidation of 2-NBA metabolism that is localized within a 24-kb chromosomal locus of strain KU-7.
Nitroreduction, catalyzed by the NAD(P)H-dependent nitroreductases, is the quintessential first step in the catabolism of a variety of structurally diverse nitroaromatic compounds. The commonly known oxygen-insensitive and type I nitroreductases are flavoproteins that mediate the sequential transfer of two electrons from NAD(P)H to the nitro group to produce nitroso and hydroxylamine derivatives (9). Ammonium ions are subsequently removed from the hydroxylamine intermediate through two possible routes: a lyase-mediated reaction with the direct elimination of ammonia to produce the corresponding catechol (e.g., degradation of 4-nitrobenzoate, 4-nitrotoluene, and 3-nitrophenol) and a mutase-mediated Bamberger-type rearrangement to produce 2-aminophenols or benzoates resulting in a much later release of ammonia (e.g., metabolism of nitrobenzene, 3-nitrophenol, 4-chloronitrobenzene, 4-nitrotoluene, and 2,4,6-trinitrotoluene) (for recent reviews, see references 20 and 51).
The metabolism of 2-nitrobenzoate (2-NBA) by Pseudomonas fluorescens strain KU-7 afforded the first example in a prokaryotic organism of the formation of 3-hydroxyanthranilate (3-HAA), a metabolite that is otherwise well known in the kynurenine pathway of tryptophan metabolism, and the biosynthesis of nicotinic acid in yeast and mammalian systems (27, 49, 53). This organism is unable to grow on 3-nitrobenzoate or nitrophenols as the sole carbon or nitrogen source (27). Recently, Colabroy and Begley (15) identified a new tryptophan catabolic pathway in Burkholderia cepacia J2315 that includes the formation of 3-HAA. Crude extracts of strain KU-7 cells converted 2-NBA to 3-HAA with the oxidation of 2 mol NADPH. In this pathway, 2-NBA is first reduced to 2-hydroxy/aminobenzoate acid (2-HABA) by an uncharacterized NADPH-dependent reductase, followed by a mutase-catalyzed conversion of 2-HABA to 3-HAA via a Bamberger-type rearrangement (27). Previously, we described the genetic locus, consisting of nbaEXHJIGFCDR, that is responsible for the conversion of 3-HAA to a Krebs cycle intermediate in strain KU-7 (49). A highlight of this pathway is the establishment of the novel identity of nbaC, encoding 3-HAA 3,4-dioxygenase, and nbaD, encoding 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase, that catalyze the ring opening of 3-HAA to form 2-amino-3-carboxymuconate-6-semialdehyde and a decarboxylation step to form 2-aminomuconate-6-semialdehyde, respectively (49). The latter compound is a convergent metabolite in the metabolism of nitrobenzene, 2-aminophenol, 3-HAA, and 4-amino-3-hydroxybenzoic acid (49, 52).
To complete the full gene complement for 2-NBA metabolism in strain KU-7, we describe here the gene localization and characterization of initial genes nbaA and nbaB, encoding 2-NBA nitroreductase and 2-HABA mutase, respectively, by nucleotide sequencing and functional analysis. In particular, a homology model of NbaA was built to probe the structure-function relationship of this protein. Additionally, we determined the chemotactic property of a methyl-accepting chemotaxis transducer-like open-reading frame (nbaY) that is located downstream of nbaA. This study has also led to the discovery of a related gene locus in the Polaromonas sp. strain JS666 genome (Joint Genome Institute database), an organism known to degrade cis-dichloroethene. The possible mobility of the nba gene locus is discussed.
(A portion of this work was presented at the American Society for Microbiology 9th International Conference on Pseudomonas, Quebec City, Canada, 6 to 10 September 2003.)
MATERIALS AND METHODS
Chemicals.
2-HABA was synthesized chemically as described previously by Bauer and Rosenthal (5). All other chemicals were of the highest purity commercially available.
Bacterial strains, culture conditions, and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1. The culture conditions for the Pseudomonas and Escherichia coli strains, their growth media, except for MS-S medium (MY medium without the yeast extract and 2-NBA but replaced with 0.3% succinate), and antibiotic selection were described previously (27, 49).
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| BL21 | F−ompT hsdSB(rB− mB−) gal dcm | Novagen |
| DH5α | supE44 thi-1 recA1 hsdR17 endA1 gyrA (Nalr) Δ(lacIZYA-argF) U169 deoR [φ80dlac Δ(lacZ)M15] | 62 |
| S17-1 | recA pro thi hsdR RP4-2-Tc::Mu-Km::Tn7 Trar+ Tpr Smr | 66 |
| XL1-Blue | recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 [F′ laclZM15 Tn10 (Tetr)] | 10 |
| P. fluorescens | ||
| KU-7 | Wild type, 2-NBA− 4-HBA+ | 27 |
| KUM-9 | orf16::Tn5-31 Tp 2-NBA− 4-HBA+ | This study |
| KUM-19 | nbaA::Tn5-31 Tp 2-NBA− 4-HBA+ | This study |
| KUM-33 | nbaB::Tn5-31 Tp 2-NBA− 4-HBA+ | This study |
| KU-ΔY | nbaY deletion mutant | This study |
| Plasmids | ||
| pKN31 | Tn5-31 Tp transposon delivery plasmid; mob RP4 Kmr Tpr | 1 |
| pK18mobsacB | Negative selection vector; Kmr | 63 |
| pREP1 | Expression vector with T7 promoter, p15A origin; Cmr | 57 |
| pSD80 | Expression vector with tac promoter, ColE1 origin,r Apr | 68 |
| pT7-5 | Expression vector with T7 promoter, ColE1 origin,r Apr | 70 |
| pUC19 | Cloning vector with lac promoter, ColE1 origin,r Apr | 74 |
| pNBA3 | 9.3-kb HindIII fragment from P. fluorescens strain KU-7 in pUC19; Apr | 49 |
| pNBA5 | 6.2-kb EcoRI fragment from P. fluorescens strain KUM-19 in pUC19; Apr Tpr | This study |
| pNBA6 | 3.8-kb BamHI fragment from P. fluorescens strain KUM-19 in pUC19; Apr Tpr | This study |
| pSDnbaA | EcoRI*-PstI* fragment containing nbaA in pSD80 | This study |
| pSDnbaA-N40A | Replacement of Asn40 by Ala in NbaA in pSDnbaA | This study |
| pSDnbaA-H63A | Replacement of His63 by Ala in NbaA in pSDnbaA | This study |
| pSDnbaA-H69A | Replacement of His69 by Ala in NbaA in pSDnbaA | This study |
| pSDnbaA-D76A | Replacement of Asp76 by Ala in NbaA in pSDnbaA | This study |
| pSDnbaA-H110A | Replacement of His110 by Ala in NbaA in pSDnbaA | This study |
| pSDnbaA-E113A | Replacement of Gln113 by Ala in NbaA in pSDnbaA | This study |
| pSDnbaB | EcoRI*-PstI* fragment containing nbaB in pSD80 | This study |
| pSDnbaAnbaB | pSDnbaA plus ribosome binding site at HindIII and NheI plus nbaB cloned at AscI and NheI | This study |
| pNbaΔY | nbaY deletion cassette in pK18mobsacB | This study |
EcoRI* and PstI* are restriction endonucleases introduced by PCR design. 2-NBA+, growth on 2-NBA: 4-HBA+, growth on 4-hydroxybenzoate; 2-NBA−, no growth on 2-NBA.
DNA manipulations, sequencing, and analysis.
Chromosomal and plasmid DNA isolation was performed according to methods described previously (49). Purification of DNA fragments, labeling with digoxigenin (DIG)-11-dUTP using the DIG DNA labeling kit, and visualization with a DIG luminescent detection kit (Roche Diagnostics K.K., Tokyo, Japan) were previously described (27, 49). Southern blotting used a positively charged nylon membrane (Hybond-N+; Amersham Biosciences Corp., Piscataway, NJ), and other DNA manipulations such as DNA ligation were described previously by Sambrook et al. (61). Restriction endonucleases and T4 DNA ligase were obtained from New England Biolabs (Beverly, MA). Automated and BigDye terminator nucleotide sequencing were used described previously (49). DNA and protein sequence analyses were carried out using GENETYX-Mac software (Software Development Co., Ltd., Tokyo, Japan) as well as BLAST programs at the National Center for Biotechnology Information (NCBI) server (2).
Transposon mutagenesis and screening for 2-NBA-negative mutant strains.
Transposon mutagenesis was carried out in order to gain access to the 2-NBA catabolic pathway gene locus by the generation of mutant derivatives of strain KU-7 that are unable to grow on 2-NBA as a sole carbon source. As previously described (49), pKN31 was used as a delivery vehicle for transposon Tn5-31Tp and E. coli strain S17-1 as a donor in conjugation with wild-type strain KU-7 for the construction of strains KUM-4, etc. (Table 1). Kanamycin (Km) (50 μg/ml) was used as a selective marker for the transposon. Transconjugants of strain KU-7 were identified following incubation at 30°C for 2 days. Mutants that were not able to grow on 2-NBA were identified by replica plating onto MS medium containing 0.2% 2-NBA (12 mM) and Km as previously described (49).
Amplification of Tn5-31Tp flanking fragments by inverse PCR (IPCR) was performed according to a method described previously by Huang et al. (30). Total DNA prepared from the Tn5-31Tp mutant of strain KU-7 was digested with BamHI to cut the middle of Tn5-31Tp and the BamHI recognition sequence flanking the transposon insertion site. Following phenol-chloroform extraction and ethanol precipitation, the digested DNA was self-ligated in a final volume of 1 ml at a concentration of 0.3 to 0.5 μg in the presence of 3 U T4 DNA ligase (New England Biolabs) overnight at 16°C. The ligation mixture was extracted with phenol-chloroform, precipitated with ethanol, and resuspended in sterile distilled H2O to a concentration of 20 ng μl−1. To amplify the flanking sequences of the Tn5-31Tp insertion, a set of two parallel IPCR reactions using the ligation mixture as a template were performed using primers BL and IR1 and BR and IR1. Primers BL and BR (5′-GGGGACCTTGCACAGATAGC and 5′-CATTCCTGTAGCGGATGGAGATC, respectively) are sequences on both sides of the unique BamHI site of the transposon; primer IR1 (5′-GAGCAGAAGTTATCATGAACG) is based on the terminal inverted repeats of Tn5-31Tp. IPCR was performed in a volume of 25 μl with 25 ng DNA of the ligation mixture, 7.5 pmol of each primer, 200 μΜ each deoxynucleoside triphosphate, 2.5 μl of GeneAmp High Fidelity 10× PCR buffer with MgCl2, and 0.25 μl of GeneAmp High Fidelity enzyme mix (Applied Biosystems). For amplification, a 2-min initial denaturation step at 94°C was followed by 30 thermal cycles of denaturation at 94°C for 15 s, annealing at 50°C for 1 min, and extension at 68°C for 8 min. The PCR product was purified from a 1.0% agarose gel, and the sequence reaction was performed with primer IR1.
Construction of an nbaY deletion mutant.
A crossover PCR deletion product of nbaY was constructed as follows, based on the same strategy as that for the PCR overlap extension mutagenesis described previously by Link et al. (47). Figure S1 in the supplemental material depicts the strategy. At first, two different asymmetric PCRs were used to generate fragments to the left and right of nbaY with the following pairs of PCR primers: nbaY-Co (5′-CGGAATTCGCGAACTGCTGGACGTGCCT-3′) and nbaY-Ci (5′-TGTTTAAGTTTAGTGGATGGGGACCTGAGTCAGCTGTTAGG-3′) and nbaY-Ni (5′-CCCATCCACTAAACTTAAACAATTACGTAAACTAGTCACCG-3′) and nbaY-No (5′-CGGAATTCTATCTCAGAAGCCGAAGCCG-3′), respectively. The left and right fragments were then annealed at their overlapping regions (double-underlined complementary sequences) and amplified by PCR as a single fragment using primers nbaY-Co and nbaY-No. The crossover PCR deletion product was subcloned into the EcoRI site of pK18mobsacB, generating pNbaΔY. To facilitate this cloning, EcoRI recognition sites were introduced into the primer design of nbaY-Co and nbaY-No (underlined above).
Plasmid pNbaΔY was transferred into strain KU-7 by conjugation with E. coli S17-1 as described above. Transconjugants were first selected on MS-S agar containing Km. These transconjugants were not able to grow on MS-S agar containing 10% sucrose since the levansucrase-encoding gene (sacB) on the pK18mobsacB vector, when expressed, would provide a lethal phenotype to the cells growing on sucrose (18, 23). To select for a double-crossover event, a single colony was grown for 18 h in nonselective MS-S medium at 30°C. The cells were plated onto MS-S agar containing 10% sucrose and incubated for 24 h at 30°C. The resulting colonies were sensitive to Km, indicating the excision of the plasmid by a second crossover event. This second crossover either restores the wild-type situation or leads to a mutant with the desired deletion. The deletion mutants were analyzed by PCR and designated strain KU-ΔY.
Subcloning of nbaA and nbaB and expression in E. coli.
The DNA fragment carrying nbaA and nbaB was amplified from KU-7 genomic DNA by using KOD-Plus-DNA polymerase (Toyobo) with the following pairs of PCR primers and the desired restriction sites (EcoRI and PstI [underlined sequences]) to facilitate subsequent cloning: 5′-CGGAATTCATGACGCACATTGCAATGTCA and 5′-AAAACTGCAGTCAGGGAGTAATCGGAAAGA for nbaB and 5′-CGGAATTCATGTCCAATGCCCCGAATGCA and 5′-AAAACTGCAGTGAGTATTGTCAGGAAGT for nbaB.
In each case, the amplified DNA fragment was purified from an agarose gel, digested with the appropriate restriction enzymes, and cloned into the linearized pSD80 tac-inducible expression vector (66). The resultant recombinant plasmids were designated pSDnbaA and pSDnbaB (Table 1). DNA sequencing was performed to exclude the possibility of mutations of the amplified genes. The plasmid carrying E. coli BL21 cells was cultivated in 100 ml of LB medium containing 100 μg of ampicillin (Ap)/ml at 30°C. When the culture reached an A600 of 0.3 to 0.4, isopropyl-β-thio-d-galactoside (IPTG) was added to a final concentration of 1 mM in the medium. The cells were further cultured for 3 h, harvested by centrifugation, washed twice with 50 mM potassium phosphate buffer (pH 7.0), resuspended with the same buffer, and sonicated by two 30-s bursts with a Braun-Sonifier 250 apparatus. After centrifugation for 30 min at 18,000 × g at 4°C, the supernatant was used for the determination of enzyme activity.
Coexpression of nbaA and nbaB in E. coli.
Two strategies were used for the coexpression of nbaA and nbaB in E. coli: first, tandem expression of the two genes on a single plasmid and, second, expression of nbaA and nbaB on two compatible plasmids. In the tandem-gene construct, the strategy was to have a single transcript producing two independently translated polypeptides with the use of a strong ribosomal binding site located between the two genes. Toward this construction, a synthetic ribosomal binding site was introduced after NbaA into the original pSDnbaA clone at the HindIII and NheI sites using primers pSD80PC5′ (5′-AGCTTTGCACATATCGAGGTGAACATCACGCCCGGGTAAGGAGGTGGCGCGCCCTCGAGG) and pSD80PC3′ (5′-CTAGCCTCGAGGGCGCGCCACCTCCTTACCCGGGCGTGATGTTCACCTCGATATGTGCAA), producing pSD80.NbaA.SD. The synthetic region contains the ribosomal binding sequence (boldfaced) as well as the restriction endonuclease sites for HindIII, SmaI, AscI, XhoI, and NheI (underlined). NbaB was amplified from the original pSD80.NbaB clone using primers NbaB5′-AscI (5′-TTGGCGCGCCATGTCCAATGCCCCGAATG) and pSD80R (5′-GTTTTATCAGACCGCTTCTGCG). The product was ligated into pSD80.NbaA.SD at AscI and NheI, creating the single-plasmid clone pSDnbaAnbaB. The final plasmid was transformed into E. coli BL21(DE3) Rosetta (Novagen) cells containing the pRARE plasmid, which encodes rare E. coli tRNA codons, resulting in enhanced recombinant protein expression otherwise limited by codon usage. Ap and chloramphenicol (Cm) were used for plasmid selection and maintenance in the same cell. Cells were grown overnight in LB with Ap/Cm, subcultured (1:100), and induced with 1 mM IPTG at an optical density of 0.5 for 5 h.
The compatible plasmid system consists of pREP1.SD.NbaA and pT7-5.SD.NbaB derived from pREP1 and pT7-5, respectively, according to methods described previously by Parales et al. (57). As described above, the synthetic ribosomal binding site was introduced into vector pSD80, producing pSD80.SD. NbaB was amplified as described above. NbaA was amplified from the original pSDnbaA clone using primers NbaA5′-AscI (5′-TTGGCGCGCCATGACGCACATTGCAATGTCAG) and pSD80R. Both NbaA and NbaB products were introduced into pSD80.SD at the AscI and NheI sites. Both SD.NbaA and SD.NbaB were amplified using primers pSD80pc5′-SmaI (5′-CACGCCCGGGTAAGGAGG) and pSD80 R and cloned into pREP1 and pT7-5, respectively, at the SmaI and PstI sites (underlined). The plasmids were transformed into JM109(DE3) that has the IPTG-inducible T7 RNA polymerase on the chromosome. Ap and Cm were used for plasmid selection and maintenance in the same cell. Cells were grown overnight in LB with Ap/Cm, subcultured (1:100), and induced with 1 mM IPTG at an optical density of 0.5 for 5 h.
A 1 mM final concentration of 2-NBA was added to the single- and double-plasmid cultures and grown overnight. The cells were centrifuged, and an aliquot (20 μl) of the supernatant was spotted onto thin-layer chromatography plates (T-6145; Sigma). Authentic standards of 2-NBA and 3-HAA from Sigma were used to match the Rf values of the substrate and product. Thin-layer chromatography was developed with ethyl acetate-hexane (1:1), dried, and observed under UV light. 2-NBA appeared purplish, and 3-HAA appeared yellow.
Enzyme activities. (i) 2-NBA nitroreductase activity.
2-NBA nitroreductase activity was measured by monitoring the decrease in absorbance at 340 nm with the conversion of NADPH to NADP+ in a spectrophotometer (27). Reaction mixtures contained 2-NBA (0.2 μmol), NADPH (0.2 μmol), potassium phosphate buffer (50 μmol, pH 7.0), and an appropriate amount of cell extracts in a final volume of 1 ml.
To identify 2-HABA as a reaction product, 0.25 μmol of 2-NBA and 0.96 μmol of NADPH were incubated with a cell extract of E. coli BL21(pSDnbaA) in 30 mM potassium phosphate buffer (pH 7.0) in a total volume of 500 μl. After 10 min of incubation at 30°C, the reaction mixture was analyzed by high-performance liquid chromatography (HPLC). Control experiments were carried out without the addition of heterologous proteins. One unit of enzyme activity was defined as the quantity of enzyme required to oxidize 1 μmol NADPH per min at 25°C. Protein concentrations were determined using the Bradford assay (7a) and bovine serum albumin (Pierce, Rockford, IL) as the protein standard.
(ii) 2-HABA mutase activity.
2-HABA mutase activity was identified by measuring the conversion of 2-HABA to 3-HAA using a preassay mixture that consisted of 0.25 μmol of 2-NBA, 0.96 μmol of NADPH, and cell extract of E. coli BL21(pSDnbaA) in 30 mM potassium phosphate buffer (pH 7.0) in a total volume of 500 μl. The reaction mixture was incubated for 10 min at 30°C; an aliquot of the E. coli BL21(pSDnbaB) cell extract was then added, and the reaction mixture was further incubated for 10 min before analysis by HPLC. One unit of activity is defined as the amount of enzyme required to convert 1 μmol of substrate in 1 min.
HPLC analysis.
A Shimadzu instrument (model LC-6AD) equipped with a UV detector operating at a wavelength of 254 nm was used. Separations were performed on a CAPCELL PAK C18 UG120 column (column size of 4.6 by 250 mm and particle size of 5 μm; Shiseido, Tokyo, Japan) with a methanol-10 mM KH2PO4 mixture (30:70 [vol/vol]) at a flow rate of 1.0 ml min−1 as a solvent.
Site-directed mutagenesis.
PCR overlap extension mutagenesis (29) was used to generate nbaA mutants using pSDnbaA as a template (Table 1). The following 27-mer primers and, in each case, their complementary mutagenic primers (not shown) were used (the underlined codons represent the alanine [A] change): N40A (5′-GAGGGGTTGTGCGCGGCCGCGCCTTAT), H63A (5′-ATCGCCGTGGATGCGTATGGTGAAGAA), H69A (5′-GGTGAAGAAAGCGCGCGTCCTGGCGAG), D76A (5′-GGCGAGCAAAAAGCGACGCTAAAAAAC), H110A (5′-GATTTCCCCTCTGCGATCTCAGAAGCC), and E113A (5′-TCTCATATCTCAGCGGCCGAAGCCGTT).
After cloning, the sequences of the cloned fragments were confirmed by DNA sequencing, and their expression was verified.
Partial purification of 2-NBA nitroreductase.
The cell extract of strain KU-7 harvested from 1.25 liters of culture was used as the starting material for the purification of 2-NBA nitroreductase. All procedures were carried out at 4°C. The cell extract was fractionated using ammonium sulfate at 30 to 50% saturation. The resulting precipitate was dissolved in 50 mM potassium phosphate buffer (pH 7.0). This enzyme solution was dialyzed against the same buffer for 12 h. The dialyzed solution was put on a DEAE-Cellulose DE52 column. The column was washed with 50 mM potassium phosphate buffer (pH 7.0) until no protein could be detected in the flowthrough, and the enzyme was subsequently eluted with a linear gradient of 0 to 0.5 M KCl in the same buffer. Active fractions were collected, pooled, and used as a partially purified 2-NBA nitroreductase.
Protein analysis.
The partially purified 2-NBA nitroreductase was subjected to sodium dodecyl sulfate (SDS)-12% polyacrylamide gel electrophoresis (PAGE) carried out by a conventional method on a Mini-PROTEAN II electrophoresis cell (Nippon Bio-Rad Laboratories, Tokyo, Japan). The gel was transferred onto a polyvinylidene difluoride membrane (MiniProBlott; Applied Biosystems Japan Ltd.) with a Mini Trans-Blot electrophoretic transfer cell (Nippon Bio-Rad laboratories) according to instructions provided by the manufacturer. The area on the membrane containing 2-NBA nitroreductase was cut out and subjected to N-terminal amino acid sequencing with a model PPSQ-21 protein sequencer (Shimadzu Co., Kyoto, Japan).
Molecular modeling of NbaA and its complexes with Ni2+, flavin mononucleotide (FMN), and NADPH.
Structural bioinformatic analysis of the NbaA sequence was carried out at the BioInfoBank MetaServer (http://bioinfo.pl/meta), which assembles state-of-the-art fold recognition methods and provides a consensus sequence-to-structure hyperscoring with the 3D-JURY metapredictor (24).
Structural manipulations were done with the SYBYL 6.6 software (Tripos, Inc., St. Louis, MO). The homology modeling program COMPOSER (7) in SYBYL was employed to delineate six structurally conserved regions (SCRs) and five intervening structurally variable loop regions (SVRs) between NbaA and its template structures. The NbaA residues in the SCRs were modeled based on the structures of the two closest homologs (an FMN binding protein and a styrene monooxygenase) (PDB accession numbers 1EJE and 1USF, respectively), while the SVRs were initially built using the PROTEIN LOOPS structure database search module in SYBYL. Based on the crystal structure of the closest NbaA homolog identified (PDB accession number 1EJE), the NbaA homodimer was then assembled, together with one FMN molecule, one divalent metal ion (Ni2+), and seven adjacent structured water molecules per each monomer. Side-chain repacking and reorientation of polar hydrogens were performed to alleviate minor steric clashes and improve hydrogen bonding. The NbaA-Ni2+-FMN dimeric model was refined by sequential runs of molecular-mechanics force-field energy minimization that allowed the gradual relaxation of (i) the SVRs, (ii) the 23-residue N-terminal segments, and (iii) all side chains.
In order to dock NADPH into the modeled NbaA-Ni2+-FMN complex, the nicotinamide mononucleotide (NMN) half of NADPH was manually positioned into its putative binding cleft on NbaA and facing the isoalloxazine ring of FMN in an orientation and conformation observed in one of the previously identified NbaA templates (a ferric reductase) complexed with FMN and NADP+ (PDB accession number 1IOS). The initial conformation of the 2′-P-AMP half of the NADPH was also built as extending outward from the pocket by analogy. The resulting NADPH binding mode was then relaxed by energy minimization. Feasible bound conformations of the solvent-exposed 2′-P-AMP moiety were subsequently explored using the Monte Carlo minimization conformational sampling procedure (44) adapted to flexible ligand docking (13, 46, 67). The NMN half of NADPH was kept as an anchor from which random dihedral angle perturbations were introduced in the 2′-P-AMP half of NADPH, followed by energy minimizations, for a total of 10,000 Monte Carlo minimization cycles.
Docking of 2-NBA to the modeled NbaA-Ni2+-FMN complex.
Docking of 2-NBA to the modeled NbaA-Ni2+-FMN complex was performed manually by first positioning its aromatic ring similarly with the nicotinamide ring in the modeled NADPH complex (resulting in parallel stacking between 2-NBA and the isoalloxazine ring of FMN). Several alternate orientations of 2-NBA with respect to its two ortho groups were then generated and refined independently by energy minimization.
All conjugate gradient energy minimizations were carried using the AMBER all-atom molecular-mechanics force field (17, 73). A distance-dependent (4Rij, where Rij is the distance between atoms i and j) dielectric, an 8-Å nonbonded cutoff, and a root-mean-square gradient of 0.05 kcal/(mol · Å) were used. The protonation state at physiological pH was adopted. Previously described AMBER-compatible atomic parameters were used for FMN (46) and NADPH (61). Partial atomic charges for 2-NBA were obtained by a two-stage restrained fitting procedure for the single-point HF 6-31G* electrostatic potential (6) calculated in GAMESS (64). A charge-delocalized model with octahedral geometry was used for Ni2+ (55, 67). During all energy minimizations, the FMN and Ni2+ as well as the SCR backbone atoms of NbaA were constrained to their initial positions using harmonic potentials with force constants of 20 kcal/(mol · Å2) and 1 kcal/(mol · Å2), respectively.
Chemotaxis assay.
The chemotactic behavior of strain KU-7 and its nbaY deletion mutant KU-ΔY was studied by swarm plate assay (26), drop assay (21), and agarose plug assay (41, 75). In the swarm plate assay, soft agar swarm plates consisted of MS medium that contained 1 mM of chemicals (2-NBA, 3-HAA, benzoate, and succinate) and 0.3% agar. Cells (optical density at 600 nm of 1.0) were inoculated at the center of the plates, and these plates were incubated at 30°C overnight. The medium used for the drop assay consisted of 50 mM phosphate buffer (pH 7.0) that contained 20 μΜ EDTA and 0.05% glycerol. Cells were grown in MS medium, pelleted, washed and suspended in drop assay medium, and finally poured into petri plates. The chemotactic response was determined by placing the paper disk containing 100 mM attractant onto the center of the plate. Agarose plug assays were carried out with slight modifications of previously described methods (41, 75). Plugs contained 2% agarose in chemotaxis buffer (50 mM phosphate buffer [pH 7.0] that contained 20 mM EDTA and 0.05% glycerol) and 10 mM 2-NBA. A drop (100 μl) of the melted agarose mixture was placed onto a petri dish. Cells were grown in MS medium, pelleted, washed and suspended in chemotaxis buffer, and finally poured into petri plates with an agarose plug. The chemotactic response was determined by the ring appearance after 20 min of incubation at room temperature.
Nucleotide sequence accession number.
The nucleotide sequence determined in this study has been deposited in the DDBJ database under accession number AB263093.
RESULTS AND DISCUSSION
Isolation of the nbaA-disrupted mutant strain by transposon mutagenesis.
In our previous study, analysis of a 19-kb DNA region of strain KU-7 that encompasses the 3-HAA meta-cleavage and regulatory gene cluster, designated nbaEXHJIGFCDR, was found to be devoid of the two initial degradative genes, nbaA and nbaB (49). We screened additional Tn5-31Tp transformants and obtained apparently new candidates that were unable to grow on 2-NBA as a sole carbon source. Some 500-bp sequences from each IPCR product were determined and used to search the nonredundant protein sequence database of the NCBI for the presence of possible target sequences. However, the majority of the transposon insertion sites, except KUM-9, KUM-19, and KUM-33 (Fig. 1), appeared to bombard the known 3-HAA meta-cleavage and regulatory gene cluster (49).
FIG. 1.

The two initial steps of 2-NBA metabolism by P. fluorescens strain KU-7 catalyzed by 2-NBA nitroreductase (NbaA) and 2-HABA mutase (NbaB) to produce 3-HAA via the formation of 2-hydroxylaminobenzoate (2-HABA). The orientation and localization of nbaA and nbaB genes and flanking ORFs are indicated by large open arrows beneath the restriction map. Sites of transposon Tn5-31Tp insertions producing the various KUM mutant strains are pinpointed. The numbering of ORFs is done according to numbering reported previously by Muraki et al. (49). Orf11, Orf12, Orf13, Orf14, and Orf16 likely constitute an ABC transporter (22) for 2-NBA uptake. Orf15 is a putative FMN reductase. nbaE encodes a semialdehyde dehydrogenase as previously described (49). The cloned DNA fragments and probe regions are indicated. The broken lines indicate the Tn5-31Tp DNA. dhfrII and neo are trimethoprim resistance- and kanamycin resistance-encoding genes, respectively. The DNA probes, labeled with DIG-11-dUTP, are a 1.8-kb HindIII-BamHI fragment (probe 1) and a PCR-amplified DNA that is a neo gene sequence in Tn5-31Tp (probe 2). The PCR primers for probe 2 are 5′-GATCAAGAGACAGGATG-3′ and 5′-CACTCCTGCAGTTCG-3′.
The insertion site of Tn5-31Tp in strain KUM-9 was found to be within a 386-codon open reading frame (ORF) (Orf16) predicted to encode an ABC transporter substrate binding protein. Its closest homolog (81.8% sequence identity) is a putative extracellular ligand binding receptor of Polaromonas sp. strain JS666 (CP000317; DDBJ accession number ABE46993). The Tn5-31Tp insertion site in strain KUM-19 was found within a sequence, designated Orf23, and subsequently identified as being NbaA, which was similar to an FMN binding protein of Methanobacterium thermoautotrophicum ΔH (14). Resting cells of strain KUM-19, grown on succinate as the carbon source, that were noninduced with the substrate did not degrade 2-NBA.
Insertion of the Tn5-31Tp transposon into strain KUM-19 was verified by Southern blot analysis of the total DNA digested with EcoRI and BamHI. As a result, only one hybridization band (a 7.4-kb EcoRI fragment and a 4.1-kb BamHI fragment) was observed in each digest (Fig. 1).
Cloning and sequencing of the Tn5-31Tp flanking regions.
Cloning of the 7.4-kb EcoRI fragment or the 4.1-kb BamHI fragment was carried out by using pUC19 as a vector. In the former case, transformants were selected on trimethoprim- and Ap-containing plates. In the latter case, Km and Ap were used as the selection markers. The resulting plasmids were designated pNBA5 and pNBA6, respectively. The pNBA5 insert contained a 4.5-kb Tn5-31Tp segment and 2.9 kb of KU-7 DNA. Plasmid pNBA6 was found to contain a 3-kb Tn5-31Tp insert and 1.1 kb of KU-7 DNA.
In parallel with the transposon mutagenesis experiment, the DNA sequence of the flanking region of the 9.3-kb HindIII fragment was determined by direct sequencing of an IPCR product. The template for IPCR was prepared from a self-ligated 5.5-kb PstI fragment. The sequence of the IPCR product revealed that there is a 13-bp gap between KU-7 DNA in pNBA6 and the 9.3-kb HindIII fragment (pNBA3).
Sequencing of the Tn5-31Tp flanking regions in pNBA5 and pNBA6 and data analysis revealed three ORFs, two divergently transcribed ORFs, orf22 and orf23, separated by a 419-bp intergenic space and, 160-bp downstream of orf23 and on the same DNA strand, orf24 (Fig. 1). Various evidences establishing these ORFs as NbaA-encoding, NbaB-encoding, and chemotactic receptor NbaY-encoding genes are provided below.
Identification of NbaA as a 2-NBA nitroreductase.
Sequence determination of the N-terminal 15-amino-acid peptide (THIAMSGLTNMQKYW) derived from a partially purified 2-NBA nitroreductase of strain KU-7 confirmed the gene assignment of the 651-bp orf23 that encodes a 216-residue polypeptide that is preceded by an appropriate ribosome binding sequence (GGAG) 6 bp from the methionine start codon. The predicted molecular size of the protein (Mr of 24,410) matched well with the experimental value of 28 kDa that was derived from SDS-PAGE of the IPTG-inducible expression of the NbaA protein in pSDnbaA (Fig. 2). By Superdex 200 gel chromatography, NbaA was found in an active fraction that eluted at a molecular mass that is slighter greater than the 44 kDa of the molecular standard chicken albumin, indicating NbaA functions as a dimer (not shown).
FIG. 2.
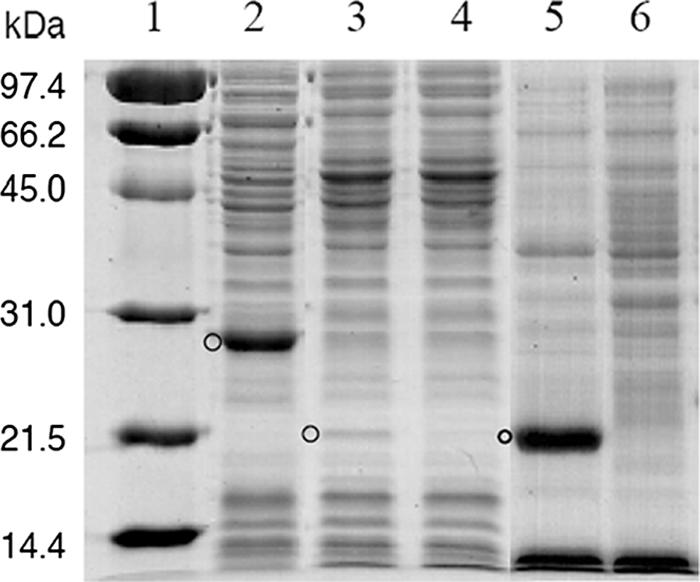
Coomassie blue-stained protein profiles of recombinant E. coli crude extracts separated by SDS-12% PAGE. Lane 1, molecular mass markers in kilodaltons; lane 2, NbaA expressed in pSDnbaA; lane 3, NbaB expressed in pSDnbaB; lane 4, E. coli cells containing the pSD80 vector only; lane 5, NbaB expressed in pT7-5SD.NbaB; lane 6, control E. coli JM109(DE3) containing the vector only. The positions of the expressed protein are indicated by circles.
The BLAST search retrieved a number of homologs of microbial origin displaying 40 to 78% sequence identity, but all are hypothetical or uncharacterized proteins. Two such sequences are derived from pollutant-degrading bacteria: a hypothetical conserved protein (77.7% identity), designated CP000317 (DDBJ accession number ABE46991), derived from a large plasmid of Polaromonas sp. strain JS666, a chlorinated-alkene-degrading organism; and another protein, Bcep 4121 (39.7%), derived from a well-known polychlorinated biphenyl degrader, Burkholderia xenovorans (formerly Burkholderia fungorum) strain LB400. However, the most useful information comes from a number of structural homologs (presented below), notably, an FMN binding protein (MTH152) (DDBJ accession number NP_275295) (31% identity) derived from Methanobacterium thermoautotrophicum ΔH (14).
Biochemical properties of NbaA.
The specific activity of NbaA toward 2-NBA was found to be 10.26 U/mg. Crude cell extracts of E. coli BL21(pSDnbaA) were able to oxidize NADPH only in the presence of 2-NBA. An investigation of the reaction stoichiometry demonstrated that the complete disappearance of 2-NBA by the cell extracts required 2 mol of NADPH per mol of 2-NBA. By HPLC analysis of the NbaA reaction product with 2-NBA and NADPH as substrates, only one product was formed in significant amounts with a retention time of 4.96 min. The expected product, 2-HABA, was identified by comparison to an authentic standard (Fig. 3).
FIG. 3.
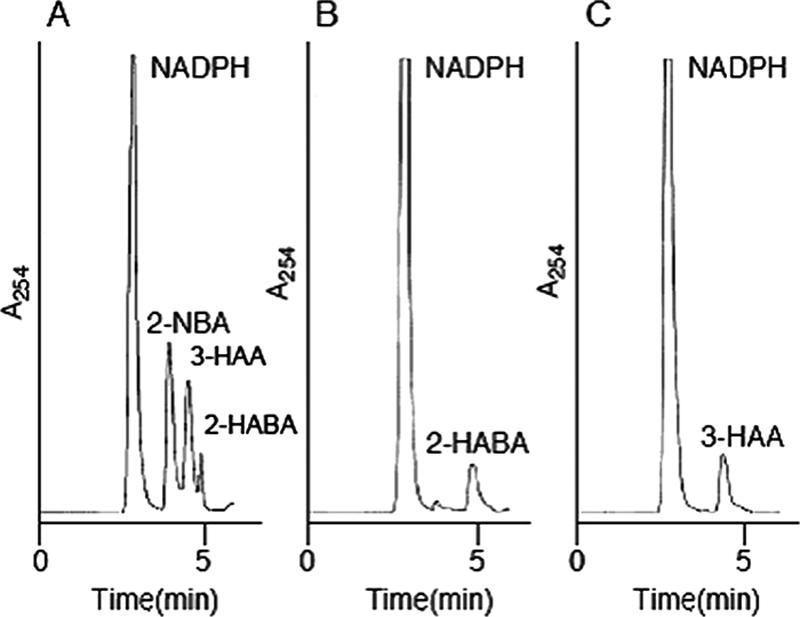
HPLC profiles of the reaction products generated by cell extracts containing NbaA and/or NbaB. (A) Authentic standards 2-NBA, 2-HABA, 3-HAA, and NADPH. (B) Identification of 2-HABA as a reaction product produced by cell extracts containing NbaA. A total of 0.25 μmol of 2-NBA and 0.96 μmol of NADPH were incubated with NbaA in 30 mM potassium phosphate buffer (pH 7.0) at a total volume of 500 μl. The reaction mixture was analyzed by HPLC after 10 min of incubation at 30°C. (C) Identification of 3-HAA as a reaction product produced by cell extracts containing NbaA and NbaB. The reaction mixture, the same as that described above (B), was incubated for 10 min at 30°C, and aliquots of cell extracts containing NbaB were then added and further incubated for 10 min before the assay. In all cases, no product formation was generated in the absence of the added heterologous enzymes.
Supplements of flavins (FMN, flavin adenine dinucleotide [FAD], and riboflavin) to crude extracts of E. coli BL21(pSDnbaA) at a final concentration of 1.0 mM resulted in increases in specific activities of 3.8-fold and 5.2-fold in the cases of FAD and FMN, respectively. The addition of riboflavin gave the same basal level as the case without the addition of FAD or FMN. Evidently, FMN was more effective than FAD, suggesting that FMN may be a physiological cofactor of NbaA.
The effect of divalent cations on NbaA activity was examined by preincubating the cell extracts containing NbaA with the chloride salts of the divalent cations, Ca, Co, Cu, Fe, Mg, Mn, Ni, and Zn, at 0.1 mM and 0.3 mM. Fe, Mg, and Mn appeared to have a potentiating effect at 0.1 mM, providing 23%, 16%, and 51% increases in activity, respectively. The addition of 0.3 mM Fe2+ and Mn2+ gave the greatest increase, 66% and 89%, respectively, whereas Ni2+ showed an increase of 17%. Ca2+ and Zn2+ seem to inhibit the enzyme to some extent. Under these experimental conditions, Mn2+ rendered the best activity to NbaA.
Modeled NbaA structure and interactions.
Structural bioinformatic searches unambiguously assigned the NbaA protein to the FMN binding split-barrel fold and specifically to the NADH:FMN oxidoreductase-like structural family (SCOP accession number b.45.1.2 or 50482) according to the SCOP database (http://scop.berkeley.edu/) (3), also classified as the flavin reductase-like domain (Pfam accession number PF01613) in the Pfam database (http://www.sanger.ac.uk/Software/Pfam/) (4). Currently, the solved structures adopting this fold include the FMN binding protein MTH152 from M. thermoautotrophicum (PDB accession number 1EJE) (14), a ferric reductase from Archeoglobus fulgidus (PDB accession numbers 1I0R and 1I0S) (12), the flavin reductase PheA2 from Bacillus thermoglucosidasius (PDB accession numbers 1RZ0 and 1RZ1) (71), a styrene monooxygenase from Thermus thermophilus (PDB accession numbers 1USC and 1USF) (unpublished data), and a probable flavoprotein from the same organism (PDB accession numbers 1WGB and 1YOA) (unpublished data). The sequence alignment of NbaA and these five structurally similar enzymes is given in Fig. 4A.
FIG. 4.

Modeled NbaA structure and interactions with FMN, NADPH, and 2-NBA. (A) Fold-recognition-based sequence alignment between NbaA and the currently structurally characterized members (identified here by their PDB accession numbers at http://www.rcsb.org/) of the NADH:FMN oxidoreductase-like structural family from the FMN-binding split-barrel fold: accession number 1EJE, Methanobacterium thermoautotrophicum FMN binding protein MTH152; accession number 1USF, Thermus thermophilus styrene monooxygenase; accession number 1IOS, Archeoglobus fulgidus ferric reductase; accession number 1RZ1, Bacillus thermoglucosidasius flavin reductase PheA2; accession number 1WGB, Thermus thermophilus probable flavoprotein. Secondary structure elements (α-helices and β-strands) of NbaA, corresponding to its three-dimensional homology model built based on this alignment, are indicated above its sequence. The NbaA residues individually mutagenized to alanine in this study are marked by closed circles that are color coded according to the activity of the corresponding mutants (red, inactive; magenta, residual activity; black, activity comparable to that of the wild type). (B) Stereo view displaying the structural model of the NbaA dimer. The trace of the NbaA main chain, rendered schematically by its secondary structure, is colored differently for the two monomers (green and cyan, respectively), with the 10-residue insertion in the β5-α2 loop relative to the other members of the fold family highlighted in red in one of the monomers. Ligands docked to NbaA are shown in only one of the two equivalent binding sites of the homodimer and are colored (blue, FMN; red, NADPH; magenta, Ni2+). (C) Stereo view zooming in the FMN binding site of NbaA. (D) Stereo view showing the modeled interactions of NADPH with the FMN-bound NbaA. (E) Stereo view detailing the putative binding mode of the 2-NBA substrate. In the latter three panels, the protein main chain is rendered as in B, select protein residues and side chains are shown explicitly, and the following color scheme is applied to the C atoms belonging to the various displayed molecules: yellow, FMN; light blue, NADPH (D); magenta, 2-NBA (E); green or cyan, protein. The other atom types are colored as follows: blue, N; red, O; yellow, S; orange, P. Hydrogen bonds are shown with yellow dashed lines, and metal ion hexacoordination is shown with black solid lines. The Ni2+ ion is represented as a magenta sphere, and its two coordinating water molecules are represented as red spheres in C.
This alignment formed the basis for the three-dimensional homology modeling of the NbaA homodimeric structure (Fig. 4B). The core of each modeled NbaA subunit is proposed to be organized around a six-stranded antiparallel β-barrel with a capping α-helix, a fold recognized as a circular permutation of the flavin binding domain of the ferredoxin reductase superfamily (34, 45, 50). However, the ferredoxin reductase-type proteins utilize a second Rossmann fold domain to bind the NAD(P)H (34). The homology to the NADH:FMN oxidoreductase-like fold family strongly suggests that the predicted single-domain structure of NbaA would provide both the flavin and NAD(P)H binding sites (12, 71).
The modeled binding mode of FMN to NbaA (Fig. 4C) is similar to that observed in other members of the NADH:FMN oxidoreductase-like fold family (12, 71). FMN fits into a well-shaped groove near the homodimer interface of NbaA. Based on this proposed model, the si face of the isoalloxazine ring is buried against the β-barrel and directly contacts the main-chain atoms of residues Ser45, Ala46, Ala60, and Asp62 and side-chain atoms of Ser45 and Lys75 via hydrogen bonds as well as Ile27, Pro43, Tyr44, Met100, Val101, Arg187, and Tyr193 by nonpolar contacts. The 3′-hydroxyl group of the ribityl moiety is modeled in hydrogen bond contact with the Lys75 side chain. The FMN phosphate group is predicted to be anchored to the β-barrel-capping α-helix of NbaA via hydrogen bonds to the Asn40, Tyr44, and Thr77 side chains and to Asp76 and Thr77 main-chain atoms.
A divalent metal ion, Ni2+, was modeled as mediating the interaction between the FMN phosphate and NbaA. This interaction has been observed in the crystal structure of the FMN-binding protein MTH152 (PDB accession number 1EJE) (14), the structural template most similar in primary sequence to NbaA. In the more distant structural homologs of NbaA, this phosphate-bridging role is fulfilled by protein side chains, e.g., the ammonium group of Lys69 in ferric reductase (PDB accession numbers 1I0R and 1I0S) (12). In the modeled NbaA-FMN complex, the octahedral coordination sphere of the Ni2+ metal ion consists of side-chain oxygen atoms from residues Asn40, Asp76, and Glu113 (Asn36, His62, and Glu99, respectively, in the FMN binding protein MTH152), two water molecules, and an oxygen atom from the FMN phosphate (Fig. 4C).
The NADPH binding mode to NbaA (Fig. 4D) was inferred from the NADP+ binding mode observed in some of the structural homologs (styrene monooxygenase, ferric reductase, and flavin reductase PheA2) (PDB accession numbers 1USF, 1IOS, and 1RZ1, respectively), primarily for the more buried NMN half of NADPH. In this binding mode, the nicotinamide ring stacks against the available re face of the FMN isoalloxazine ring. The modeled C4 (nicotinamide)-N5 (isoalloxazine) distance of ∼3.5 Å is compatible with a direct hydride transfer from NADPH to FMN (12, 71). The amide group of nicotinamide is predicted to form hydrogen bonds with the side chains of Asp62 and Arg153 as well as Tyr49 from the second subunit in the NbaA homodimer. In this plausible model, the NMN phosphate would be stabilized by a salt bridge with Arg187.
Multiple sterically feasible binding modes may be expected for the solvent-exposed 2′-P-AMP fragment of NADPH due to the fairly wide opening at the top of the FMN and NMN binding cleft of NbaA. Although the low-energy conformations obtained from conformational sampling indeed revealed variability in the binding mode of the adenosine portion of NADPH, it also appeared that the adenine moiety has a tendency to interact with the β5-α2 loop of NbaA (Fig. 4B and D). The resolution of our modeled structure in this solvent-exposed region does not allow speculations on specific intermolecular interactions. Interestingly, the β5-α2 loop of NbaA includes a 10-residue insertion relative to the other members of the NADH:FMN oxidoreductase-like structural family (Fig. 4A). Thus, it resembles the corresponding loop of the ferredoxin reductase superfamily members that has been shown to be able to interact, via one of its aromatic residues, with the adenine moiety of FAD (8, 19). The β5-α2 loop of NbaA does include an aromatic residue, His69, which might therefore represent a potential site for interaction with the adenine moiety of NADPH.
Proposed NbaA reaction mechanism.
A structural model for the mode of 2-NBA substrate binding to NbaA assumes the ping-pong mechanism in which the oxidized coenzyme leaves its binding site and is replaced by the substrate that contacts the enzyme-bound reduced flavin. This assumption is supported by the available data for NbaA structural homologs such as ferric reductase (12) and flavin reductase PheA2 (71), where steric constraints impede the simultaneous binding of the substrate and nicotinamide, thus suggesting a ping-pong kinetic mechanism. Indeed, biochemical data have recently shown that the flavin reductase PheA2 catalyzes the NADH-dependent reduction of free flavins by a ping-pong bisubstrate-biproduct mechanism (36). Other nitroaromatic reductases, although not displaying significant sequence or structural similarities to NbaA, have been shown to act according to a ping-pong catalytic mechanism. For example, the structures of nitroaromatic reductases from E. coli and Enterobacter cloacoe complexed with substrate analogs, inhibitors, and dinitrobenzoate prodrugs show that these ligands replace the NAD(P)H nicotinamide in order to form parallel stacking with the isoalloxazine moiety of enzyme-bound FMN or reduced FMN (28, 33, 48). Furthermore, a recent kinetic study of E. coli nitroaromatic reductase demonstrates that the nitroso intermediate becomes the substrate in a subsequent enzyme-catalyzed reduction reaction leading to the hydroxylamine product (59). A plausible ping-pong mechanism for a reaction catalyzed by NbaA is depicted in Fig. 5.
FIG. 5.

Proposed ping-pong mechanistic action of 2-NBA nitroreductase in the formation of 2-HABA.
Our model of 2-NBA bound to NbaA (Fig. 4E) is consistent with the observed orientation of the substrate nitrophenyl moiety against the nitroreductase-bound flavin (33). According to this putative binding mode, the aromatic substrate forms a parallel stacking with the isoalloxazine ring. The nitro group of 2-NBA would contact the isoalloxazine rings above the N5 nitrogen atom, where it is predicted to be anchored by polar interactions with the Arg25 and Arg187 positively charged side chains of NbaA. These interactions remain compatible with the binding of the 2-nitrosobenzoate intermediate, but not with the 2-hydroxyaminobenzoate product, due to electrostatic repulsions between positively charged hydrogen atoms. In the proposed model, the ortho-carboxylate substituent of 2-NBA is complementary to its putative binding site on NbaA by forming a salt bridge with the side chain of Arg153 and hydrogen bonds with Thr19 and Tyr49 (from the second monomer), which may account for the NbaA specificity toward 2-NBA. For example, a similar carboxylate binding motif (i.e., Arg, Ser, and Tyr) is present in p-hydroxybenzoate hydroxylase (65).
Site-directed mutagenesis of predicted NbaA active-site residues.
Structural modeling analysis of relevant NbaA complexes suggests how specific residues may contribute to the catalytic activity of the enzyme and thus forms the basis for mutational analyses. We first probed the predicted metal binding residues Asn40, Asp76, and Glu113 by site-directed mutagenesis. We also mutated residue His69 from the 10-residue insertion loop β5-α2 predicted to interact with the adenosine portion of NAD(P)H. Mutations of two other histidine residues, His63 and His110, predicted to be solvent exposed and not to contribute to NbaA activity (via neither ligand binding nor dimerization), were also tested. Hence, we generated six NbaA variants that in each case resulted in an alanine replacement. Individual mutations of the three predicted divalent metal ion binding-site residues (N40A, D76A, and E113A) resulted in a complete loss of the NbaA activity (Table 2). These results are in agreement with our model showing that the metal ion would act as a bridge between the FMN phosphate and the protein, thus being essential for FMN binding (Fig. 4C). The H69A replacement resulted in an enzyme that retained only 2 to 3% of wild-type NbaA activity but that was nonetheless still active (Table 2). In a recent study of another NADH:flavin oxidoreductase, a similar drop in activity was observed for a histidine residue that represents an NADH-interacting site (60). Taken together, these observations are in agreement with the predicted NAD(P)H-interacting role of His69 in NbaA. On the other hand, the H63A and H110A substitutions retained 72 to 73% and 33 to 44% of the original activity, respectively, indicating that these residues do not contribute substantially to NbaA activity. For all active mutants, it was shown that the addition of exogenous FMN potentiated the various enzyme activities about three- to fivefold. We also noted that the amounts of the variant NbaA proteins produced, as evident by Coomassie-stained SDS-PAGE analysis, are comparable to that of the native NbaA (results not shown). Certainly, additional biochemical data from purified enzyme and direct experimental structural evidence will be required for a refined characterization of the NbaA catalytic residues and mechanisms.
TABLE 2.
NbaA activities in cell-free extracts containing NbaA or its mutant proteins and effect of FMN on their activitiesa
| NbaA | Sp act
|
|||
|---|---|---|---|---|
| Without FMN
|
With FMN
|
|||
| Mean U/mgb ± SD | % | Mean U/mg ± SD | % | |
| Wild type | 10.26 ± 0.23 | 100 | 31.73 ± 0.37 | 100 |
| N40A | ND | ND | ||
| H63A | 7.44 ± 0.17 | 73 | 22.85 ± 0.33 | 72 |
| H69A | 0.20 ± 0.01 | 2 | 0.96 ± 0.03 | 3 |
| D76A | ND | ND | ||
| H110A | 3.38 ± 0.01 | 33 | 13.99 ± 0.38 | 44 |
| E113A | ND | ND | ||
FMN was added to the cell-free extracts containing NbaA and its mutants at a concentration of 5.0 mM, and it was preincubated for 5 min on ice prior to the assay. ND, not detected.
One unit is the amount (micromoles) of NADPH oxidized per minute. Results are the means of three independent experiments, with standard deviations noted.
Identification of NbaB as 2-HABA.
The deduced 181-amino-acid sequence of NbaB showed 35% identity to the 4-hydroxylaminobenzoate lyase (PnbB) sequences of three known degradative organisms: Pseudomonas putida TW3, degrading 4-nitrotoluene (32), and Pseudomonas sp. strain YH102 and Ralstonia pickettii YH105, degrading 4-nitrobenzoate (76). Higher identities were observed with four hypothetical proteins in the genomes of Rhodoferas ferrireducens DSM 15236 (38%), Azoarcus sp. strain EbN1 (39.3%), Burkholderia fungorum LB400 (40%), and Polaromonas sp. strain JS666 (66.5%). A multiple sequence alignment of the PnbB lyases with NbaB (see Fig. S2 in the supplemental material) revealed that the conserved sequences are distributed evenly throughout the polypeptide with the exception that NbaB (and its JS666 homolog) appears to have an N-terminal extension of 14 to 20 residues, and two sequence gaps (amino acids 110 to 111 and 142 to 143 [NbaB numbering]) are needed for an optimal sequence alignment. The sequence similarity between PnbB and NbaB provides a plausible basis for the second action of PnbB in the 4-nitrobenzoate pathway of P. putida strain TW3 in the production of 4-amino-3-hydroxybenzoate from 4-nitrobenzoate besides the better known product, protocatechuate (31). Evidently, 4-amino-3-hydroxybenzoate is an analog of 2-amino-3-hydroxybenzoate, i.e., 3-HAA.
The expression of a 22-kDa protein band, although weak, derived from plasmid pSDnbaB confirmed the expected molecular weight of NbaB, calculated to be 19,554 (Fig. 2). No enhanced protein band of this size was detectable in the control cells containing the pSD80 vector only. However, better expression of NbaB was seen in the pT7-5SD.NbaB construct transformed in E. coli JM109(DE3) (Fig. 2). This level was just as good as the expression level of NbaA in pSDnbaA. On the other hand, NbaA was not readily expressed in the pREP1.NbaA construct as in pSDnbaA.
By HPLC analysis of the enzyme reaction product of NbaB with 2-NBA and NADPH in the presence of NbaA, the reaction product, a peak at a retention time of 4.40 min, was identified to be 3-HAA by comparison to an authentic standard (Fig. 3). 3-HAA production, by the appearance of a yellow spot under UV illumination, was also observed by thin-layer chromatography (not shown).
NbaY (Orf24) gene disruption and chemotaxis.
NbaY (Orf24) consists of a sequence coding for 544 amino acid residues. The primary structure showed 29% sequence identity to NahY from P. putida G7, a known methyl-accepting chemotactic transducer that recognizes naphthalene (25). An updated BLAST search showed 62 to 63% sequence homology to putative chemotaxis sensory transducers, CP000094 (DDBJ accession numberABA74342) and CP000076 (accession number AAY92385), each derived from P. fluorescens strains PfO-1 and Pf-5, respectively. A multiple sequence alignment is shown in Fig. S3 in the supplemental material. The greatest sequence conservation occurs within a region designated the highly conserved domain, which is typical of chemoreceptor proteins (39). In NbaY, two possible transmembrane segments (amino acids 13 to 35 and 190 to 212) are predicted (38).
To test whether NbaY serves as a possible chemoreceptor for 2-NBA, we constructed an nbaY deletion mutant designated strain KU-ΔY. The use of three independent assays by their ring appearance in the case of the wild type compared to the mutant supported the involvement of NbaY in 2-NBA chemotaxis (Fig. 6). In liquid culture, strain KU-ΔY grew just as readily as the wild type when 2-NBA was used as the sole carbon source. At this time, we have not tested whether any of the 2-NBA metabolites would serve as a chemoattractant. A detailed chemotaxis study is outside our present scope.
FIG. 6.

NbaY is necessary for 2-NBA chemotaxis, as determined by (A) swarm plate assay, (B) drop assay, and (C) modified agarose plug assay.
To the best of available knowledge, NahY and PcaK (a transporter and chemoreceptor protein from P. putida PRS2000 that is encoded as part of the beta-ketoadipate pathway regulon for aromatic acid degradation) are two of the existing characterized chemoreceptor genes for aromatic metabolism (reviewed in references 54 and 58). On the other hand, there have been several studies illustrating chemotaxis of a variety of nitroaromatic degradative organisms toward compounds such as nitrotoluene, dinitrotoluene, TNT, etc. (43, 54, 56, 58). But in all cases, no pathway-associated gene, like NahY in naphthalene degradation, PcaK in benzoate metabolism, or NbaY as in this study, has been ascribed to the observed chemotaxis phenomena. Hence, NbaY is a prototype chemoreceptor for nitroaromatics. The relevance of chemotaxis in pollutant-degrading bacteria to bioremediation strategy has been reviewed previously (54, 58).
Concluding remarks.
For the first time, a homology model of 2-NBA nitroreductase is available, which adds to our limited knowledge on the structure and function of nitroreductases in nitroaromatic metabolism for which a limited number of genetic and/or biochemical studies are available (11, 35, 40, 51, 69). NbaA is a newly discovered representative of a family of homodimeric NADH:FMN oxidoreductase-like fold proteins that is different from the nitro/flavin reductase family. The latter comprises the major oxygen-insensitive nitroreductase (NfsA) from E. coli NfsA (37), flavin reductase of Vibrio harveyi (42), PnrA of TNT-degrading P. putida JLR11 (11), and NitA and NitB of another TNT-degrading strain, Clostridium acetobutylicum ATCC 824 (40). It remains to be seen whether the 10-residue insertion loop between strand β5 and helix α2 (amino acids 65 to 74) is unique to NbaA that attacks an ortho-substituted nitroaromatic substrate. It is unfortunate that we are as yet unable to assign a function or substrate to the MTH152 FMN binding protein of M. thermoautotrophicum (14). However, the structure relationship of MTH152 to NbaA suggests that aromatic compounds should be tested as possible substrates.
The availability of NbaA- and NbaB-encoding genes paves a plausible biotechnological route for the production of 3-HAA, a known antioxidant and reductant. However, for the efficient substrate uptake of aromatic acids such as 2-NBA, this recombinant route might require the involvement of the numerous ABC transporter permease and outer membrane porin sequences, etc., that are possibly encoded by ORFs 16, 14, 13, 12, and 11 (immediately downstream of nbaB) and Orf17, which is downstream of the nbaE gene cluster (49) (DDBJ database accession number AB088043).
This study completes the gene elucidation of 2-NBA metabolism in strain KU-7 in a 24-kb chromosomal locus that comprises a chemoreceptor gene (nbaY) on one end of the biodegradative gene cluster and a putative transcriptional regulator (nbaR) of the LysR type on the other end that is followed by an orf17 of unknown function (49) (Fig. 7). Interestingly, in the current microbial genome sequence database (Joint Genome Institute database), we have identified the presence of a very similar gene cluster, exhibiting high sequence identities (in the range 63% to 87%), that is encoded on a plasmid of 360,405 bp in the draft genome sequence of Polaromonas sp. strain JS666, a strain that has been studied for its degradation of chlorinated alkenes such as cis-dichloroethene but no other compounds (16). Evidently, the gene organization of the core nba biodegradative gene cluster (i.e., from nbaA to nbaR) is the same in the two strains. Notable differences are in the lengths of the various intergenic spaces and especially the presence of a putative integrase and insertion sequences IS3 and IS911 found at both ends of the JS666 gene locus in the regions otherwise occupied by nbaY and orf17 in strain KU-7 (Fig. 6). This observation leads to the possibility that the nba gene locus constitutes a transferable or transposable “genomic island” or integrative and conjugative elements, as frequently found among catabolic pathways of pollutant-degrading bacteria (72).
FIG. 7.

Comparative organization and homology between the nba gene locus in P. fluorescens strain KU-7 and the uncharacterized ORFs encoded on a large plasmid in Polaromonas sp. strain JS666. Major differences in the coding capacities are notably at the extremities of the core biodegradative genes and in the spacing of the intergenic regions.
Supplementary Material
Acknowledgments
We thank A. Nakazawa for providing pKN31 and the National Bioresource Project (National Institute of Genetics, Japan) for plasmid pK18mobsacB.
This research was financially supported in part by the Kansai University Research grants, a Grant-in-Aid for Encouragement of Scientists, 2006 (H.I.), and a High-Tech Research Center Project for Private Universities, matching fund subsidy from the Ministry of Education, Culture, Sports, Science, and Technology, 2002-2006 (Y.H.).
We dedicate this paper to the memory of Tai Tokuyama.
Footnotes
Published ahead of print on 2 February 2007.
This publication is issued as NRCC number 49029.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Abe, M., M. Tsuda, M. Kimito, S. Inoue, A. Nakazawa, and T. Nakazawa. 1996. A genetic analysis system of Burkholderia cepacia: construction of mobilizable transposon and cloning vector. Gene 174:191-194. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreeva, A., D. Howorth, S. E. Brenner, T. J. P. Hubbard, C. Chothia, and A. G. Murzin. 2004. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 32:D226-D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bateman, A., L. Coin, R. Durbin, R. D. Finn, V. Hollich, S. Griffiths-Jones, A. Khanna, M. Marshall, S. Moxon, E. L. L. Sonnhammer, D. J. Studholme, C. Yeats, and S. R. Eddy. 2004. The Pfam protein families database. Nucleic Acids Res. 32:D138-D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauer, H., and S. M. Rosenthal. 1944. 4-Hydroxylaminobenzenesulfonamide, its acetyl derivatives and diazotization reaction. J. Am. Chem. Soc. 66:611-614. [Google Scholar]
- 6.Bayly, C. I., P. Cieplak, W. Cornell, and P. A. Kollman. 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97:10269-10280. [Google Scholar]
- 7.Blundell, T., D. Carney, S. Gardner, F. Hayes, B. Howlin, T. Hubbard, J. Overington, D. A. Singh, B. L. Sibanda, and M. Sutcliffe. 1988. Knowledge-based protein modelling and design. Eur. J. Biochem. 172:513-520. [DOI] [PubMed] [Google Scholar]
- 7a.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 8.Bruns, C. M., and P. A. Karplus. 1995. Refined crystal structure of spinach ferredoxin reductase at 1.7 Å resolution: oxidized, reduced and 2′-phospho-5′-AMP bound states. J. Mol. Biol. 247:125-145. [DOI] [PubMed] [Google Scholar]
- 9.Bryant, C., and W. D. McElroy. 1991. Nitroreductases, p. 291-304. In F. Muller (ed.), Chemistry and biochemistry of flavoenzymes, vol. II. CRC Press, Boca Raton, FL. [Google Scholar]
- 10.Bullock, W. O., J. M. Fernandez, and J. M. Stuart. 1987. XL-1 Blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques 5:376-379. [Google Scholar]
- 11.Caballero, A., J. J. Lazaro, J. L. Ramos, and A. Esteve-Nunez. 2005. PnrA, a new nitroreductase-family enzyme in the TNT-degrading strain Pseudomonas putida JLR11. Environ. Microbiol. 7:1211-1219. [DOI] [PubMed] [Google Scholar]
- 12.Chiu, H., E. Johnson, I. Schroder, and D. C. Rees. 2001. Crystal structures of a novel ferric reductase from the hyperthermophilic archaeon Archaeoglobus fulgidus and its complex with NADP+. Structure 9:311-319. [DOI] [PubMed] [Google Scholar]
- 13.Chowdhury, S. F., J. Sivaraman, J. Wang, G. Devanathan, P. Lachance, H. Qi, R. Menard, J. Lefebvre, Y. Konishi, M. Cygler, T. Sulea, and E. O. Purisima. 2002. Design of noncovalent inhibitors of human cathepsin L. From the 96-residue proregion to optimized tripeptides. J. Med. Chem. 45:5321-5329. [DOI] [PubMed] [Google Scholar]
- 14.Christendat, D., A. Yee, A. Dharamsi, Y. Kluger, A. Savchenko, J. R. Cort, V. Booth, C. D. Mackereth, V. Saridakis, I. Ekiel, G. Kozlov, K. L. Maxwell, N. Wu, L. P. McIntosh, K. Gehring, M. A. Kennedy, A. R. Davidson, E. F. Pai1, M. Gerstein, A. M. Edwards, and C. H. Arrowsmith. 2000. Structural proteomics of an archaeon. Nat. Struct. Biol. 7:903-909. [DOI] [PubMed] [Google Scholar]
- 15.Colabroy, K. L., and T. P. Begley. 2005. Tryptophan catabolism: identification and characterization of a new degradative pathway. J. Bacteriol. 187:7866-7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coleman, N. V., T. E. Mattes, J. M. Gossett, and J. C. Spain. 2002. Biodegradation of cis-dichloroethene as the sole carbon source by a β-proteobacterium. Appl. Environ. Microbiol. 68:2726-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179-5197. [Google Scholar]
- 18.Dedonder, R. 1966. Levansucrase from Bacillus subtilis. Methods Enzymol. 8:500-505. [Google Scholar]
- 19.Deng, Z., A. Aliverti, G. Zanetti, A. K. Arakaki, J. Ottado, E. G. Orellano, N. B. Calcaterra, E. A. Ceccarelli, N. Carrillo, and P. A. Karplus. 1999. A productive NADP+ binding mode of ferredoxin-NADP+ reductase revealed by protein engineering and crystallographic studies. Nat. Struct. Biol. 6:847-853. [DOI] [PubMed] [Google Scholar]
- 20.Esteve-Nunez, A., A. Caballero, and J. L. Ramos. 2001. Biological degradation of 2,4,6-trinitrotoluene. Microbiol. Mol. Biol. Rev. 65:335-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fahrner, K. A., S. M. Block, S. Krishnaswamy, J. S. Parkinson, and H. C. Berg. 1994. A mutant hook-associated protein (HAP3) facilitates torsionally induced transformations of the flagellar filament of Escherichia coli. J. Mol. Biol. 238:173-186. [DOI] [PubMed] [Google Scholar]
- 22.Fath, M., and R. Kolter. 1993. ABC transporters: bacterial exporters. Microbiol. Rev. 57:995-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gay, P., D. Le Coq, M. Steinmetz, T. Berkelman, and C. I. Kado. 1985. Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol. 164:918-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginalski, K., A. Elofsson, D. Fischer, and L. Rychlewski. 2003. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 19:1015-1018. [DOI] [PubMed] [Google Scholar]
- 25.Grimm, A. C., and C. S. Harwood. 1999. NahY, a catabolic plasmid-encoded receptor required for chemotaxis of Pseudomonas putida to the aromatic hydrocarbon naphthalene. J. Bacteriol. 181:3310-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harwood, C. S., N. N. Nichols, M.-K. Kim, J. L. Ditty, and R. E. Parales. 1994. Identification of the pcaRKF gene cluster from Pseudomonas putida: involvement in chemotaxis, biodegradation, and transport of 4-hydroxybenzoate. J. Bacteriol. 176:6479-6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa, Y., T. Muraki, T. Tokuyama, H. Iwaki, M. Tatsuno, and P. C. K. Lau. 2000. A novel degradative pathway of 2-nitrobenzoate via 3-hydroxyanthranilate in Pseudomonas fluorescens strain KU-7. FEMS Microbiol. Lett. 190:185-190. [DOI] [PubMed] [Google Scholar]
- 28.Haynes, C. A., R. L. Koder, A.-F. Miller, and D. W. Rodgers. 2002. Structures of nitroreductase in three states. J. Biol. Chem. 277:11513-11520. [DOI] [PubMed] [Google Scholar]
- 29.Horton, R. M. 1995. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol. Biotechnol. 3:93-99. [DOI] [PubMed] [Google Scholar]
- 30.Huang, G., L. Zhang, and R. G. Birch. 2000. Rapid amplification and cloning of Tn5 flanking fragments by inverse PCR. Lett. Appl. Microbiol. 31:149-153. [DOI] [PubMed] [Google Scholar]
- 31.Hughes, M. A., M. J. Baggs, J. Al-Dulayymi, M. S. Baird, and P. A. Williams. 2002. Accumulation of 2-aminophenoxazin-3-one-7-carboxylate during growth of Pseudomonas putida TW3 on 4-nitro-substituted substrates requires 4-hydroxylaminobenzoate lyase (PnbB). Appl. Environ. Microbiol. 68:4965-4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes, M. A., and P. A. Williams. 2001. Cloning and characterization of the pnb genes, encoding enzymes for 4-nitrobenzoate catabolism in Pseudomonas putida TW3. J. Bacteriol. 183:1225-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johansson, E., G. N. Parkinson, W. A. Denny, and S. Neidle. 2003. Studies on the nitroreductase prodrug-activating system. Crystal structures of complexes with the inhibitor dicoumarol and dinitrobenzamide prodrugs and of the enzyme active form. J. Med. Chem. 46:4009-4020. [DOI] [PubMed] [Google Scholar]
- 34.Karplus, P. A., M. J. Daniels, and J. R. Herriott. 1991. Atomic structure of ferredoxin-NADP+ reductase: prototype for a structurally novel flavoenzyme family. Science 251:60-66. [PubMed] [Google Scholar]
- 35.Kim, H. Y., and H. G. Song. 2005. Purification and characterization of NAD(P)H-dependent nitroreductase I from Klebsiella sp. C1 and enzymatic transformation of 2,4,6-trinitrotoluene. Appl. Microbiol. Biotechnol. 68:766-773. [DOI] [PubMed] [Google Scholar]
- 36.Kirchner, U., A. H. Westphal, R. Muller, and W. J. van Berkel. 2003. Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem. 278:47545-47553. [DOI] [PubMed] [Google Scholar]
- 37.Kobori, T., H. Sasaki, W. C. Lee, S. Zenno, K. Saigo, M. E. P. Murphy, and M. Tanokura. 2001. Structure and site-directed mutagenesis of a flavoprotein from Escherichia coli that reduces nitrocompounds. J. Biol. Chem. 276:2816-2823. [DOI] [PubMed] [Google Scholar]
- 38.Krogh, A., B. Larsson, G. von Heijne, and E. L. L. Sonnhammer. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567-580. [DOI] [PubMed] [Google Scholar]
- 39.Kuroda, A., T. Kumano, K. Taguchi, T. Nikata, J. Kato, and H. Ohtake. 1995. Molecular cloning and characterization of a chemotactic transducer gene in Pseudomonas aeruginosa. J. Bacteriol. 177:7019-7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kutty, R., and G. N. Bennett. 2005. Biochemical characterization of trinitrotoluene transforming oxygen-insensitive nitroreductases from Clostridium acetobutylicum ATCC 824. Arch. Microbiol. 184:158-167. [DOI] [PubMed] [Google Scholar]
- 41.Lanfranconi, M. P., H. M. Alvarez, and C. A. Studdert. 2003. A strain isolated from gas oil-contaminated soil displays chemotaxis towards gas oil and hexadecane. Environ. Microbiol. 5:1002-1008. [DOI] [PubMed] [Google Scholar]
- 42.Lei, B., M. Liu, S. Huang, and S.-C. Tu. 1994. Vibrio harveyi NADPH-flavin oxidoreductase: cloning, sequencing, and overexpression of the gene and purification and characterization of the cloned enzyme. J. Bacteriol. 176:3536-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leungsakul, T., B. G. Keenan, B. F. Smets, and T. K. Wood. 2005. TNT and nitroaromatic compounds are chemoattractants for Burkholderia cepacia R34 and Burkholderia sp. strain DNT. Appl. Microbiol. Biotechnol. 69:321-325. [DOI] [PubMed] [Google Scholar]
- 44.Li, Z., and H. A. Scheraga. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA 84:6611-6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liepinsh, E., M. Kitamura, T. Murakami, T. Nakaya, and G. Otting. 1998. Common ancestor of serine proteases and flavin-binding domains. Nat. Struct. Biol. 5:102-103. [DOI] [PubMed] [Google Scholar]
- 46.Lin, L. Y., T. Sulea, R. Szittner, V. Vassilyev, E. O. Purisima, and E. A. Meighen. 2001. Modeling of the bacterial luciferase-flavin mononucleotide complex combining flexible docking with structure-activity data. Protein Sci. 10:1563-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Link, A. J., D. Phillips, and G. M. Church. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 179:6228-6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lovering, A. L., E. I. Hyde, P. F. Searle, and S. A. White. 2001. The structure of Escherichia coli nitroreductase complexed with nicotinic acid: three crystal forms at 1.7 Å, 1.8 Å and 2.4 Å resolution. J. Mol. Biol. 309:203-213. [DOI] [PubMed] [Google Scholar]
- 49.Muraki, T., M. Taki, Y. Hasegawa, H. Iwaki, and P. C. K. Lau. 2003. Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7. Appl. Environ. Microbiol. 69:1564-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murzin, A. G. 1998. Probable circular permutation in the flavin-binding domain. Nat. Struct. Biol. 5:101. [DOI] [PubMed] [Google Scholar]
- 51.Nishino, S. F., and J. C. Spain. 2004. Catabolism of nitroaromatic compounds, p. 575-608. In J. L. Ramos. (ed.), Pseudomonas, vol. 3. Kluwer Academic Publishers, New York, NY. [Google Scholar]
- 52.Orii, C., S. Takenaka, S. Murakami, and K. Aoki. 2004. A novel coupled enzyme assay reveals an enzyme responsible for the deamination of a chemically unstable intermediate in the metabolic pathway of 4-amino-3-hydroxybenzoic acid in Bordetella sp. strain 10d. Eur. J. Biochem. 271:3248-3254. [DOI] [PubMed] [Google Scholar]
- 53.Pandey, G., D. Paul, and R. K. Jain. 2003. Branching of o-nitrobenzoate degradation pathway in Arthrobacter protophormiae RKJ100: identification of new intermediates. FEMS Microbiol. Lett. 229:231-236. [DOI] [PubMed] [Google Scholar]
- 54.Pandey, G., and R. K. Jain. 2002. Bacterial chemotaxis toward environmental pollutants: role in bioremediation. Appl. Environ. Microbiol. 68:5789-5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pang, Y. P., K. Xu, J. E. Yazal, and F. G. Prendergas. 2000. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 9:1857-1865. [PMC free article] [PubMed] [Google Scholar]
- 56.Parales, R. E. 2004. Nitrobenzoates and aminobenzoates are chemoattractants for Pseudomonas strains. Appl. Environ. Microbiol. 70:285-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parales, R. E., M. D. Emig, N. A. Lynch, and D. T. Gibson. 1998. Substrate specificities of hybrid naphthalene and 2,4-dinitrotoluene dioxygenase enzyme systems. J. Bacteriol. 180:2337-2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parales, R. E., and C. S. Harwood. 2002. Bacterial chemotaxis to pollutants and plant derived aromatic molecules. Curr. Opin. Microbiol. 5:266-273. [DOI] [PubMed] [Google Scholar]
- 59.Race, P. R., A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton, and E. I. Hyde. 2005. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 280:13256-13264. [DOI] [PubMed] [Google Scholar]
- 60.Russell, T. R., and S. C. Tu. 2004. Aminobacter aminovorans NADH:flavin oxidoreductase His140: a highly conserved residue critical for NADH binding and utilization. Biochemistry 43:12887-12893. [DOI] [PubMed] [Google Scholar]
- 61.Ryde, U. 1999. Redesign of the coenzyme specificity in L-lactate dehydrogenase from Bacillus stearothermophilus using site-directed mutagenesis and media engineering. Protein Eng. 12:851-856. [DOI] [PubMed] [Google Scholar]
- 62.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 63.Schafer, A., A. Tauch, W. Jager, J. Kalinowski, G. Thierbach, and A. Puhler. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69-73. [DOI] [PubMed] [Google Scholar]
- 64.Schmidt, M. W., K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis, and J. A. Montgomery. 1993. General atomic and molecular electronic structure system. J. Comput. Chem. 14:1347-1363. [Google Scholar]
- 65.Schreuder, H. A., P. A. Prick, R. K. Wierenga, G. Vriend, K. S. Wilson, W. G. Hol, and J. Drenth. 1989. Crystal structure of the p-hydroxybenzoate hydroxylase-substrate complex refined at 1.9 A resolution. Analysis of the enzyme-substrate and enzyme-product complexes. J. Mol. Biol. 208:679-696. [DOI] [PubMed] [Google Scholar]
- 66.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 67.Sivaraman, J., R. S. Myers, L. Boju, T. Sulea, M. Cygler, V. J. Davisson, and J. D. Schrag. 2005. Crystal structure of Methanobacterium thermoautotrophicum phosphoribosyl-AMP cyclohydrolase HisI. Biochemistry 44:10071-10080. [DOI] [PubMed] [Google Scholar]
- 68.Smith, S. P., K. R. Barber, S. D. Dunn, and G. S. Shaw. 1996. Structural influence of cation binding to recombinant human brain S100b: evidence for calcium-induced exposure of a hydrophobic surface. Biochemistry 35:8805-8814. [DOI] [PubMed] [Google Scholar]
- 69.Somerville, C. C., S. F. Nishino, and J. C. Spain. 1995. Purification and characterization of nitrobenzene nitroreductase from Pseudomonas pseudoalcaligenes JS45. J. Bacteriol. 177:3837-3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tabor, S., and C. C. Richardson. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82:1074-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van den Heuvel, R. H. H., A. H. Westphal, A. J. R. Heck, M. A. Walsh, S. Rovida, W. J. H. van Berkel, and A. Mattevi. 2004. Structural studies on flavin reductase PheA2 reveal binding of NAD in an unusual folded conformation and support novel mechanism of action. J. Biol. Chem. 279:12860-12867. [DOI] [PubMed] [Google Scholar]
- 72.Van der Meer, J. R., and V. Sentchilo. 2003. Genomic islands and the evolution of catabolic pathways in bacteria. Curr. Opin. Biotechnol. 14:248-254. [DOI] [PubMed] [Google Scholar]
- 73.Wang, J., R. M. Wolf, J. W. Caldwell, P. A. Kollman, and D. A. Case. 2004. Development and testing of a general amber force field. J. Comput. Chem. 25:1157-1174. [DOI] [PubMed] [Google Scholar]
- 74.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]
- 75.Yu, H. S., and M. Alam. 1997. An agarose-in-plug bridge method to study chemotaxis in the archaeon Halobacterium salinarum. FEMS Microbiol. Lett. 156:265-269. [DOI] [PubMed] [Google Scholar]
- 76.Zylstra, G. J., S. W. Bang, L. M. Newman, and L. L. Perry. 2000. Microbial degradation of mononitrophenols and mononitrobenzoates, p. 145-160. In J. C. Spain, J. B. Hughes, and H.-J. Knackmuss (ed.), Biodegradation of nitroaromatic compounds and explosives. Lewis Publishers, Boca Raton, FL.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.