

Abstract
The GTPase dynamin I is required for synaptic vesicle (SV) endocytosis. Our observation that dynamin binds to the SV protein synaptophysin in a Ca2+-dependent fashion suggested the possibility that a dynamin/synaptophysin complex functions in SV recycling. In this paper we show that disruption of the dynamin/synaptophysin interaction by peptide injection into the squid giant synapse preterminal results in a decrease in transmitter release during high-frequency stimulation, indicating an inhibition of SV recycling. Electron microscopy of these synapses reveals a depletion of SVs, demonstrating a block of vesicle retrieval after fusion. In addition, we observed an increase in clathrin-coated vesicles, indicating that the peptide does not block clathrin-dependent endocytosis. We conclude that the dynamin/synaptophysin complex functions in a clathrin-independent mechanism of SV endocytosis that is required for efficient synaptic transmission.
Local recycling of synaptic vesicles (SVs) at nerve terminals is essential to synaptic function. In the classical model of SV endocytosis, the vesicle collapses into the plasma membrane following transmitter release and is then retrieved at a site away from the active zone by a clathrin-dependent mechanism involving the GTPase dynamin (1). The importance of clathrin-dependent recycling at synapses is well established (2–6). A second model of SV recycling, called rapid endocytosis or “kiss-and-run,” postulates that endocytosis is closely coupled to exocytosis (7–9). In this model, the SV does not collapse into the plasma membrane following transmitter release, but instead is rapidly retrieved at the active zone by a clathrin-independent mechanism. Although several reports indicate that secretory granules as well as SVs can be endocytosed very quickly (10–15), there is no data indicating that rapid, clathrin-independent endocytosis of SVs is important to synaptic function.
Dynamin I interacts with the C-terminal cytoplasmic tail of synaptophysin (SYP) at Ca2+ concentrations (≈150 μM) found only at sites of SV fusion (C.D. and E.B.Z., unpublished observations). This finding suggested the possibility that SYP recruits dynamin to the vesicle membrane during rapid endocytosis at the active zone rather than during clathrin-dependent endocytosis, which takes place away from active zones (16) where Ca2+ concentration is not expected to be elevated. In this study, we employ the squid giant synapse to demonstrate a function for the dynamin/SYP complex in vesicle retrieval.
Materials and Methods
Immunoblots.
Squid optic lobe extracts were immunoblotted with a polyclonal Ab against the last 13 aa of human SYP (SV063; Stressgen Biotechnologies, Victoria, Canada) at 1 μg/ml, a mAb against the C terminus of bovine SYP (SY38; Boehringer Mannheim) at 0.5 μg/ml, a mAb against the proline-rich domain of rat dynamin I (D25520; Transduction Laboratories, Lexington, KY) at 0.5 μg/ml, and a polyclonal Ab (anti-Pan 65) against a conserved region of the dynamin I N-terminal domain (17) (gift of M. McNiven) at 1:1000 dilution.
Electrophysiology.
Experiments were performed at the Marine Biological Laboratory in Woods Hole, MA, using the giant synapse of the stellate ganglion of squid (Loligo Pealii). The isolation of the stellate ganglion and the electrophysiological techniques used have been described previously (18, 19). The glutathione S-transferase (GST)—SYP fusion protein contains the cytoplasmic C-terminal tail of SYP (aa 219–307), to which dynamin binds (C.D. and E.B.Z., unpublished observations). GST and GST–SYP were purified on glutathione-agarose beads, eluted with 10 mM glutathione, and dialyzed against PBS (pH 8.0). The proteins were then conjugated to FITC by incubation with FITC succinimidyl ester (Molecular Probes) in sodium bicarbonate buffer (pH 8.3). FITC-labeled proteins were separated from free dye by extensive dialysis against PBS (pH 7.4). The injection fluid contained GST or GST–SYP at ≈1.5 mg/ml (≈37.5 μM GST–SYP) in 0.5 M K+Acetate and 0.1 M Hepes (pH 7.2). The proteins were pressure injected into the largest (most distal) presynaptic terminal digit. The exact location of injection and the diffusion and steady-state distribution of the labeled proteins in the presynaptic terminal were monitored by using a fluorescence microscope attached to a Hamamatsu (Middlesex, NJ) camera system. In all experiments a correlation was made between the localization of the fluorescent proteins and the electrophysiological findings. Data were obtained from 13 GST—SYP-injected synapses and from 7 GST-injected synapses. In every case, GST–SYP had a large effect on transmitter release, relative to GST.
Electron Microscopy.
The ganglia were removed from the electrophysiology recording chamber, fixed by immersion in gluteraldehyde, postfixed in osmium tetroxide, stained in block with uranium acetate, dehydrated, and embedded in resin Embed 812 (EM Science). Ultrathin sections were collected on Pyoloform (Ted Pella, Redding, CA) and carbon-coated single sloth grids and contrasted with uranyl acetate and lead citrate. Morphometry and quantitative analysis of the SVs were performed with a Zidas digitizing system (Zeiss) interfaced with a Macintosh G3 computer. Electron micrographs were taken at an initial magnification of ×16,000 and ×31,500 and photographically enlarged to a magnification of ×40,000 and ×79,000 for SV and clathrin-coated vesicle (CCV) counting, respectively. Vesicle density at the synaptic active zones was determined as the number of vesicles per μm2, on an average area of 0.8 μm2 per active zone. CCV density was determined within the limits of the presynaptic terminal, on an average terminal area of 3.3 μm2. Approximately 30 terminals and 40 active zones were counted for each synapse represented in Table 1.
Table 1.
Quantitation of the effects of GST–SYP on SV number, docked SV number, and CCV number during high-frequency stimulation
| Vesicles | Docked vesicles | Coated vesicles | |
|---|---|---|---|
| GST-1 | 98.13 ± 12.3 | 14.11 ± 1.64 | 0.84 ± 0.06 |
| GST-2 | 71.2 ± 5.29 | 7.96 ± 0.63 | 0.77 ± 0.06 |
| SYP-1 | 34.58 ± 2.87 | 3.42 ± 0.4 | 2.33 ± 0.3 |
| SYP-2 | 22.77 ± 3.48 | 3.27 ± 0.49 | 1.76 ± 0.2 |
| Average GST | 84.66 ± 8.79 | 11.03 ± 1.13 | 0.8 ± 0.06 |
| Average SYP | 28.67 ± 3.17 | 3.34 ± 0.44 | 2.45 ± 0.25 |
Vesicle numbers are the mean number of synaptic vesicles per μm2, clustered at active zones (mean ± SEM). Docked vesicles are defined as those within 0.1 vesicle diameters of the plasma membrane at the active zone. Coated vesicle numbers are the mean number of CCV per μm2. The GST-1 and SYP-1 synapses were stimulated 25 min after injection for 1 min continuously (50 Hz, 750-ms trains with a 1.5-s interval between trains), allowed to rest for 1 min, and then given one final train of action potentials before fixation. The GST-2 and SYP-2 synapses were stimulated for 3 min continuously 50 min after injection and then fixed. Average GST and average SYP are averages of the values from the two GST-injected and GST–SYP-injected synapses, respectively.
Results
Disruption of the Dynamin/SYP Interaction Causes an Inhibition of SV Recycling.
We tested the involvement of the dynamin/SYP complex in SV recycling by blocking the interaction in the squid giant synapse preterminal and assessing the effect this has on neurotransmitter release. Injection of a fusion protein (GST–SYP) containing the dynamin-binding region of SYP (the C-terminal cytoplasmic tail) should compete with endogenous SYP for dynamin binding, thereby blocking recruitment of dynamin to SV membranes. If this resulted in an inhibition of SV endocytosis, the vesicle pool would be depleted during high-frequency stimulation, resulting in a decrease in transmitter release. To ensure the relevance of disrupting a dynamin/SYP complex in squid, we confirmed that both proteins are expressed in squid, are exactly the same size as the mammalian proteins, and share multiple epitopes with the mammalian proteins (Fig. 1). We assessed the effects of GST or GST–SYP on transmitter release during trains of high-frequency stimulation (Fig. 2). At ≈1, 5, 10, 15, and 25 min after injection, the synapses were stimulated as follows: 1 train of action potentials at 50 Hz for 750 ms, 1 min of rest, 1 min of continuous stimulation with the 50 Hz, 750-ms trains separated by 1.5-s intervals, 1 min of rest, and 1 final train of action potentials at 50 Hz for 750 ms. By the end of the 1-min continuous stimulation (given at 15 min after injection), GST–SYP caused a significant inhibition of transmitter release during the action potential trains, relative to GST (Fig. 2B, Middle). Even during the final, single train of action potentials, after a full minute of rest, transmitter release in the presence of GST–SYP was reduced (Fig. 2B, Bottom). The reduction in transmitter release was not due to a modification of the amplitude or duration of the presynaptic action potential, which was unaffected by GST–SYP (Fig. 2A). GST–SYP did not inhibit the vesicle fusion mechanism because the postsynaptic response to single stimuli was unaffected (Fig. 2A). In addition, the kinetics of the inhibition of the postsynaptic response during a train of action potentials suggest a reduction in vesicle availability rather than an impairment of vesicle fusion (Fig. 2B). Thus, GST–SYP inhibits the ability of the synapse to maintain transmitter release during high-frequency stimulation and inhibits recovery of release capability during a 1-min rest period following stimulation. One possible explanation of these effects is that GST–SYP blocks SV endocytosis, resulting in a depletion of the vesicle pool.
Figure 1.

Dynamin and SYP are expressed in squid. Squid optic lobe extract was subjected to immunoblot with (A) a polyclonal Ab against the C terminus of human SYP (Left) in the absence (−) or presence (+) of the antigenic peptide or a mAb against the C terminus of bovine SYP (Right) and (B) a mAb against the proline-rich domain (PRD) of rat dynamin (Left) or a polyclonal Ab against a conserved region of the dynamin N-terminal domain (Right).
Figure 2.
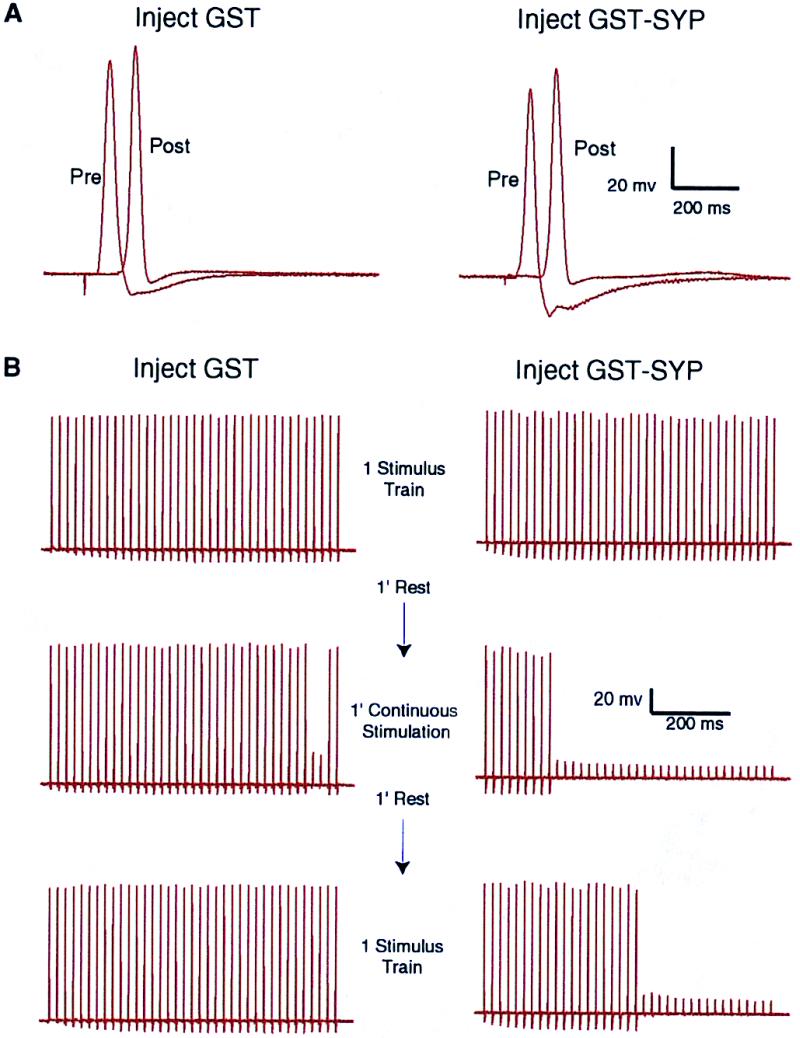
Injection of GST–SYP into the squid giant synapse preterminal results in a decrease in neurotransmitter release during high-frequency stimulation. Synapses were injected with GST or GST–SYP and stimulated at ≈1, 5, 10, 15, and 25 min after injection as follows: 1 train of action potentials at 50 Hz for 750 ms, 1 min of rest, 1 min of continuous stimulation with the 50 Hz, 750-ms trains separated by 1.5-s intervals, 1 min of rest, and 1 final train of action potentials at 50 Hz for 750 ms. (A) The traces show the presynaptic and postsynaptic potentials in response to a single stimulus, given ≈15 min after injection, just before the high-frequency stimulation shown in B. (B) The traces show the postsynaptic potential during high-frequency stimulation at ≈15 min after injection of GST or GST–SYP.
To quantify the magnitude of the effect of GST–SYP on transmitter release, we performed postsynaptic voltage-clamp experiments. In Fig. 3 A and B, the effects of GST or GST–SYP on the amplitude of the postsynaptic current during the final, single train of action potentials, given after 1 min of continuous stimulation and 1 min of rest, are compared at various times after injection. At ≈30 min after injection, in the presence of GST–SYP, but not GST, there was a dramatic reduction in transmitter release during the final stimulus train, indicating a failure to recover release capability following the 1-min continuous stimulation (Fig. 3 A and B, Bottom). GST–SYP had no effect on the presynaptic action potential (Fig. 3B, Top). Fig. 3 C and D illustrate the progressive nature of the effect of GST–SYP. The traces depict the amplitude of the postsynaptic current as a function of time during the final train of action potentials at various times after injection. In the presence of GST–SYP there was a decrease in amplitude and an increase in the rate of decline of the postsynaptic current with each successive set of stimulus trains (Fig. 3D, Top). These changes were much more pronounced than those observed in the presence of GST (Fig. 3C, Top). The rate of decline of the postsynaptic current during the final train of action potentials can be described by two time constants. The very rapid initial decline in current amplitude during a stimulus train (Fig. 3 C and D, Middle) presumably reflects depletion of the docked and primed pool of SVs. Once this pool is depleted, SVs can become available for release through priming of already-docked vesicles. The decline of this second phase of release during a stimulus train (Fig. 3 C and D, Bottom) was much slower. Quantitation of the areas under the curves in Fig. 3 C and D (Middle and Bottom), which is a measure of the relative amounts of transmitter released during the stimulus trains, allows a direct assessment of the effects of GST and GST–SYP on the two components of the release process (Fig. 3E). In the presence of GST–SYP, the fast component of release was reduced by ≈90% (compared with no reduction in the presence of GST), whereas the slow component was reduced by ≈60% (compared with a 20% reduction in the presence of GST). Thus, GST–SYP dramatically inhibits the ability of synapses to recover release capability following high-frequency stimulation. This inhibition could be explained by a block of vesicle retrieval, resulting in a decrease in the number of vesicles available to refill the docked pool.
Figure 3.

Injection of GST–SYP into the squid giant synapse preterminal inhibits recovery of release capability following high-frequency stimulation. (A and B) Synapses were injected with GST or GST–SYP and stimulated at ≈1, 5, 10, 15, and 25 min after injection as follows: 1 train of action potentials at 50 Hz for 750 ms, 1 min of rest, 1 min of continuous stimulation with the 50 Hz, 750-ms trains separated by 1.5-s intervals, 1 min of rest, and 1 final train of action potentials at 50 Hz for 750 ms. Transmitter release during the final train of action potentials is a measure of the recovery of release capability following the 1-min stimulation. The Middle and Bottom panels show the amplitude of the postsynaptic current during the final train of action potentials, given at the indicated times after injection. The Top panels illustrate the lack of effect of GST–SYP on the amplitude and duration of the presynaptic action potential, measured ≈30 min after injection. (C and D) The traces in the Top panels show the amplitude of the postsynaptic current as a function of time during the final train of action potentials, given at the indicated times after injection of GST or GST–SYP. The curves were fit to records like those shown in A and B (Middle and Bottom panels) by using a double exponential fitting method, i.e., the rate of decline of the current amplitude during the final train of action potentials is described by two time constants. The traces in the Middle panels depict the fast component of the decline, whereas the traces in the Bottom panels depict the slow component of the decline. (E) The relative areas under the curves in C and D (Middle and Bottom panels) were quantitated, giving a measure of the relative amounts of transmitter released during the stimulus trains. The relative areas for the fast and slow components of the release process at the indicated times after injection are shown. (F) The synapse was injected with GST–SYP and stimulated according to the protocol described above (except at 100 Hz, with a 2-s interval between stimulus trains during the 1-min continuous stimulation) at 1, 6, 11, 16, 26, 31, 36, and 46 min after injection. The synapse was then stimulated once more after ≈3 h of rest. The traces show the amplitude of the postsynaptic current as a function of time during the final train of action potentials given at the indicated times after injection.
Fig. 3F illustrates the reversibility of the effects of GST–SYP injection. The traces depict the amplitude of the postsynaptic current during the final train of action potentials given at the indicated times after injection. At 46 min after injection, the amplitude of the postsynaptic current was dramatically reduced (Fig. 3F). After ≈3 h of rest, which allows for dilution of GST–SYP by diffusion in the axon, the synapse was stimulated again (3 h, 48 min). The inhibition of transmitter release was completely reversed (Fig. 3F). Thus, the effects of GST–SYP on SV recycling (Figs. 2 and 3) are not the result of irreversible damage to synapses.
Disruption of the Dynamin/SYP Interaction Results in a Depletion of the SV Pool During High-Frequency Stimulation.
The inhibitory effects of GST–SYP on transmitter release during high-frequency stimulation could result from inhibition of any step in the SV recycling pathway, e.g., endocytosis, refilling of vesicles with transmitter, or docking. A block of vesicle retrieval, but not a block of the other steps in recycling, would result in a depletion of the vesicle pool. To assess whether GST–SYP blocks SV endocytosis, synapses injected with GST or GST–SYP were stimulated at high-frequency, fixed immediately, and processed for electron microscopy. Two effects of GST–SYP, relative to GST, were apparent. First, there was a large reduction in the size of the total vesicle pool (Fig. 4, Table 1) and in the number of docked vesicles (Fig. 4, Table 1). Note the regularly spaced clusters of SVs at active zones in GST-treated synapses (Fig. 4 C and D) and their virtual absence in GST–SYP treated synapses (Fig. 4 A and B). This finding indicates that the inhibition of transmitter release by GST–SYP reflects a block of SV endocytosis and a depletion of the vesicle pool. Thus, formation of a dynamin/SYP complex is required for SV endocytosis.
Figure 4.

Injection of GST–SYP into the squid giant synapse preterminal results in a depletion of the vesicle pool and an increase in the number of CCVs during high-frequency stimulation. (A and B) Electronmicrographs from cross sections of a GST–SYP injected synapse. (A) A low magnification image showing several active zones with very small SV clusters. Numerous CCVs are indicated by the arrows. (B) A higher magnification image of the same synapse showing an active zone with a small number of SVs and numerous CCVs (arrows) at some distance from the active zone. Note the numerous uncoated vesicles directly behind the CCVs at the membrane. The Inset in B shows clear examples of CCVs which had pinched off from the plasma membrane in GST–SYP treated synapses. (C and D) Electronmicrographs from cross sections of a GST-injected synapse. (C) A low magnification image showing several active zones with much larger vesicle clusters. A few CCVs (arrows) are seen between the active zones. (D) A higher magnification image showing two active zones with their typical cluster of SVs.
In addition, there was a large increase in the number of CCVs (Fig. 4, Table 1), indicating that GST–SYP does not block clathrin-dependent endocytosis, and therefore that the dynamin/SYP complex functions in a clathrin-independent mechanism of endocytosis. We interpret the increased number of CCVs as reflecting an increased rate of CCV formation. Presumably, the block of rapid endocytosis causes a greater number of the fused SVs to be recycled by the clathrin-dependent pathway. A model depicting our interpretation of the effects of GST–SYP on the vesicle pool and on transmitter release is shown in Fig. 5.
Figure 5.

Model for the effects of GST–SYP on the vesicle pool and on neurotransmitter release. In control synapses stimulated at high-frequency (Upper), many vesicles are recycled by rapid endocytosis (RE) and some collapse into the membrane and are retrieved by clathrin-mediated endocytosis (CME). When rapid endocytosis is blocked (Lower), more of the fused SVs are forced to recycle via CCVs. Because the clathrin pathway is slower, the result is a temporary redistribution of SV membrane from the active zone to the clathrin-dependent recycling pathway. The depletion of vesicles at the active zone results in a decrease in transmitter release.
Discussion
The data presented in this report indicate that the dynamin/SYP complex functions in a mechanism of SV endocytosis that is apparently clathrin-independent. In addition, our data indicate that this mechanism is required for efficient synaptic transmission, because the clathrin-dependent recycling pathway is not sufficient, under the conditions of our experiments, to maintain an adequate supply of releasable vesicles.
Our conclusion that the dynamin/SYP complex mediates clathrin-independent endocytosis is derived from the morphological data in Fig. 4, which shows an increase in the number of CCVs in the presence of GST–SYP. We interpret this increase as reflecting an increased rate of CCV formation. An alternate interpretation, because many of the CCVs are close to the plasma membrane, would be that their accumulation reflects a block of fission from the membrane. We view this possibility as being extremely unlikely for several reasons. First, many of the CCVs have pinched off from the plasma membrane (see Inset in Fig. 4B for particularly clear examples of pinched off CCVs in GST–SYP treated synapses). Thus, the morphological phenotype we observe is quite distinct from that observed when dynamin recruitment to sites of CCV formation is blocked by an amphiphysin SH3 domain peptide (4). In that study, there were virtually no CCVs separated from the plasma membrane.
Second, even in control synapses, the CCVs are very close, or attached to, the plasma membrane (Fig. 4C, arrows). This finding suggests that once a CCV has pinched off, it uncoats very rapidly. Thus, the localization of many CCVs close to the membrane in GST—SYP-treated synapses does not necessarily indicate a block of CCV fission. This conclusion is supported by the presence of numerous uncoated vesicles directly behind the CCVs in the GST—SYP-treated synapses (Fig. 4B). The very close proximity of these uncoated vesicles to the CCVs at the membrane, and the fact that they are not at active zones, strongly suggest that they arose via uncoating of newly pinched off CCVs. Again, it is instructive to compare our phenotype with that observed after injection of the amphiphysin SH3 domain (4). In that study, there were no uncoated vesicles behind the accumulated coated pits at the membrane, consistent with a block of CCV fission. The only uncoated vesicles observed were clustered at active zones. Thus, it is very difficult to explain the large number of uncoated vesicles in the GST—SYP-treated synapses, localized away from the active zone and directly behind the CCVs at the membrane, if one postulates that clathrin-dependent endocytosis is arrested at the coated pit stage.
Third, the interaction of dynamin with SYP requires Ca2+ concentrations found only at sites of vesicle exocytosis (C.D. and E.B.Z., unpublished observations). Although the injected GST–SYP fusion protein was distributed diffusely throughout the entire preterminal, it should only bind dynamin transiently at vesicle release sites immediately after Ca2+ influx. Thus, it is highly unlikely that GST–SYP could interfere with dynamin function (i.e., bind dynamin) during fission of CCVs, which occurs away from active zones (16) and which is not dependent on elevated Ca2+ concentration (20). Our data are therefore most consistent with an increased rate of formation of CCVs in GST–SYP treated synapses, and not with a block of fission of CCVs.
Other Evidence for Clathrin-Independent Endocytosis.
Several lines of evidence suggest, albeit indirectly, the existence of a clathrin-independent mechanism of SV endocytosis. First, the number of CCVs present in nerve terminals is not correlated with the extent of stimulation (21), suggesting that a clathrin-independent pathway operates in parallel. Our own data also indicate a lack of correlation between CCV number and stimulation intensity. The number of CCVs/μm2 in the GST-1 and GST-2 synapses was essentially the same, despite the fact that the GST-2 synapse was stimulated much more extensively just before fixation (Table 1).
Second, studies of the kinetics of SV retrieval indicate that rapid endocytic events, with a time constant of ≈2 s, can be detected (11, 14). Although such studies do not directly address the molecular mechanisms underlying endocytosis, they at least suggest the possibility of a clathrin-independent mechanism, because clathrin-dependent endocytosis at synapses appears to proceed with a time constant of ≈20 s (22).
Third, at frog motor nerve terminals, the kinase inhibitor staurosporine appears to promote, via an unknown mechanism, kiss-and-run recycling (23). In staurosporine-treated terminals, vesicle fusion is accompanied by neurotransmitter release but not by release of the styryl dye FM1-43 (23). This finding suggests a rapid opening and closing of the fusion pore, which does not allow escape of FM1-43. During a 10-Hz, 5-min stimulation in the presence of staurosporine, very few vesicles incorporated horseradish peroxidase present in the medium, suggesting that recycling was not via collapse of vesicles into the plasma membrane and internalization in CCVs (23).
The Role of Ca2+ in Endocytosis.
In our model of rapid endocytosis, the high Ca2+ concentration at SV release sites triggers vesicle retrieval by promoting recruitment of dynamin to the vesicle by SYP. The role of Ca2+ in endocytosis has been somewhat controversial. One report indicates that Ca2+ inhibits endocytosis in goldfish retinal bipolar neurons (24). However, such inhibition is not seen in other systems (22, 25). In fact, Ca2+ appears to promote rapid endocytosis of both SVs (14) and chromaffin granules (10, 12, 15).
Recently, it has been shown that high Ca2+ concentrations (≈30 μM) inhibit the GTPase activity of dynamin (26) and inhibit the ability of dynamin to vesiculate liposomes in vitro (27). Inhibition at such a high Ca2+ concentration is clearly relevant to the rapid endocytic mechanism proposed here and not to dynamin function in clathrin-dependent recycling. This inhibition of dynamin GTPase activity was proposed to “shunt” the site of vesicle fission away from the active zone, where Ca2+ levels are not so high (27). However, because high Ca2+ microdomains disappear very rapidly (≈1 ms) (28), it seems unlikely that the inhibition would shift the site of vesicle retrieval very far from the release site. Instead, given the high intrinsic GTPase activity of dynamin (29), it is possible that high Ca2+ serves to keep dynamin “active” (GTP bound) while it forms a ring in preparation for fission. As soon as the Ca2+ microdomain dissipates, endocytosis could proceed.
Regulation of Endocytosis at Synapses.
Given that synapses have two endocytic mechanisms at their disposal, an important issue is how the use of these pathways is regulated. Two studies suggest that the frequency of stimulation determines which endocytic mechanism is used (11, 14). Synaptic activity might induce modifications of proteins involved in the endocytic mechanisms, resulting in enhancement or inhibition of their function. In fact, several proteins involved in clathrin-dependent endocytosis (including dynamin) undergo activity-dependent dephosphorylation (30–33), which may activate this pathway (34–36). As mentioned above, the kinase inhibitor staurosporine appears to promote rapid endocytosis, suggesting that this pathway is regulated by the phosphorylation state of a protein(s) (23). A major challenge for the future will be to determine whether rapid endocytosis is modulated by synaptic activity and to define signaling pathways involved in such modulation.
The data presented in this study indicate that synapses have evolved a highly specialized mechanism of endocytosis, which relies on the high Ca2+ concentration at sites of SV fusion and on the SV-specific protein SYP. Our results suggest that rapid, clathrin-independent endocytosis, by efficiently maintaining the pool of releasable vesicles, plays a major role in ensuring reliable synaptic transmission.
Acknowledgments
We thank M. McNiven for anti-dynamin Ab, Gabriel M. Arisi for morphometrical analysis, M. Barry for helpful discussions, and Carlos Hernandez for computational assistance. This work was supported by Grant AG13620 from the National Institutes of Health-National Institute on Aging to E.B.Z., by Grant 13742 from the National Institutes of Health-National Institute of Neurological Disorders and Stroke to R.L., and by FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo, Brazil) Grants 97/3026-6 and 11097-0 to J.E.M.
Abbreviations
- SV
synaptic vesicle
- GST
glutathione S transferase
- SYP
synaptophysin
- CCV
clathrin-coated vesicle
References
- 1.Cremona O, De Camilli P. Curr Opin Neurobiol. 1997;7:323–330. doi: 10.1016/s0959-4388(97)80059-1. [DOI] [PubMed] [Google Scholar]
- 2.Maycox P R, Link E, Reetz A, Morris S A, Jahn R. J Cell Biol. 1992;118:1379–1388. doi: 10.1083/jcb.118.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Gaitan M, Jackle H. Cell. 1997;88:767–776. doi: 10.1016/s0092-8674(00)81923-6. [DOI] [PubMed] [Google Scholar]
- 4.Shupliakov O, Low P, Grabs D, Gad H, Chen H, David C, Takei K, De Camilli P, Brodin L. Science. 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- 5.Zhang B, Koh Y H, Beckstead R B, Budnik V, Ganetzky B, Bellen H J. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 6.Morgan J R, Zhao X, Womack M, Prasad K, Augustine G J, Lafer E M. J Neurosci. 1999;19:10201–10212. doi: 10.1523/JNEUROSCI.19-23-10201.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meldolesi J, Ceccarelli B. Philos Trans R Soc London B. 1981;296:55–65. doi: 10.1098/rstb.1981.0171. [DOI] [PubMed] [Google Scholar]
- 8.Valtorta F, Fesce R, Grohovaz F, Haimann C, Hurlbut W P, Iezzi N, Torri Tarelli F, Villa A, Ceccarelli B. Neuroscience. 1990;35:477–489. doi: 10.1016/0306-4522(90)90323-v. [DOI] [PubMed] [Google Scholar]
- 9.Palfrey H C, Artalejo C R. Neuroscience. 1998;83:969–989. doi: 10.1016/s0306-4522(97)00453-3. [DOI] [PubMed] [Google Scholar]
- 10.Neher E, Zucker R S. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-m. [DOI] [PubMed] [Google Scholar]
- 11.von Gersdorff H, Matthews G. Nature (London) 1994;367:735–739. doi: 10.1038/367735a0. [DOI] [PubMed] [Google Scholar]
- 12.Artalejo C R, Henley J R, McNiven M A, Palfrey H C. Proc Natl Acad Sci USA. 1995;92:8328–8332. doi: 10.1073/pnas.92.18.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M. Nature (London) 1997;389:509–512. doi: 10.1038/39081. [DOI] [PubMed] [Google Scholar]
- 14.Klingauf J, Kavalali E T, Tsien R W. Nature (London) 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- 15.Ales E, Tabares L, Poyato J M, Valero V, Lindau M, Alvarez de Toledo G. Nat Cell Biol. 1999;1:40–44. doi: 10.1038/9012. [DOI] [PubMed] [Google Scholar]
- 16.Heuser J E, Reese T S. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henley J R, Krueger E W, Oswald B J, McNiven M A. J Cell Biol. 1998;141:85–99. doi: 10.1083/jcb.141.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Llinas R, Steinberg I Z, Walton K. Biophys J. 1981;33:289–321. doi: 10.1016/S0006-3495(81)84898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llinas R, Sugimori M, Silver R B. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- 20.Gad H, Low P, Zotova E, Brodin L, Shupliakov O. Neuron. 1998;21:607–616. doi: 10.1016/s0896-6273(00)80570-x. [DOI] [PubMed] [Google Scholar]
- 21.Torri-Tarelli F, Haimann C, Ceccarelli B. J Neurocytol. 1987;16:205–214. doi: 10.1007/BF01795304. [DOI] [PubMed] [Google Scholar]
- 22.Ryan T A, Smith S J, Reuter H. Proc Natl Acad Sci USA. 1996;93:5567–5571. doi: 10.1073/pnas.93.11.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henkel A W, Betz W J. J Neurosci. 1995;15:8246–8258. doi: 10.1523/JNEUROSCI.15-12-08246.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Gersdorff H, Matthews G. Nature (London) 1994;370:652–655. doi: 10.1038/370652a0. [DOI] [PubMed] [Google Scholar]
- 25.Wu L G, Betz W J. Neuron. 1996;17:769–779. doi: 10.1016/s0896-6273(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 26.Liu J P, Zhang Q X, Baldwin G, Robinson P J. J Neurochem. 1996;66:2074–2081. doi: 10.1046/j.1471-4159.1996.66052074.x. [DOI] [PubMed] [Google Scholar]
- 27.Cousin M A, Robinson P J. J Neurosci. 2000;20:949–957. doi: 10.1523/JNEUROSCI.20-03-00949.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugimori M, Lang E J, Silver R B, Llinas R. Biol Bull. 1994;187:300–303. doi: 10.2307/1542286. [DOI] [PubMed] [Google Scholar]
- 29.Schmid S L, McNiven M A, De Camilli P. Curr Opin Cell Biol. 1998;10:504–512. doi: 10.1016/s0955-0674(98)80066-5. [DOI] [PubMed] [Google Scholar]
- 30.Liu J P, Sim A T, Robinson P J. Science. 1994;265:970–973. doi: 10.1126/science.8052858. [DOI] [PubMed] [Google Scholar]
- 31.Bauerfeind R, Takei K, De Camilli P. J Biol Chem. 1997;272:30984–30992. doi: 10.1074/jbc.272.49.30984. [DOI] [PubMed] [Google Scholar]
- 32.Wigge P, Kohler K, Vallis Y, Doyle C A, Owen D, Hunt S P, McMahon H T. Mol Biol Cell. 1997;8:2003–2015. doi: 10.1091/mbc.8.10.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, Slepnev V I, Di Fiore P P, De Camilli P. J Biol Chem. 1999;274:3257–3260. doi: 10.1074/jbc.274.6.3257. [DOI] [PubMed] [Google Scholar]
- 34.Marks B, McMahon H T. Curr Biol. 1998;8:740–749. doi: 10.1016/s0960-9822(98)70297-0. [DOI] [PubMed] [Google Scholar]
- 35.Slepnev V I, Ochoa G C, Butler M H, Grabs D, Camilli P D. Science. 1998;281:821–824. doi: 10.1126/science.281.5378.821. [DOI] [PubMed] [Google Scholar]
- 36.Lai M M, Hong J J, Ruggiero A M, Burnett P E, Slepnev V I, De Camilli P, Snyder S H. J Biol Chem. 1999;274:25963–25966. doi: 10.1074/jbc.274.37.25963. [DOI] [PubMed] [Google Scholar]