

Abstract
Objective:
The aim of this study was to determine the frequency of MYH mutations in one large population of polyposis patients without APC mutation identified.
Summary Background Data:
Familial adenomatous polyposis (FAP) is the most known inherited colorectal cancer syndrome. In 70% to 80% of polyposis patients, an APC mutation is found. Patients with polyposis but no APC mutation are considered as APC-muted patients and followed as their relatives accordingly. Biallelic mutation of MYH has been found to responsible of colorectal polyposis and cancer in an autosomal recessive pattern of inheritance.
Methods:
Between 1978 and 2004, 433 patients were operated for polyposis. A mutation on APC was identified in 322 patients. Among the remaining patients, 44 were identified as possible MYH-muted patients and contacted, and 31 signed informed consent. Clinical data were obtained from the patients’ medical notes. Germline mutation of MYH was searched by sequencing the whole gene. To confirm the deleterious effects of biallelic MYH mutation, transversions on K-ras and APC were searched.
Results:
There were 9 women and 22 men with a mean age of 53.9 years (range, 22–68 years) at the time of diagnosis. The mean number of polyps was 62.8 (range, 11–266). Eighteen patients (58.1%) had a colorectal cancer. We found biallelic MYH mutation in 6 patients (19.3%; 95% confidence interval, 5.2%–33.5%) and 5 (83.3%) had transversions in K-ras and/or APC.
Conclusion:
MYH is a new gene responsible for about 1.4% of all adenomatous polyposis and about 20% of adenomatous polyposis without APC mutation identified. Search for MYH biallelic mutation in these patients should be systematic as it changes their and relatives'surveillance.
Patients with FAP without APC mutation identified are considered as APC-muted patients and survey as their relatives accordingly. Within these patients, 20% were identified to have MYH biallelic mutation. Such mutation being responsible of a recessive disease, relatives’ surveillance can dramatically be reduced.
Colorectal cancer (CRC) is the third most frequent cancer in Western countries.1 Inherited factors are thought to play a major role in sporadic colorectal carcinogenesis as more than 25% of all CRC are associated with some family history.2 However, well-established CRC predisposal genes as APC,3 MLH1, or MHS24 account for only a minority of cases. Nevertheless, they are implicated in 2 well-known genetic syndromes representing 3% to 5% of all CRC: familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC). FAP is the commonest adenomatous polyposis syndrome with an autosomal dominant pattern of inheritance. Classic FAP is characterized by the development of multiple adenomas (at least 100) during the second decade with a high risk of developing CRC;5 when FAP is diagnosed after the age of 40 years, 73% of patients have already developed CRC.6 Extracolonic manifestations of FAP include congenital hypertrophy of the retinal pigmented epithelium (CHRPE), desmoid tumors, and duodenal, periampullary or ampullary adenoma. Some patients develop attenuated FAP (AFAP), which is characterized by the presence of fewer polyps (<100), proximal colonic predominance of the polyps and the later age of onset of polyp and CRC development.7
Direct sequencing of the APC gene is considered to be the most accurate diagnostic test for FAP or AFAP.8 APC mutations are responsible for 70% to 80% of the classic forms of FAP but are found in less than 30% in AFAP.9–11 Possible explanations are germinal mosaïsm and large genetic deletions, which are not identified by classic techniques. Until recently, patients without mutation were considered as APC carriers and surveyed accordingly. Theirs relatives (siblings and offspring) had the same surveillance, which is associated with considerable cost as well as anxiety. The endoscopy protocol includes a full colonoscopy every year after the teenage years to search for adenomatous polyps.
In 2002, Al-Tassan et al12 discovered that the biallelic mutation of MYH increased the risk of CRC. MYH13 is a base excision repair (BER) gene like MTH1 or OGG1. It's a DNA glycosylase responsible for the removal of adenines from DNA that have been mispaired with 8-hydroxiguanine (8oxoG). 8oxoG is a nucleotide product of oxidative reaction and can readily mismatch with adenine.14,15 If the BER system is deficient, as in the case of biallelic mutation of MYH, the mispaired adenines will lead to an accumulation of somatic transversions G:C→T:A in specific growth-regulatory genes such as APC or K-ras. Previous studies have shown that patients with mutations in MYH have numerous polyps (but never thousands) and some extracolonic features as osteomas, duodenal polyps, and CHRPE.16 The clinical differentiation between patients with de novo APC mutation (20% of all FAP patients17) and MYH mutated patients can be difficult as there is no polyposis history in either of the cases.
The aim of our study was to determine the frequency of MYH biallelic mutations in one large population of polyposis patients without APC mutation.
PATIENTS AND METHODS
Patients
Between January 1978 and September 2004, 433 patients were operated for colorectal polyposis at our institution. Among these patients, 351 had genetic testing for APC mutation. Patients with proven APC mutation were not included in our study. For patients not tested for APC mutation, personal and familial history was reviewed. Patients with a family history evoking an APC mutation (≥3 first-degree relatives developing colorectal polyposis) were also excluded from the study. The remaining patients were contacted and invited to participate in the study. Approval from the Commission National de l'Informatique et des Libertés (French National Data Processing Agency) was obtained (Bulletin Officiel de la Ville de Paris, May, 15, 2004).
Age, gender, cancer family history, and details of surgical procedures were obtained by data collection from patients’ medical notes (family history of cancer being recorded by surgeons for each patient and routinely detailed in the patient's notes). The family history of all patients included in the present study was also checked by direct interview.
DNA Extraction
For each patient, tumor DNA and normal control DNA were extracted from frozen tissue sections using the QiampKit (Qiagen Inc, Santa Clarita, CA). For tumor DNA, only those areas containing >70% tumor cells were used. The corresponding normal control DNA for each patient was extracted from normal colonic tissue, which was checked by a histopathologist to ensure absence of tumor cells in the sample.
PCR Amplification and Sequencing
Sixteen primer pairs were used to amplified the coding region and exon/intron junctions of MYH (GenBank ID: 4595) as previously described by Al-Tassan et al.12 PCR was performed on normal DNA to find germline mutation. To confirm the deleterious effects of biallelic MYH mutation, we looked for transversions on K-ras for every patient with available tumor DNA and on APC for every patient with a biallelic mutation on MYH. We amplified the first exon of K-ras (GenBank ID: 3845) and the mutation cluster region (codon 1250 to 1550) of APC (GenBank ID: 324) with 2 primers pairs (APC1: codon 1182 to 1415, APC2: codon 1390 to 1590). PCR reactions were performed in 50-μL reaction mixtures containing 60 ng of template DNA, 50 μmol/L of each oligonucleotide primer pair, 1.25 unit of AmpliTaq Gold DNA polymerase (Applied Biosystems, Applera, Courtaboeuf, France), and 0.2 mmol/L of dNTP (Invitrogen, Life Technologies, Cergy-Pontoise, France). Amplifications were realized in a Thermocycler GeneAmp PCR System 2700 (Applied Biosystems). The sequences of the primers used, the length of the PCR products, the MgCL2 concentration, and the annealing temperature are shown in Table 1. PCR products were run on a 2% standard agarose gel (InVitrogen), then eluted and purified using the QIAquick kit (Qiagen) according to the manufacture protocol. DNA fragments were then sequenced in both directions by MillGen Biotechnologies (Prologue Biotech, Labege, France).
TABLE 1. Primers of MYH, K-ras and APC, Annealing Temperature

Immunohistochemical Staining
Immunohistochemical staining for MSH2 and MLH1 was performed as previously described.18
RESULTS
Patients
In the 433 colonic polyposis patients operated at our institution between 1978 and 2004, including 199 relatives from 82 FAP families, APC sequencing was performed in 351 patients and a mutation was identified in 322 (92%). In 29 patients, no mutation was identified. Patients without APC mutation were statistically older than patients with a mutation (35.7 years vs. 28.0 years, P = 0.0367). Eighty-two patients had not been tested.
Among the 111 patients without an APC mutation, 47 had a family history of FAP and were excluded from the study. Of 64 patients, 11 had died and 9 were lost to follow-up. The remaining 44 patients were approached to participate in the study and 31 (70.4%) signed informed consent.
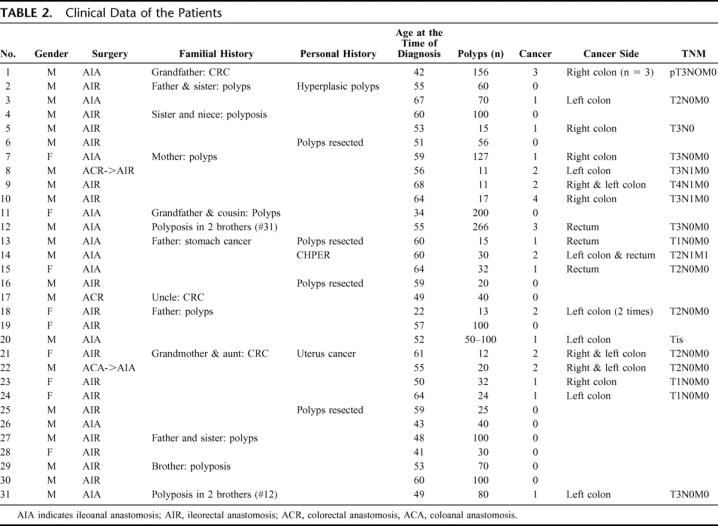
Clinical data of the patients are detailed in Table 2. There were 9 women and 22 men, and the mean age at the time of diagnosis was 53.9 years (±7.3; median, 55; range, 22–68 years). The mean number of polyps was 62.8 (±44.5; median, 40; range, 11–266). Eighteen patients (58.1%) were operated on as they had a cancer. Cancers were in the left colon or rectum in 58.1% (18 of 31 cancers). Two patients had extracolonic manifestations: patient 14 had hypertrophy of the pigmented retinal epithelium and 2 patients (patients 3 and 14) had duodenal polyps.
TABLE 2. Clinical Data of the Patients
MYH Analysis
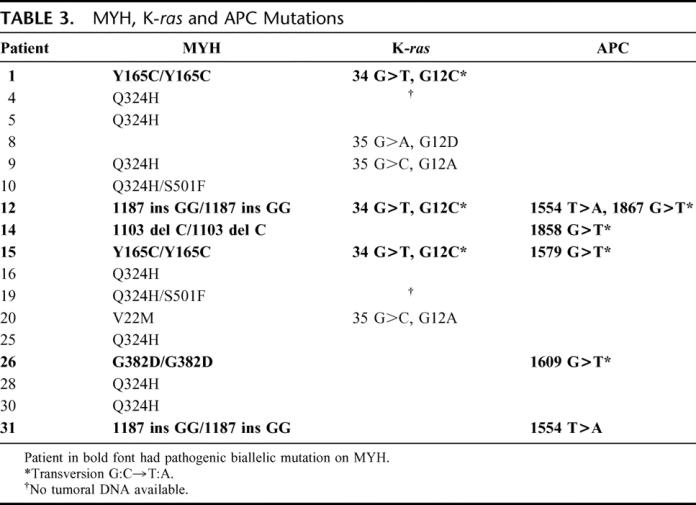
In 6 patients, a biallelic MYH mutation was found (19.3%; 95% CI, 5.2%–33.5%) (Fig. 1, Table 3). The most frequently reported missense changes Y165C and G382D were found in 3 patients. One or 2 MYH polymorphisms were identified in 10 patients (32.2%): Q324H (n = 9; 29.0%), V22M (n = 1; 3.2%) and S501F (n = 2; 6.4%) (Table 3).

FIGURE 1. MYH biallelic mutations.
TABLE 3. MYH, K-ras and APC Mutations
K-ras and APC Analysis
Amplification of the first exon of K-ras was performed in 24 patients. Six patients were found to have a mutation (25%). Three patients who had a biallelic mutation of MYH also had a mutation of K-ras, and in all cases it was a transversion G:C→T:A. The 3 others mutations on K-ras happened in patients without a pathogenic variant of MYH, and none of them was a transversion (Table 3). The study of APC in the 6 patients muted on MYH showed 4 transversions and 2 other mutations (two T>A in the 2 brothers, patients 12 and 31) (Table 3). All transversions observed in K-ras or APC occurred in GAA or GAAAA sequences. With these 2 sequences, we found in 5 patients (83.3%) with a biallelic mutation on MYH, 1 or 2 transversions in K-ras and/or APC.
Immunohistochemical Staining
Among the 31 patients who were included in this study, only 1 patient (patient 31) lost expression of MLH1.
Clinical Characteristics of Biallelic MYH Mutation Patients
There was 5 males and 1 female patients that demonstrated a biallelic MYH mutation. The mean number of polyps was 100 (median, 60; range, 30–266). One third of the patients with MYH mutation had more than 100 polyps. Among the 22 patients with more than 20 polyps, the frequency of biallelic mutation on MYH was 27.2% (95% CI, 8.2%–35.5%), and none of the patients with less than 20 polyps was found to have a MYH biallelic mutation. The mean age of patients with MYH mutation at the time of the operation was 52.2 years (median, 52 years; range, 42–64 years). Two extracolonic features were seen in 1 patient: duodenal polyposis and CHRPE. Ten cancers occurred in 5 of the 6 patients (85.6%) with biallelic MYH mutation. The cancers were mainly localized on the left colon (n = 2 in the colon, n = 5 in the rectum).
DISCUSSION
Beside classic FAP with a germline mutation of APC, some patients have numerous adenomatous polyps, a high risk of CRC, and 2 wild-type copies of APC. Since 2002, the MYH gene, which belongs to the BER system involved in the DNA repair of oxidative lesion, is known to predispose to recessive inheritance of numerous adenomatous polyps.12 The aim of our work was to study the frequency of MYH mutations in one large single-center population of polyposis patients without APC mutation and in a further step to adapt the surveillance offered to the relatives of newly diagnosed polyposis patients without APC mutation.
Among our 31 patients without an APC mutation, 6 (19.3%) had a biallelic germline mutation of MYH. Two of them had more than 100 polyps. Given this observation, the number of polyps cannot be viewed as a pathognomonic feature of FAP any more. Five of the 6 patients had CRC; they were not known to have colorectal polyposis and only diagnosed when they became symptomatic. Our group6 has shown that in 141 patients with FAP and mutation of APC, 71 were symptomatic at the time of the operation (as were the MYH patients in this study). The mean age of those patients was 40 years and 32 (45%) had a cancer at the time of the operation. It therefore appears that polyps and cancers develop later in patients with biallelic mutation of MYH than in those with APC mutation. One patient had extracolonic features: CHRPE (n = 1) and duodenal polyps (n = 1). Thus, confronted with a patient with adenomatous polyposis, the 2 main clinical features that should evoke MYH biallelic mutation are the age at the time of diagnosis and the absence of a family history of polyposis, especially in siblings and offspring. Concerning sex ratio, if in our study 5 of the 6 patients with biallelic MYH mutation were males, it seems that this observation is purely incidental. Thus, gender should not be considered for MYH mutation search. If no mutation is found at APC testing, MYH genetic testing should be undertaken.
Most investigators explore only the exons 7 and 1319,20 where the 2 most frequent mutations in MYH Y165C and G382D occur. We would have diagnosed only 3 of our 6 patients with MYH biallelic mutation had we followed this strategy. We therefore chose to explore the entire coding sequence of MYH. Moreover, recently novel mutations of other exons have been described21 as well as specific mutations of MYH in different ethic populations such as E466X in Indian cases,22 which demonstrates the importance of screening the whole coding sequence of MYH.
Among our 6 patients muted on MYH, 5 had typical transversions on APC and/or K-ras demonstrating the pathogenicity of biallelic MYH mutation. Patient 31 was the only one in whom no transversion could be identified. However, in this patient, a loss of expression was identified by immunohistochemical staining, demonstrating a tumor with microsatellite instability phenotype. A search for mutation or methylation of hMLH1 was performed and showed no abnormality. Study of the DNA sequence of hMLH1 (GenBank 29729888) shows the presence of several GAA sequences that could be the site of transversion and then explain the inactivation of hMLH1 by the MYH pathway. Moreover, patient 31 is the brother of patient 12 (both patients having the same MYH mutation: 1187insGG/1187insGG) who was found to have a microsatellite stable tumor with normal expression of hMLH1 and hMSH2 but transversions on K-ras and APC. Thus, biallelic MYH mutation could induce tumorogenesis by the 2 pathways known in CRC, mainly by inactivation of APC and K-ras and in some case also by inactivating hMLH1 and thereby the mismatch repair system.
Despite the search of germline mutation in MYH, there is still no explanation for the polyposis in 25 of the 31 patients (80.6%) included in this study. These patients with no genetic etiology are still considered as APC mutant and they and their families are submitted to intensive clinical and endoscopy follow-up. Alternate splicing modifications of APC or MYH and unknown genes implicated in the colonic cancers may be the explanation for these polyposis cases. Lipton et al23 have recently proven the involvement of germline mutation in BMPR1A in adenomatous polyposis. This gene belongs to the TGF-β family known to cause the juvenile polyposis syndrome.
Depending on number and location of the polyps, patients with MYH biallelic mutations should undergo total colectomy or restorative proctocolectomy, just like APC-muted patients. As previously described,16 we found 1 patient with duodenal polyposis; surveillance of the upper gastrointestinal tract for duodenal polyps should therefore be continued. The main difference with FAP concerns the siblings and offspring. When an APC mutation is identified, the risk for each child to have FAP is 50%, whereas it is less than 1% (heterozygote frequency24 divided by 2) for a child of a MYH biallelic muted parent. For the siblings of an APC muted patient, the risk is 50%, compared with 25% with a MYH biallelic muted parent. Moreover, identification of biallelic mutation on MYH in a patient with FAP or AFAP phenotype but without APC mutation allows specific investigation of the relatives. Surveillance of siblings may then be modified accordingly. Nevertheless, the real risk for monoallelic carrier of pathogenic mutation of MYH is still unknown. As in every disease with a recessive pattern of transmission, heterozygous patients should be healthy, but Kambara et al25 showed a higher level of loss of heterozygosity in 1p where the locus of MYH is localized and a higher level of transversion in APC and K-ras in patients with a single pathogenic variant of MYH. There was, however, no significant difference of CRC rates between the general population and the monoallelic carriers of MYH.
Reducing the number of unnecessary colonoscopies would save money and avoid the discomfort of bowel preparation, the risks of anesthesia, and of the examination itself as well as any associated anxiety. For those polyposis patients with neither mutation of APC nor MYH, other gene mutations should be searched for, but until another explanation is found they have to be surveyed as if they were APC mutants.
CONCLUSION
MYH is a gene responsible for 1.4% of all adenomatous polyposis and 20% of adenomatous polyposis without mutation of the APC gene. The risk of transmission to offspring is less than 1% as it is a recessive inherited disease. Colonoscopic surveillance can thus be dramatically reduced in the offspring of the index patient. For these reasons, we propose searching for MYH biallelic mutations to all patients with colorectal polyposis without APC mutation.
Discussions
Dr. Neil Mortensen: Thank you for a beautifully presented and explained paper. Life used to be so simple. FAP was all about abnormalities of the APC gene, and now we find that is not the case. While APC gene abnormalities are the cause in about 80% of patients with FAP, you've looked at that other 20% and you've only found a small proportion having MYH abnormalities. I wonder if you could please tell us the answers to 2 questions?
Are there patients within the APC group who also have MYH gene abnormalities, which would, if you like, give a more severe phenotype, perhaps?
Do you think that there is, within that residual, let's say 15% to 18% of patients in whom you haven't found any abnormality, the place for some kind of international collaboration using GNY searches on huge numbers of patients to try and find out what's going on in those last 15% to 18%?
Dr. Yann Parc: No, we did not find any MYH mutations in patients already with an APC mutation; however, we tested only a few patients with an APC mutation. In the literature, such association of mutations has not been reported and, yes, we are ready to collaborate in a study to find other genes. A family case has been reported in which the BRMA gene was considered to be responsible for the polyposis. However, much larger series are required to confirm this finding and allow the identification of other genes that could be responsible for colorectal polyposis.
Dr. Mario Morino: This is a very interesting study but, in the abstract, you have proposed a different follow-up strategy for these patients and you did not speak of this point in your presentation. Do you think there are clinical implications or have you changed your mind compared with the conclusions of your abstract?
Dr. Yann Parc: No, we have not changed our mind. For the patient with APC mutation identified, you know which patients will have to be surveyed and treated because you can find the at risk parents as they have the same mutation. In such cases, the risk for the children of the patient to have the disease is 50%, and it is the same for the siblings. When you have found no APC mutation identified, you have to consider all the parents at risk, and so you have to survey them with a colonoscopy every year. When, you find an MYH biallelic mutation, you can search for the same mutation in the siblings and you know that the risk for siblings to have the same biallelic mutations is only 25%. For the children, this risk is inferior to 1% as the risk that other parent is one allele of MYH mutated is less than 1%. You can therefore dramatically change the surveillance of the parents of these patients. It will reduce the total number of colonoscopies and the stress induced by such surveillance.
Footnotes
Reprints: Yann Parc, MD, PhD, Department of Digestive Surgery, Hôpital Saint-Antoine, Assistance Publique-Hôpitaux de Paris, Université Pierre et Marie Curie, Paris VI., 184 rue du Faubourg Saint-Antoine, 75012, Paris, France. E-mail: yann.parc@sat.ap-hop-paris.fr.
REFERENCES
- 1.Landis SH, Murray T, Bolden S, et al. Cancer statistics, 1999. CA Cancer J Clin. 1999;49:8–31. [DOI] [PubMed] [Google Scholar]
- 2.Johns LE, Houslton RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96:2292–2303. [DOI] [PubMed] [Google Scholar]
- 3.Kinzler VW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. [DOI] [PubMed] [Google Scholar]
- 5.Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–818. [PMC free article] [PubMed] [Google Scholar]
- 6.Penna C, Tiret E, Kartheuser AH, et al. Value of screening of familial adenomatous polyposis for prevention of colorectal cancer. Gastroenterol Clin Biol. 1992;16:210–214. [PubMed] [Google Scholar]
- 7.Lynch HT, Smyrk T, McGinn T, et al. Attenuated familial adenomatous polyposis (AFAP): a phenotypically and genotypically distinctive variant of FAP. Cancer. 1995;76:2889–2892. [DOI] [PubMed] [Google Scholar]
- 8.Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology. 2001;121:198–213. [DOI] [PubMed] [Google Scholar]
- 9.Lamlum H, Al Tassan N, Jaeger E, et al. Germline APC variants in patients with multiple colorectal adenomas, with evidence for the particular importance of E1317Q. Hum Mol Genet. 2000;9:2215–2221. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong JG, Davies DR, Guy SP, et al. APC mutations in familial adenomatous polyposis families in the Northwest of England. Hum Mutat. 1997;10:376–380. [DOI] [PubMed] [Google Scholar]
- 11.Heinimann K, Thompson A, Locher A, et al. Nontruncating APC germ-line mutations and mismatch repair deficiency play a minor role in APC mutation-negative polyposis. Cancer Res. 2001;61:7616–7622. [PubMed] [Google Scholar]
- 12.Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colo-rectal tumors. Nat Genet. 2002;30:227–232. [DOI] [PubMed] [Google Scholar]
- 13.Slupska MM, Baikalov C, Luther WM, et al. Cloning and sequencing a human homolog (MYH) of the Escherichia coli MutY gene whose function is required for the repair of oxidative damage. J Bacteriol. 1996;178:3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531:231–251. [DOI] [PubMed] [Google Scholar]
- 15.Ames BN, Gold LS. Endogenous mutagens and the causes of aging and cancer. Mutat Res. 1991;250:3–16. [DOI] [PubMed] [Google Scholar]
- 16.Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis ans germ-Line mutations in MYH. N Engl J Med. 2003;348:791–799. [DOI] [PubMed] [Google Scholar]
- 17.Grady WM. Genetic testing for high-risk colon cancer patient. Gastroenterology. 2003;124:1574–1594. [DOI] [PubMed] [Google Scholar]
- 18.Rigau V, Sebbagh N, Olschwang S, et al. Microsatellite instability in colorectal carcinoma: the comparison of immunohistochemistry and molecular biology suggests the interest of hMSH6 immunostaining. Arch Pathol Lab Med. 2003;127:694–700. [DOI] [PubMed] [Google Scholar]
- 19.Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delgga germline mutations of the myh gene in Italian patients with adenomatous polyposis coli and colorectal adenomas Int J Cancer. 2004;109:680–684. [DOI] [PubMed] [Google Scholar]
- 20.Enholm S, Hienonen T, Suomalainen A, et al. Proportion and phenotype oh MYH-associated colorectal neoplasia in a population-based series of Finnish colorectal cancer patients. Am J Pathol. 2003;163:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alhopuro P, Parker AR, Lehtonen R, et al. A novel functionally deficient MYH variant in individuals with colorectal adenomatous polyposis. Hum Mutat. 2005;26:393. [DOI] [PubMed] [Google Scholar]
- 22.Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. [DOI] [PubMed] [Google Scholar]
- 23.Lipton L, Sieber OM, Thomas HJW, et al. Germline mutations in the TGF-β and Wnt pathways are a rare cause of the multiple adenoma phenotype. J Med Genet. 2003;40:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Baudhuin LM, Boardman LA, et al. MYH mutations in patients with attenuated and classic polyposis and with young-onset colorectal cancer without polyps. Gastroenterology. 2004;127:9–16. [DOI] [PubMed] [Google Scholar]
- 25.Kambara T, Whitehall VLJ, Spring KJ, et al. Role of inherited defects of MYH in the development of sporadic colorectal cancer. Genes Chromosomes Cancer. 2004;40:1–9. [DOI] [PubMed] [Google Scholar]