

Abstract
Demyelination contributes to the loss of function consequent to central nervous system (CNS) injury. Enhanced remyelination through transplantation of myelin-producing cells may offer a pragmatic approach to restoring meaningful neurological function. An unlimited source of cells suitable for such transplantation therapy can be derived from embryonic stem (ES) cells, which are both pluripotent and genetically flexible. In this paper we show that oligodendrocyte cultures can be reliably produced from retinoic acid-induced ES cells and that these oligodendrocytes can myelinate axons in vitro. Methods were further developed for generating highly enriched cultures of oligodendrocytes through an additional culturing step, producing an intermediate “oligosphere” stage. To test whether ES cells can survive, migrate, and differentiate into mature myelin-producing cells in areas of demyelination in the adult CNS, ES cells were transplanted into the dorsal columns of adult rat spinal cord 3 days after chemical demyelination. In the demyelination site, large numbers of ES cells survived and differentiated primarily into mature oligodendrocytes that were capable of myelinating axons. Furthermore, when oligosphere cells were transplanted into the spinal cords of myelin-deficient shiverer (shi/shi) mutant mice, the ES cell-derived oligodendrocytes migrated into the host tissue, produced myelin and myelinated host axons. These studies demonstrate the ability of ES cell-derived oligodendrocytes to myelinate axons in culture and to replace lost myelin in the injured adult CNS. Transplantation of ES cells may be a practical approach to treatment of primary and secondary demyelinating diseases in the adult CNS.
Recovery in central nervous system (CNS) disorders is hindered by the limited ability of the vertebrate CNS to regenerate lost cells, replace damaged myelin, and re-establish functional neural connections. In many CNS disorders, including multiple sclerosis, stroke, spinal cord injury, and other trauma, demyelination of intact axons (1–4) is an important factor contributing to loss of function. Previous studies suggest that substantial recovery of function might be achieved through remyelination of otherwise intact axons (5). As a therapeutic modality, functional recovery through remyelination may prove to be a pragmatic approach to regeneration.
Ethical considerations and a lack of a reliable source for undifferentiated pluripotent cells have limited the application of neural transplantation studies in humans. Embryonic stem cells (ES cells) provide a partial solution to these problems because they are genetically normal, pluripotent, capable of indefinite replication (6), and have been derived from several vertebrate species including mice (7, 8) and humans (9, 10, 50). ES cells are also the most flexible stem cell for genetic engineering. Double allele knockouts in single ES cells are possible and such genetic capacities are providing powerful scientific tools (11, 12). Studies examining the regulation of ES cell differentiation into CNS cells are in their infancy. Although ES cells have been shown in culture to differentiate into multiple CNS cell types (13–22), methods for reliably producing oligodendrocytes from retinoic acid-induced ES cells have not been developed. Recently, ES cell-derived oligodendrocytes have been shown to myelinate in the immature CNS (22).
The purpose of the present studies was threefold: (i) to develop methods for producing enriched cultures of ES cell-derived oligodendrocytes, (ii) to determine whether these cells could myelinate axons in vitro, and (iii) to determine whether ES cells would survive transplantation into adult spinal cord (both injured and uninjured), migrate, differentiate into oligodendrocytes and myelinate axons. As an injury model, localized chemical demyelination was induced in the dorsal column white matter of rats while sparing passing axons (23–26). Myelin-deficient shiverer (shi/shi) mutant mice, lacking the gene to produce myelin basic protein (MBP) (27–31), were used as a noninjury model where transplanted oligodendrocytes and newly produced myelin could be identified by immunoreactivity for MBP.
Materials and Methods
Animals and Care.
Homozygous adult (shi/shi) shiverer mice (8–12 weeks old, 17–21 g; 30 mice total) and female Long-Evans rats (8–12 weeks old, 240–270 g; 20 rats total) were obtained from The Jackson Laboratory and Simonsen Laboratories (Gilroy, CA), respectively. Interventions were in accordance with the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, EHEW Pub. No. 78-23, Revised 1978) and the Guidelines and Policies for Rodent Survival Surgery (Animal Studies Committee of Washington University School of Medicine). Anesthesia was induced by ketamine/medetomidine (75:0.5 mg/kg for rats, 75:1 mg/kg for mice; i.p.) and reversed with atipamezole (1.0 mg/kg, s.c.). All animals received (i) cyclosporine (10 mg/kg, s.c.) 24 h before transplantation and daily thereafter, (ii) antibiotics (enrofloxacin, 2.5 mg/kg, s.c.) before surgery and daily for 3–5 days, and (iii) saline (2–5 ml i.p.) and nutritional supplements (Nutri-Cal, EVSCO Pharmaceuticals, Buena, NJ) for 3–5 days after surgery.
ES Cell Culture.
ROSA26 (32) or D3 mouse ES cells were used in all experiments, and were differentiated by using the 4−/4+ retinoic acid protocol (15, 33). ES cells were grown in embryonic stem cell induction medium (ESIM, containing DMEM, newborn calf serum, FBS, and nucleosides) for 4 days and ESIM plus retinoic acid (500 nM) for 4 additional days (15, 32). Induced ES cells formed floating clusters of cells, termed embryoid bodies (EBs). After 8 days in vitro (DIV), the 4−/4+ stage EBs were partially dissociated (5 min at 37°C, 0.25% trypsin with EDTA) and resuspended in ESIM for transplantation or were further triturated to single cell suspension for plating in vitro in modified SATO medium (DMEM with BSA, pyruvate, progesterone, putrescine, thyroxine, triiodothryonine, insulin, transferrin, sodium selenite, amino acids, neurotrophin 3, ciliary neurotrophic factor, and Hepes) (34, 35) with 5% equine serum and 5% FCS.
“Oligosphere” Culture.
To produce oligospheres, dissociated 4−/4+ stage EBs were grown in flasks containing modified SATO medium. After 4 DIV, the flasks were gently shaken to suspend loosely adhering cells (primarily oligodendrocytes), while astrocytes remained adhering to the flask. Suspended cells were transferred to new flasks in SATO medium and grown for an additional 2 days. After 6 DIV, oligospheres were partially dissociated (as above) and resuspended in SATO medium for transplantation or completely dissociated for plating in oligosphere-conditioned medium (derived from the shaking step) for in vitro studies.
Demyelination.
Demyelination was chemically induced in rats by using characterized methods (23–26). After a T10 laminectomy, ethidium bromide (1 μl of 0.1% ethidium bromide in 0.9% saline) or lysophosphatidyl choline (LPC) [lysolecithin (Sigma); 2 μl of 1.0% LPC in 0.9% saline] was injected at 0.5-mm depth in the dorsal column over a 10-min period by using a stereotaxic microinjector (Stoelting) and a 30-μm tip glass pipette attached to a 5-μl Hamilton syringe. Three days later, the demyelinated areas were transplanted with partially dissociated EBs or control medium. Animals were killed 1 and 4 weeks after transplantation (n = 5 each with equal numbers of rats that received vehicle medium sham transplantation).
Preparation of Cells for Transplantation.
Prelabeled (see cell tracking methods below) 4−/4+ stage EBs or oligospheres were prepared as described above (33) to produce suspensions of small clusters of cells. Cell density was adjusted to 50,000 viable cells per μl by using a hemocytometer.
Transplantation.
Demyelination injury rats received transplants of 125,000 cells from partially dissociated 4−/4+ EBs or medium vehicle. A 50- to 100-μm tip diameter glass pipette was stereotaxically advanced 0.5 mm into the dorsal column white matter. By using a stereotaxic microinjector, 2.5 μl of the ES cell suspension or control vehicle medium was injected at a rate of 0.25 μl/min. The needle was left in place for 5 min and then withdrawn, and the laminectomy site was covered with artificial dura. In the second model, shiverer mice were transplanted with 100,000 oligosphere cells or vehicle medium (n > 6 each) at the T8 and T10 level (1 μl at two sites 0.35 μm below the dura). Histologic examination was completed at 2 weeks (n = 20) and 4 weeks (n = 10) after transplantation.
Cell Tracking and Immunohistochemistry.
Several methods were used to accurately track ES cells after transplantation: (i) lacZ transgene [ROSA26 line (32)], (ii) BrdUrd DNA prelabeling in vitro, (iii) fluorescent Cell Tracker Orange (Molecular Probes) prelabeling in vitro, and (iv) mouse-specific Abs. ES cells were pulse labeled with BrdUrd (10 μM; Boehringer Mannheim) for 24 h on the third to fourth day of the 4−/4+ protocol (33, 36). Partially trypsin-dissociated 4−/4+ EBs were incubated with stable fluorescent marker Cell Tracker Orange for 20 min, washed, incubated for another 20 min, and then washed before transplantation. Cell Tracker Orange diffuses into cells and is transformed into a fixable, membrane-impermeant form in the cytoplasm.
Mouse-specific Abs were used to detect the mouse ES cells in the rat demyelination experiments: anti-M2 [labels mouse glia > neurons (37)], and anti-EMA [labels mouse neurons > glia (38)]. Abs used to identify the oligodendrocyte lineage included [see McDonald et al. (39) for details of Ab classification]: anti-NG2 for oligodendrocyte progenitors (Chemicon; labels NG2 chondroitin sulfate proteoglycan on the cell surface), anti-O4 for immature oligodendrocytes (Boehringer Mannheim; labels O4 sulfatides on the surface of developing oligodendrocytes), anti-O1 (Boehringer Mannheim; marks O1 sulfatide on mature oligodendrocytes), and anti-adenamatous polyposis coli (APC) (Calbiochem; labels cytoplasmic product of the adenomatous polyposis coli tumor suppressor gene product, found in cell bodies of mature oligodendrocytes) were used to identify mature oligodendrocytes (40). Anti-MBP (Boehringer Mannheim; labels an integral membrane protein in myelin) was used to label terminally mature, myelin-producing oligodendrocytes. Homozygous shiverer (shi/shi) mice are devoid of MBP because they lack the functional gene. Thus, the presence of MBP+ myelin in these animals following transplantation provides a useful marker for confident identification of transplanted oligodendrocytes (27, 28). Anti-neuron-specific nuclear antigen (Chemicon; identifies a neuron-specific nuclear protein in postmitotic cells), anti-neuron-specific enolase (Chemicon; reacts with neuron-specific enolase), and anti-β-tubulin III (Sigma; reacts with neuron specific tubulin) were used to identify neurons.
Electron Microscopy (EM).
EBs and cultures were processed by using standard methods (41). Samples were viewed with a Hitachi S-450 scanning EM operated at 20 KV accelerating voltage and JEOL 100CX transmission EM.
Results and Discussion
ES cells are theoretically capable of differentiating into any cell type, and previous work has shown that they can be neural- induced in vitro by using a number of protocols (16, 20–22). Retinoic acid exposure conditions have been developed to restrict differentiation down a neural lineage, and such treatment has been shown to produce cultures containing primarily CNS cells (15). ES cells cultured by using a 4−/4+ retinoic acid protocol (15) have been shown to develop into oligodendrocytes after transplantation into the injured spinal cord (33). This protocol was used as the basis for methods developed to reliably produce both mixed cultures of oligodendrocytes, neurons and astrocytes as well as enriched oligosphere-derived cultures containing predominantly oligodendrocytes.
ES cells grown under the 4−/4+ protocol form floating clusters of cells, termed EBs. Ultrastructural scanning EM examination of EBs revealed that the cells on the surface were covered with extensive microprocesses analogous to the extraembryonic visceral endoderm layer (Fig. 1 A and B). Immunohistochemical studies of EBs showed limited expression of markers of differentiated neural cells (Fig. 1 C–F, Table 1), and the majority were nestin-positive, a marker of neural precursors. Ultrastructural EM evidence suggested that a substantial number of EB cells exhibit features of apoptotic death (Fig. 1B). Consistent with these data, chromatin condensation (visible in Hoechst stained nuclei) and immunoreactivity to the activated form of caspase 3 were present in 6 ± 4% (n = 6 EBs) of cells within 4−/4+ stage EBs [Table 1; anti-CM-1, IPO13, Idun Pharmaceuticals, La Jolla, CA (42)].
Figure 1.
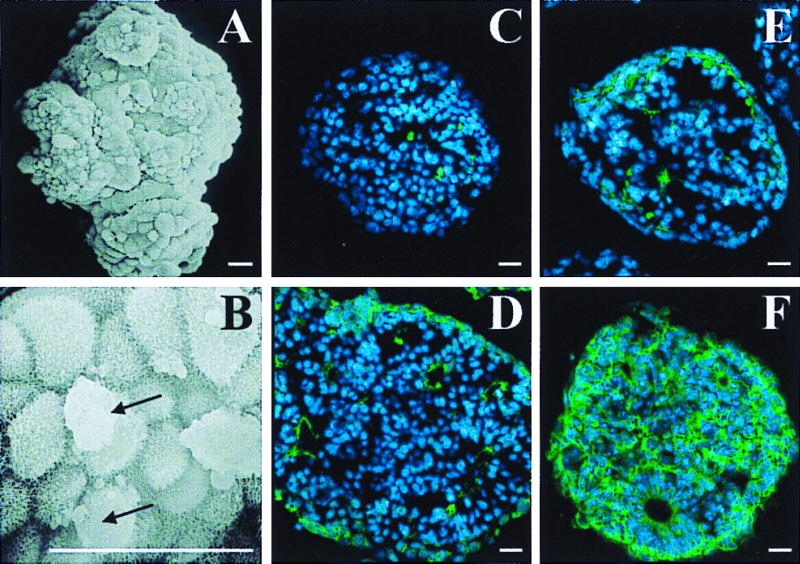
(A) Scanning EM shows 4−/4+ stage EBs, characterized as floating clusters of undifferentiated cells. (B) Multiple small processes cover the surfaces of the outer layer cells. Membrane blebbing, consistent with apoptotic cell death, occurs in a fraction of the EB cells (arrows in B). Immunoreactivity (green) indicates that the vast majority of EB cells possess immunomarkers for neural precursors (anti-nestin; F), whereas few express markers consistent with differentiated neurons (anti-NeuN; C), astrocytes (anti-GFAP; D), and oligodendrocytes (anti-O1; E) (see Table 1). Nuclei are counterstained with Hoechst 33342 (blue). Scale bars = 20 μm.
Table 1.
Expression of markers of differentiated neural cells and activated caspase 3 in EBs and oligospheres
| Cell type | CM1, % | NeuN, % | Nestin, % | GFAP, % | O4, % | O1, % |
|---|---|---|---|---|---|---|
| EB | 6 ± 4 | 6 ± 2 | 89 ± 2 | 13 ± 1 | 38 ± 3 | 35 ± 5 |
| Oligosphere | 10 ± 5 | 27 ± 4 | 9 ± 4 | 6 ± 2 | 51 ± 7 | 54 ± 9 |
Data are given as cell-type percent as indicated by immunoreactivity to phenotypic markers in EBs and oligospheres (mean ± SEM, n = 5 for each).
Plating of dissociated 4−/4+ stage EBs in neurobasal media (Gibco/BRL, catalogue no. 21103-049) produced mixed cultures of neurons, astrocytes, and oligodendrocytes (Fig. 2). Like primary cultures, ES cell-derived type-I astrocytes formed a confluent layer, the other cells grew on top, and neurons grew in small clusters with large bundles of axons radiating outward. Mixed cultures grew best in SATO-defined medium supplemented with serum, and could be maintained for at least 1 month. Inhibition of cell division (10-5 M cytosine arabinoside), as used in previous studies of cultured ES cell-derived neurons (15), greatly limited oligodendrocyte viability (27 ± 5 no inhibition vs 0.5 ± 0.2 with inhibition, number of O1-positive cells per ×200 field, n = 4, P < 0.05, independent Student's t test).
Figure 2.

Mixed cultures of neurons, astrocytes, and oligodendrocytes can be produced by plating dissociated 4−/4+ stage EBs in SATO-defined medium (34, 35) supplemented with 5% FCS and 5% equine serum. Examples of neurons (anti-β-tubulin III; A), type-I astrocytes (anti-GFAP; B), type-II astrocytes (anti-GFAP; C), and mature oligodendrocytes (anti-O1; D) are seen in mixed cultures (DIV 9). Scale bars = 20 μm.
By using immunohistochemical markers as well as scanning and transmission EM, it was observed that ES cell-derived oligodendrocytes could rapidly and consistently myelinate axons in culture (Fig. 3). Individual oligodendrocytes that simultaneously wrap multiple axons and multiple segments of single axons could be easily identified by using fluorescent Abs directed against components of myelin (O1, MBP). Transmission EM verified that the wrapping observed in the immunocytochemical studies indeed represented myelination. By 9 DIV, evidence of immature axonal myelin profiles with two to five loosely wrapped layers and prominent inner/outer tongue processes were common (Fig. 3D). Surprisingly, more mature, highly wrapped (10–15 layers), compact myelin profiles could already be seen by 9 DIV. Previous studies, using primary-derived cultures, suggest that early forms of myelination typically do not appear until 21 DIV, and that development of compact mature myelin profiles typically takes 4–6 weeks. Evidence of early axonal degeneration was also evident in many myelin profiles. This is likely a reflection of the nondepolarizing conditions used in our culture system, which limit neuronal viability past 14 DIV. In the absence of axons, oligodendrocytes formed sheets of myelin, similar to cultures of primary oligodendrocytes (39).
Figure 3.

(A) ES cell-derived oligodendrocytes are capable of myelinating multiple axons in culture. O1 immunoreactivity (green) is superimposed on a phase-contrast image in a mixed ES cell-derived neuronal/glial culture (9 DIV). White arrows indicate axons and red arrows indicate O1 immunoreactive wrapped axon segments. (B) Scanning EM shows oligodendrocyte (O) and passing axons (black arrows). Higher magnification (box from B) demonstrates early axon wrapping (red arrows) by an oligodendrocyte process (C), which is similar to early phases of myelination described in studies using video microscopy (49). Transmission EM shows myelin profiles typical of early myelination in 9 DIV culture (D and E).
To produce cultures containing primarily oligodendrocytes, an additional culturing step was developed. Dissociated 4−/4+ stage EBs were transferred into flasks containing 5 ml of oligosphere medium, which promotes survival and proliferation of oligodendrocyte progenitors. After 4 DIV, nonadhering cells, consisting primarily of oligodendrocyte precursors, were passed into fresh oligosphere medium at a 1:1 ratio. The few astrocytes generated in this culture adhered to the flask and were not passed. Free-floating spherical cell clusters were formed over 6 days. We termed these clusters oligospheres because later plating produced cultures containing primarily oligodendrocytes. Immunohistochemical studies suggest that oligospheres contain substantial numbers of immature and mature oligodendrocytes, but few astrocytes (Fig. 4, Table 1). In contrast to the high percentage of nestin-positive cells in EBs, only minorities of oligosphere cells were nestin positive. Oligospheres also appeared to contain substantially more cells that immunolabeled for markers of neurons (anti-neuron-specific enolase, NeuN, and neurofilament), although these cells typically did not have processes or ultrastructure suggestive of differentiated neurons. Plating dissociated oligospheres in oligosphere-conditioned medium selected for oligodendrocytes and yielded cultures comprised of 92 ± 7% oligodendrocytes (n = 4). Despite the neuron marker expression in oligosphere cells, very few neurons were observed in any derived cultures.
Figure 4.

Oligospheres are floating cell clusters produced from EBs through an additional culture step and yield highly enriched cultures of oligodendrocytes when plated in oligosphere-conditioned medium. Oligospheres are immunoreactive for early markers of neurons (anti-NeuN; A), oligodendrocytes (anti-O4; C, anti-O1; D), neural precursors (anti-nestin; E) but few astrocytes (anti-GFAP; B) (see Table 1). Oligosphere-derived cultures are enriched for O4 (positive) oligodendrocytes (5 DIV; F). Hoechst 33342 (blue). Scale bars = 50 μm.
To evaluate differentiation of ES cells in vivo, in the “injured” adult CNS partially dissociated 4−/4+ stage EBs were transplanted into the dorsal column of rat spinal cord 3 days after chemical demyelination. Successful engraftment in the demyelinated region was evident in 9 of 10 rats when examined 1 week after transplantation. This was indicated by immunostaining with anti-mouse-specific Abs (Fig. 5 A and D), and by an increased cell density demonstrated by Hoechst 33342 labeling in animals receiving transplants. In rats that received a sham vehicle medium transplant, axons of passage were largely spared, as shown previously (23–26), and a paucity of Hoechst 33342 nuclear labeling was present at the site of demyelination. At the lesion site in rats receiving transplants, ES cells differentiated primarily into oligodendrocytes (anti-APC CC-1, anti-MBP; Fig. 5B), but not astrocytes [anti-glial fibrillary acidic protein (GFAP)] or neurons (anti-NeuN; data for neurons not shown). A normal low density of host oligodendrocytes (determined by anti-APC, MBP, CNPase, O1) was evident outside the area of demyelination in rats that received transplantation of ES cells or control medium. The host immunoreactive oligodendrocytes are not evident in the low magnification views of Fig. 5 because of the need to prevent over-saturation of high immunoreactivity at the transplantation sites. Enhanced anti-GFAP reactivity was consistently observed at the lesion borders in rats that received both ES cell and vehicle medium transplantation, indicating the association with host reactive astrocytes (Fig. 5E). Lack of double labeling for mouse-specific Abs further supported the host origin of the reactive astrocytes (data not shown). Although immature cells can exhibit GFAP immunolabeling, the GFAP labeling here represented primarily astrocytes based on additional morphological characteristics and lack of labeling for other phenotype markers. Histologic evidence of acute graft rejection was present in 1 of 10 rats that received ES cell transplantation.
Figure 5.

Transplanted 4−/4+ stage EB cells are capable of differentiating into myelin forming oligodendrocytes in demyelinated adult rat spinal cord. Partial coronal cross-sections (dorsal surface downward) from rats that received sham-transplantation (A–C) or ES cell transplantation (D–F and G–I, two separate animals) demonstrate immunoreactivity for anti-M2 (A, D, and G), anti-APC (B and E) and anti-GFAP (H). Anti-M2 is a mouse-specific Ab for recognition of transplanted mouse ES cells (37), whereas anti-APC labels oligodendrocytes (40). Increased nuclear density at the site of transplantation is indicated by corresponding Hoechst 33342 staining (arrows; F and I) compared with control (C). ES cell-derived oligodendrocytes primarily occupy the zone of demyelination, whereas there is a corresponding paucity of GFAP immunoreactivity in this area (arrows; D, E, G, and H). High magnification shows a native MBP immunoreactive cell (green) from the ventral column distant from the site of transplantation (white arrow, J) and a probable transplanted MBP immunoreactive cell from the center of the area of transplant (white arrow, K). Transmission EM shows early M2-positive myelin (black arrows indicate DAB precipitate particles associated with M2 immunoreactivity) produced by a transplanted ES cell-derived oligodendrocytes (L), and early M2-negative native myelin (M) in ultrathin sections without EM staining. Typical EM myelin contrast staining was not done to be able to visualize the diaminobenzidine (DAB) precipitate associated with anti-M2 immunoreactivity. Scale bars: A–I = 1 mm; J and K = 20 μm; L and M = 0.5 μm.
A second study was performed to assess the potential for ES cell transplants to differentiate into oligodendrocytes and myelinate in the dysmyelinated adult CNS. Dissociated oligospheres were transplanted into the thoracic spinal cord of adult shiverer (shi/shi) mice, which lack a functional gene for MBP, an essential component of myelin required for compact mature myelin formation. Transplanted cells were tracked by prelabeling the oligospheres with the fluorescent marker Cell Tracker Orange and BrdUrd, or by detecting MBP expressed by transplanted cells. MBP immunoreactivity is always absent in the host shiverer mutant CNS. Two weeks after transplantation, ES cell-derived cell tracker orange (positive) (Fig. 6 A and B) and MBP (positive) (Fig. 6 C and D) oligodendrocytes were found in white matter. ES cell-derived oligodendrocytes conformed to the organization that oligodendrocytes normally respect in white matter: they would align with host intrafascicular oligodendrocytes and myelinate axons (Fig. 6 C–G). Because homozygous shiverer mice do not exhibit MBP immunoreactivity (Fig. 6E) (27, 28), all MBP immunoreactivity could be attributed to the transplanted ES cell-derived oligodendrocytes. In mice that received oligosphere transplantation, but not in mice that received sham transplantation, MBP immunoreactivity was evident in a radial longitudinal gradient paralleling the white matter tract. Gross MBP immunoreactivity was consistently observed for 0.5 mm surrounding the sites of transplantation, and individual MBP positive cells could be found at sites more distant (2–3 mm) when examined in rats that received transplantation 1 month earlier. Occasionally small central areas of confluent MBP immunoreactivity also colabeled with Abs against extracellular matrix molecules (laminin, fibronectin; data not shown) and were not considered myelin products.
Figure 6.

ES oligosphere-derived cells can migrate and myelinate axons when transplanted into dysmyelinated spinal cords of adult shiverer mice, which lack the gene to produce MBP (27–31). Transplanted cells were identified by Cell Tracker Orange epifluorescence (red) or immunoreactivity for MBP (green). Hoechst 33342 (blue). Cell Tracker Orange-labeled cells were found to align with native intrafascicular oligodendrocytes in white matter (A and B). An ES cell-derived (MBP immunoreactive) oligodendrocyte (asterisk) with longitudinally oriented processes (white arrows) is shown in C and D. Red arrows mark probable myelination around an adjacent axon (C). Little MBP immunoreactivity is present in white matter of a longitudinal spinal cord section from a mouse that received sham transplantation (E). A gradient of MBP immunoreactivity is centered on the site of ES cell transplantation (F). (G) High magnification shows intrafascicular oligodendrocyte nuclei (blue) and MBP immunoreactivity (green) characteristic of axonal myelination (white arrows; refs. 43 and 44) in white matter from a mouse that received ES cell transplantation. The spatial distribution of MBP immunoreactivity, 1 month after ES cell transplantation, is shown at low magnification (H) with corresponding Hoechst 33342 counterstaining (I). White arrows indicate the center of the transplant. Transmission EM shows four loose wraps of myelin, which represents the maximal number of layers typically seen around axons in control animals (red arrow, J), and 9 or greater compact wraps around axons from the area of the transplant (red arrow, K). shiverer mutant mice lack a functional MBP gene that is required to form mature compact myelin; therefore, the presence of mature compact myelin is a gold standard for transplant oligodendrocyte associated myelin. Scale bars: A–I = 10 μm; J and K = 0.3 μm.
The longitudinal parallel arrays of MBP immunoreactivity, outlining spaces occupied by axons in white matter, are consistent with myelination and are similar to those observed in previous studies of myelination (43, 44) (Fig. 6 E–G). Immunocytochemical evidence of myelination was confirmed by transmission EM. Ultrastructurally, shiverer mice lack normal myelination; most axons are not myelinated or contain only one to three loose wraps of myelin (Fig. 6J). MBP is required for compact myelin formation. Therefore, the presence of multilayered, compact myelin has been used as the gold standard for demonstrating myelination from transplanted oligodendrocytes. In shiverer mice that received transplants of oligospheres 1 month earlier, such evidence was present (Fig. 6K).
We did not observe any ES cell-derived tumor formation in this study or in our previous studies (33). The pluripotential of ES cells poses a risk for forming normal tissues but in the wrong place (e.g., teratomas). Therefore, it is not surprising that ES cells have produced teratomas under specialized conditions. Because retinoic acid is a strong neural induction signal, we have used it to restrict the cells to a neural lineage to help avoid abnormal tissue formation in the CNS after transplantation. Compared with nonretinoic acid differentiation protocols, the 4−/4+ protocol yields a high percentage of neural cells. However, the culture system is very time sensitive and exposure intervals must be accurate. Transplantation studies of longer duration will be required to adequately assess for the risk of tumor formation. A heartening feature of ES cells is that they can be assessed for genetic normalcy by generating a chimeric animal after implantation into blastocysts.
The present studies demonstrate that ES cells can be used to reliably generate mixed and enriched cultures of oligodendrocytes and that these oligodendrocytes are capable of producing myelin and capable of myelinating axons in vitro. In addition, transplanted retinoic acid induced ES cells can: (i) preferentially differentiate into oligodendrocytes in areas of demyelination, and (ii) myelinate host axons in the dysmyelinated spinal cord. Our study demonstrates the ability of ES cell-derived oligodendrocytes to myelinate in vitro and to show that ES cells survive and myelinate axons in the mature and injured CNS after transplantation. Our findings in the mature CNS are particularly relevant because the most common disorders that are targets for therapeutic strategies of remyelination occur predominantly in adults. In particular, we demonstrate that injured, demyelinated areas of the adult CNS may preferentially stimulate oligodendrocyte differentiation/survival. Since the completion of these studies, another group has reported ES cell-derived oligodendrocyte myelination after transplantation in the immature CNS of myelin-deficient rats (22). A more lengthy induction protocol, not employing retinoic acid, was used that yielded mixed cultures of oligodendrocytes and astrocytes.
Remyelination is a potential mechanism underlying the rapid recovery of locomotor function we observed when dissociated 4−/4+ stage ES cells were transplanted 9 days after moderate spinal cord contusion injury in rats (33). Significant recovery of locomotion was first evident 2 weeks after transplantation. Oligodendrocytes represented the largest identifiable population of differentiated ES cell-derived cells in that study. Transplantation of oligosphere cells in the contusion injury model may provide a useful test of this hypothesis.
The present study suggests that local conditions in the lesioned CNS can select for differentiation or survival of particular types of ES cell-derived neural cells. When ES cells are transplanted into a contusion-injured spinal cord, they differentiate into substantial numbers of oligodendrocytes and astrocytes and <10% neurons (33). In contrast, it is shown here that primary demyelination lesions, sparing passing axons, preferentially support ES cell-derived oligodendrocytes. This observation is compatible with previous demonstrations that CNS isolated progenitors differentiate into different neuronal phenotypes based on their transplantation site in the CNS (45–47).
One important advantage of the use of ES cells for transplantation over primary cells, or other neural progenitors, is their flexibility for genetic engineering. To date, over a dozen double allele knock-ins/knock-outs in single ES cells have been completed (e.g., refs. 11 and 12). This finding provides a powerful scientific and therapeutic tool. ES cells can be genetically modified to produce pro-regenerative factors, such as neurotrophin 3, which has been shown to promote axonal regeneration and myelination (43, 48). These tools will be important for studying the mechanisms that regulate oligodendrocyte differentiation and myelination as well as for developing accurate and rapid methods for quantifying myelin.
Remyelination is a promising and pragmatic approach for repairing the damaged CNS, and strategies are coming within our reach that may offer meaningful recovery of function, such as improved bowel and bladder control, or limb movement. The availability of human ES cells (9, 10, 50) and the possibility of producing autologous ES cells by nuclear transfer provide exciting possibilities for the treatment of many human diseases including those involving demyelination such as multiple sclerosis, Alzheimer's disease, leukodystrophies, and CNS trauma.
Acknowledgments
We thank D. I. Gottlieb for critical discussions, Dr. D. C. Beebe, Dr. C. Xang, and R. Purcell for technical assistance, and A. Lu and J. Y. Norris for expert care of the spinal cord injured animals. This work was supported by grants from the National Institute of Neurological Disorders and Stroke, National Institutes of Health (NS01931 and NS37927), as well as the Keck Foundation and National Football League Charities.
Abbreviations
- CNS
central nervous system
- EB
embryoid body
- ES cell
embryonic stem cell
- ESIM
embryonic stem cell induction medium
- MBP
myelin basic protein
- DIV
days in vitro
- EM
electron microscopy
- GFAP
glial fibrillary acidic protein
- APC
antigen-presenting cell
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gledhill R F, Harrison B M, McDonald W I. Nature (London) 1973;244:443–444. doi: 10.1038/244443a0. [DOI] [PubMed] [Google Scholar]
- 2.Griffiths I R, McCulloch M C. J Neurol Sci. 1983;58:335–349. doi: 10.1016/0022-510x(83)90093-x. [DOI] [PubMed] [Google Scholar]
- 3.Blight A R. Cent Nerv Syst Trauma. 1985;2:299–315. doi: 10.1089/cns.1985.2.299. [DOI] [PubMed] [Google Scholar]
- 4.Bunge R P, Puckett W, Becerra J L, Marcillo A, Quencer R M. Adv Neurol. 1993;59:75–89. [PubMed] [Google Scholar]
- 5.Waxman S G, Utzschneider D A, Kocsis J D. Prog Brain Res. 1994;100:233–243. doi: 10.1016/s0079-6123(08)60790-6. [DOI] [PubMed] [Google Scholar]
- 6.Suda Y, Suzuki M, Ikawa Y, Aizawa S. J Cell Physiol. 1987;133:197–201. doi: 10.1002/jcp.1041330127. [DOI] [PubMed] [Google Scholar]
- 7.Evans M J, Kaufman M H. Nature (London) 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 8.Martin G R. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomson J A, Itskovitz-Eldor J, Shapiro S S, Waknitz M A, Swiergiel J J, Marshall V S, Jones J M. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 10.Shamblott M J, Axelman J, Wang S, Bugg E M, Littlefield J W, Donovan P J, Blumenthal P D, Huggins G R, Gearhart J D. Proc Natl Acad Sci USA. 1998;95:13726–13731. doi: 10.1073/pnas.95.23.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilder P J, Kelly D, Brigman K, Peterson C L, Nowling T, Gao Q-S, McComb R D, Capecchi M R, Rissino A. Dev Biol. 1997;192:614–629. doi: 10.1006/dbio.1997.8777. [DOI] [PubMed] [Google Scholar]
- 12.Hakem R, Hakem A, Duncan G S, Henderson J T, Woo M, Soengas M S, Elia A, de la Pompa J L, Kagi D, Khoo W, et al. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 13.Wobus A M, Holzhausen H, Jakel P, Schoneich J. Exp Cell Res. 1984;152:212–219. doi: 10.1016/0014-4827(84)90246-5. [DOI] [PubMed] [Google Scholar]
- 14.Wobus A M, Grosse R, Schoneich J. Biomed Biochim Acta. 1988;47:965–973. [PubMed] [Google Scholar]
- 15.Bain G, Kitchens D, Yao M, Gottlieb D I. Dev Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- 16.Fraichard A, Chassande O, Bilbaut G, Dehay C, Savatier P, Samarut T. J Cell Sci. 1995;108:3181–3188. doi: 10.1242/jcs.108.10.3181. [DOI] [PubMed] [Google Scholar]
- 17.Strubing C, Ahnert-Hilger G, Shan J, Wiedenmann B, Hescheler J, Wobus A M. Mech Dev. 1995;53:275–287. doi: 10.1016/0925-4773(95)00446-8. [DOI] [PubMed] [Google Scholar]
- 18.Okabe S, Forsberg-Nilsson K, Spiro A C, Segal M, McKay R D. Mech Dev. 1996;59:89–102. doi: 10.1016/0925-4773(96)00572-2. [DOI] [PubMed] [Google Scholar]
- 19.Finley M F, Kulkarni N, Huettner J E. J Neurosci. 1996;16:1056–1065. doi: 10.1523/JNEUROSCI.16-03-01056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinsmore J, Ratliff J, Deacon T, Pakzaban P, Jacoby D, Galpern W, Isacson O. Cell Transplant. 1996;5:131–143. doi: 10.1177/096368979600500205. [DOI] [PubMed] [Google Scholar]
- 21.Brustle O, Spiro A C, Karram K, Choudhary K, Okabe S, McKay R D. Proc Natl Acad Sci USA. 1997;94:14809–14814. doi: 10.1073/pnas.94.26.14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brustle O, Jones K N, Learish R D, Karram K, Choudhary K, Wiestler O D, Duncan I D, McKay D G. Science. 1999;285:754–756. doi: 10.1126/science.285.5428.754. [DOI] [PubMed] [Google Scholar]
- 23.Hall S M. J Cell Sci. 1972;10:535–546. doi: 10.1242/jcs.10.2.535. [DOI] [PubMed] [Google Scholar]
- 24.Blakemore W F. Neuropathol Appl Neurobiol. 1976;2:21–39. [Google Scholar]
- 25.Waxman S G, Kocsis J D, Nitta K C. J Neurol Sci. 1979;44:45–53. doi: 10.1016/0022-510x(79)90221-1. [DOI] [PubMed] [Google Scholar]
- 26.Blakemore W F, Crang A J. J Neurol Sci. 1985;70:207–223. doi: 10.1016/0022-510x(85)90088-7. [DOI] [PubMed] [Google Scholar]
- 27.Dupouey R, Jacque C, Bourre J M, Cesselin F, Privat A, Baumann N. Neurosci Lett. 1979;12:113–118. doi: 10.1016/0304-3940(79)91490-3. [DOI] [PubMed] [Google Scholar]
- 28.Gumpel M, Baumann N, Raoul M, Jacque C. Neurosci Lett. 1983;37:307–311. doi: 10.1016/0304-3940(83)90449-4. [DOI] [PubMed] [Google Scholar]
- 29.Sidman R L, Conover C S, Carson J H. Cytogenet Cell Genet. 1985;39:241–245. doi: 10.1159/000132151. [DOI] [PubMed] [Google Scholar]
- 30.Molineaux S M, Engh H, de Ferra F, Hudson L, Lazzarini R A. Proc Natl Acad Sci USA. 1986;83:7542–7546. doi: 10.1073/pnas.83.19.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gout O, Gansmuller A, Baumann N, Gumpel M. Neurosci Lett. 1988;87:195–199. doi: 10.1016/0304-3940(88)90169-3. [DOI] [PubMed] [Google Scholar]
- 32.Friedrich G, Soriano P. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 33.McDonald J W, Liu X Z, Qu Y, Liu S, Mickey S K, Turetsky D, Gottlieb D I, Choi D W. Nat Med. 1999;5:1410–1412. doi: 10.1038/70986. [DOI] [PubMed] [Google Scholar]
- 34.Bottenstein J E, Sato G H. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raff M C, Miller R H, Noble M. Nature (London) 1983;303:390–396. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- 36.Gage F H, Coates P W, Palmer T D, Kuhn H G, Fisher L J, Suhonen J O, Peterson D A, Suhr S T, Ray J. Proc Natl Acad Sci USA. 1995;92:11879–11883. doi: 10.1073/pnas.92.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagenaur C, Schachner M. J Supramol Struct Cell Biochem. 1981;15:335–346. doi: 10.1002/jsscb.1981.380150404. [DOI] [PubMed] [Google Scholar]
- 38.Baumrind N L, Parkinson D, Wayne D B, Heuser J E, Pearlman A L. Dev Dyn. 1992;194:311–325. doi: 10.1002/aja.1001940407. [DOI] [PubMed] [Google Scholar]
- 39.McDonald J W, Levine J M, Qu Y. NeuroReport. 1998;9:2757–2762. doi: 10.1097/00001756-199808240-00014. [DOI] [PubMed] [Google Scholar]
- 40.Bhat R V, Axt K J, Fosnaugh J S, Smith K J, Johnson K A, Hill D E, Kinzler K W, Baraban J M. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 41.Mulvey M A, Lopez-Boado Y S, Wilson C L, Roth R, Parks W C, Heuser J, Hultgren S J. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 42.Srinivasan A, Roth K A, Sayers R O, Shindler K S, Wong A M, Fritz L C, Tomaselli K J. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- 43.McTigue D M, Horner P J, Stokes B T, Gage F H. J Neurosci. 1998;18:5354–5365. doi: 10.1523/JNEUROSCI.18-14-05354.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demerens C, Stankoff B, Zalc B, Lubetzki C. Neurology. 1999;52:346–350. doi: 10.1212/wnl.52.2.346. [DOI] [PubMed] [Google Scholar]
- 45.Vicario-Abejon C, Cunningham M G, McKay R D G. J Neurosci. 1995;15:6351–6363. doi: 10.1523/JNEUROSCI.15-10-06351.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brustle O, Maskos U, McKay R D G. Neuron. 1995;15:1275–1285. doi: 10.1016/0896-6273(95)90007-1. [DOI] [PubMed] [Google Scholar]
- 47.Shuhonen J O, Peterson D A, Ray J, Gage F H. Nature (London) 1996;383:624–627. doi: 10.1038/383624a0. [DOI] [PubMed] [Google Scholar]
- 48.Grill R, Murai K, Blesch A, Gage F H, Tuszynski M H. J Neurosci. 1997;17:5560–5572. doi: 10.1523/JNEUROSCI.17-14-05560.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asou H, Hamada K, Sakota T. Cell Struct Funct. 1995;20:59–70. doi: 10.1247/csf.20.59. [DOI] [PubMed] [Google Scholar]
- 50.Reubinoff B E, Pera M F, Fong C Y, Trounson A, Bongso A. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]