

Abstract
Mutations within the gene encoding methyl CpG binding protein 2 (MECP2) cause the autism-spectrum neurodevelopmental disorder Rett Syndrome (RTT). MECP2 recruits histone deacetylase to methylated DNA and acts as a long-range regulator of methylated genes. Despite ubiquitous MECP2 expression, the phenotype of RTT and the Mecp2-deficient mouse is largely restricted to the postnatal brain. Since Mecp2-deficient mice have a defect in neuronal maturation, we sought to understand how MECP2/Mecp2 mutations globally affect histone modifications during postnatal brain development by an immunofluorescence approach. Using an antibody specific to acetylated histone H3 lysine 9 (H3K9ac), a bright punctate nuclear staining pattern was observed as MECP2 expression increased in early postnatal neuronal nuclei. As neurons matured in juvenile and adult brain samples, the intensity of H3K9ac staining was reduced. Mecp2-deficient mouse and RTT cerebral neurons lacked this developmental reduction in H3K9ac staining compared to age-matched controls, resulting in a significant increase in neuronal nuclei with bright H3K9ac punctate staining. In contrast, trimethylated histone H3 lysine 9 (H3K9me3) localized to heterochromatin independent of MeCP2, but showed significantly reduced levels in Mecp2 deficient mouse and RTT brain. Autism brain with reduced MECP2 expression displayed similar histone H3 alterations as RTT brain. These observations suggest that MeCP2 regulates global histone modifications during a critical postnatal stage of neuronal maturation. These results have implications for understanding the molecular pathogenesis of RTT and autism in which MECP2 mutation or deficiency corresponds with arrested neurodevelopment.
Keywords: histone, acetylation, methylation, MeCP2, Rett syndrome, autism, brain, neuron
INTRODUCTION
Rett syndrome (RTT) is an autism-spectrum neurodevelopmental disorder caused by mutations in the gene encoding methyl CpG binding 2 (MECP2) on Xq28 (1). RTT is an X-linked dominant disorder in which females heterozygous for MECP2 mutation show apparently normal development in early infancy, but around 6–18 months develop deceleration of head growth, ataxia, seizures, loss of verbal and motor skills, and autistic features (2). Mecp2-deficient mice recapitulate the delayed postnatal onset and neurological impairments of RTT and are a useful model for understanding the molecular pathogenesis (3–5). Interestingly, the phenotype of Mecp2 deficiency is limited to postnatal brain development (3, 4), suggesting that MeCP2 is only essential in this tissue and developmental stage. MeCP2 expression increases in individual neurons during postnatal brain development and is a considered a marker for mature neurons (6–10). Further support for MeCP2 involvement in neuronal maturation comes from studies of cultured neurons (11) and olfactory bulb (12, 13) showing neuronal immaturity as a result of Mecp2 deficiency. A recent electrophysiology study of hippocampal neurons from Mecp2-deficient mice showed neuronal defects in long-term potentiation and synaptogenesis (14). These studies all point to a precise role for MeCP2 during neuronal maturation.
MeCP2 is one member of a family of methyl-binding domain proteins with affinity for methylated CpG dinucleotides (15). Although MeCP2 was originally characterized as a transcriptional repressor of methylated genes (16, 17), recent studies have suggested more diverse roles for MeCP2 in chromatin dynamics. MeCP2 associates with both histone deacetylase (HDAC) activity (16) and histone methyltransferase activity that directs the silencing mark for histone H3, lysine 9 (H3K9) (18). Acetylation of H3K9 (H3K9ac) is a mark of active chromatin while a reciprocal pattern of trimethylation of the same residue (H3K9me3) often correlates with repression (19). MeCP2 has been also shown to interact with the maintenance DNA methyltransferse Dnmt1 (20) and Brahma, a protein related to DNA helicases (21). MeCP2 can also physically compact chromatin when added to histone-bound or naked DNA in vitro (22). Furthermore, MeCP2 has been demonstrated to regulate silent chromatin loop formation in the imprinted Dlx5/Dlx6 locus (23).
Mecp2308/y mutant mouse brains exhibit elevated histone H3 acetylation by immunoblot (5), as expected given MeCP2’s role in HDAC recruitment (16). Other immunoblot results from MECP2 mutant cell lines or blood cells have demonstrated inconsistent changes in histone modifications, however (24–26). Although some studies have characterized the effect of MeCP2 on histone modifications at specific genes (27–29), binding sites for MeCP2 also include repetitive sequences and heterochromatin throughout the genome (30–32). The role for MeCP2 in global chromatin organization and changes to specific histone modifications during neuronal maturation has not been well characterized.
Therefore, in order to bring together both biochemical and cell biological aspects of MeCP2 function in the developing nervous system, we have employed an immunofluorescent approach to examine histone H3K9 modifications in the developing mammalian brain with and without MECP2/Mecp2 mutation or deficiency. Here we demonstrate dynamic changes in the localization and level of H3K9ac in early postnatal mouse and human brain. Mecp2-deficient mouse and RTT brain samples appear to be arrested at an immature stage of neuronal maturation that retains a bright punctate pattern of H3K9ac staining. In contrast, H3K9me3 staining remains colocalized with heterochromatin in Mecp2-deficient neurons, but at a lower level. These results bring together several previous findings regarding MeCP2’s function and may help explain a precise role for MeCP2 in the dynamic changes in histone modifications during neuronal maturation.
METHODS AND MATERIALS
Mouse Tissue Microarray
Wildtype C57BL/6J and Mecp2tm1.1Bird/y tissue for multiple age time points were obtained, fixed and embedded in paraffin and sampled as described previously (33). The mouse tissue microarray included triplicate 600 μm cores of grey matter from cerebral cortex of eight age-matched male time points for both wild-type (wt) and Mecp2tm1.1Bird/y: embryonic day 19 (E19), postnatal day 1 (P1), P7, P28, P35, P49, P56, and P70 for wild-type only.
Human Tissue Microarray
Tissues were received frozen and were fixed, embedded and arrayed as described previously (34). The human tissue microarray contained triplicate 600 μm cores of frontal cortex, Broadman Area 9 (< 30 h post-mortem). Tissues on the tissue microarray included: 1157 (20 d), 1321 (62 d), 125 (76 d), 1055 (96 d), 135 (120 d), 1257 (2 y), 1065 (15 y), 1027 (22 y), 602 (27 y), 1029 (29 y), RTT 1238 (2 y), and AUT 797 (9 y) from University of Maryland Brain and Tissue Bank for Developmental Disorders. Additional samples B4192 (46 y), B4503 (56 y), and RTT 4312 (24 y) were obtained from Harvard Brain Tissue Resource Center and 3835 (9 y) from University of Miami, Brain Endowment Bank. Fragile X (25 y) brain was a gift from Drs. P. J. and R. J. Hagerman.
Immunofluorescence
Paraffin blocks containing tissue microarrays were cut into 5 μm sections on glass slides and baked overnight at 56°C. Slides underwent four 5 m xylene washes, two 5 m 100% EtOH washes, were dried on a slide warmer at 50°C, then placed in 1x DAKO antigen retrieval solution at 95°C for 1 h, cooled to RT, and washed for 5 m in 0.2x SSC. Primary antibodies were diluted in IF buffer (1x PBS, 0.5% Tween, 0.01% FCS), incubated on slides at 37°C for 2 h, washed three times for 5 m in 1x PBS/0.5% Tween with agitation. Secondary antibodies were diluted in IF buffer (along with 250 μg/ml RNAse and 7 μg/ml propidium iodide for LSC only) and incubated on slides at 37°C for 1 h followed by three additional 5 m washes in 1x PBS/0.5% Tween with agitation. Slides were mounted either with 5 μg/ml DAPI in Vectashield (for fluorescent microscopy) or 7 μg/ml PI in 50% glycerol/50% 1x PBS (for LSC). Antibodies sources and dilutions were as follows: anti-H3K9ac, anti-H3K9me3, anti-H3ac, anti-H4ac (Upstate, rabbit polyclonal) at 1:100; anti-MeCP2 (C-terminal, chicken polyclonal, described previously (35) at 1:10,000; anti-histone H1 (Upstate, mouse polyclonal) at 1:100; anti-NeuN (Chemicon, mouse polyclonal) at 1:100; goat-anti-rabbit-IgG-Oregon Green (Molecular Probes) at 1:100; donkey anti-chicken-IgY-Cy5 (Jackson Immunochemicals) at 1:100; donkey-anti-mouse-IgG-Cy3 (Jackson Immunochemicals) at 1:5,000; and goat-anti-mouse-IgG-Cascade Blue (Molecular Probes) at 1:100. Control nonspecific IgY (Aves) or rabbit and mouse IgG (Upstate) were used at equivalent or higher concentrations as the primary antibodies with the same secondary reagents. All above antibodies stained significantly above control background (data not shown).
Fluorescent Microscopy
Slides stained for anti-H3K9ac or anti-H3K9me3, anti-MeCP2, anti-NeuN (for blind scoring of H3K9ac patterns on human tissue microarray) and counterstained with DAPI were visualized using both 63x and 100x oil immersion lenses on an Axioplan 2 fluorescence microscope (Carl Zeiss, Inc, NY) equipped with a Sensys CCD camera (Photometrics, Tucson, AZ), appropriate fluorescent filter sets, and automated xyz stage controls. The microscope and peripherals were controlled by a Macintosh running IPLab Spectrum (Scanalytics, Vienna, VA) software with Multiprobe, Zeissmover, and 3D extensions. Images were captured for blue, green, and red filters at one edge of the specimen, then repeated at 0.4 micron sections through the depth of the tissue. Each image stack was digitally deconvolved to remove out-of-focus light using HazeBuster software (Vaytek, Fairfield, IA). Following haze removal, image stacks for each fluorophore were merged and stacked to create a two-dimensional image representing all of fluorescence within the section. Adobe Photoshop software was used to resize and adjust brightness and contrast of images, but identical settings were used for all image comparisons within the same figure, with the exception of Figure 4 where 2 y control and RTT1238 images were taken with a 63x objective and RT4312 was taken with a 100x objective. Blind scoring for bright H3K9ac punctate staining in NeuN positive cells was conducted with the identity of the microarray cores unknown to the investigator.
Figure 4.
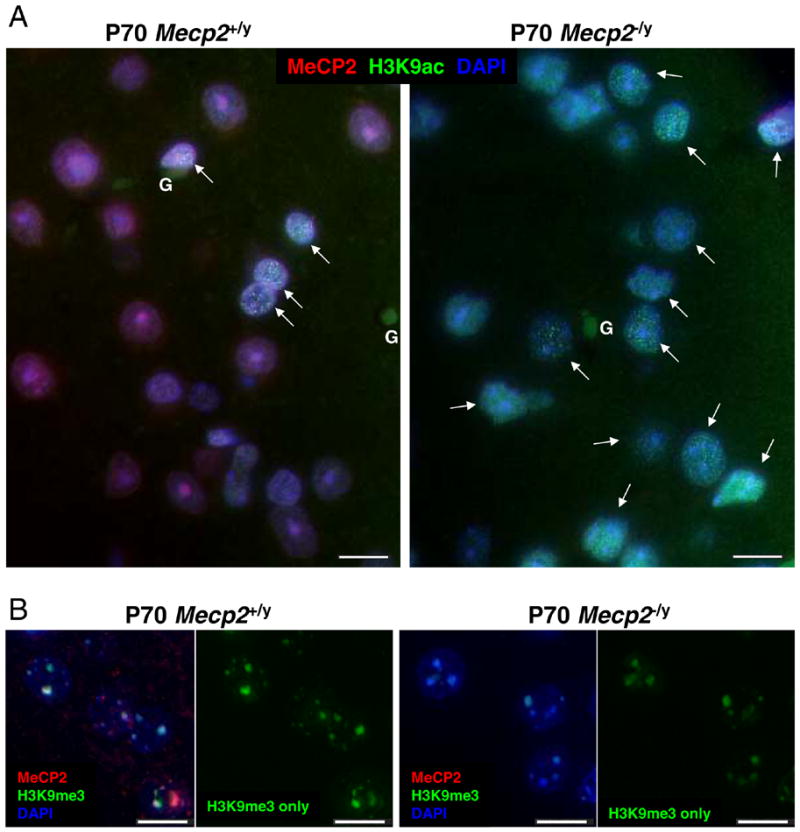
A) More nuclei in Mecp2 deficient mouse cortex show “transitioning” pattern of bright H3K9ac punta than in wild-type controls. Representative images from adult (P70) wild-type and Mecp2 deficient cortex. Sections were stained with anti-MeCP2 (red fluorescence), anti-H3K9ac (green fluorescence) and nuclei counterstained with DAPI (blue fluorescence). A greater number of Mecp2−/y mutant neuronal nuclei showed a bright punctate H3K9ac staining pattern (arrows) compared to the age-matched wild-type control. B) H3K9me3 localization is not altered by Mecp2 deficiency, but fluorescence is lower in Mecp2−/y neuronal nuclei. Representative images from adult (P70) wild-type and Mecp2 deficient cortex. Sections were stained with anti-MeCP2 (red fluorescence), anti-H3K9me3 (green fluorescence) and nuclei counterstained with DAPI (blue fluorescence). H3K9me3 colocalized with MeCP2 to DAPI-bright heterochromatin in wild-type neuronal nuclei and localization of H3K9me3 was unchanged in Mecp2-deficient nuclei. Lower fluorescence was observed for H3K9me3 foci in Mecp2−/y nuclei compared to controls, consistent with LSC results (Fig. 3). Glial cells (G) are indicated. Scale bars represent 10 microns.
LSC
Slides were scanned using a Compucyte Laser Scanning Cytometer (CompuCyte Inc., Cambridge, MA), as described previously (7). Nuclei, stained red with propidium iodide, were contoured within each core and fluorescence was measured for each nucleus for all channels (red, green, blue and long red). Data was analyzed using WinCyte software from Compucyte and used to create all LSC figures.
RESULTS
Differences in H3K9ac staining patterns during human postnatal brain development
Since the localization pattern of the transciptionally activating histone modification H3K9ac has not previously been described in neurons, we determined the immunofluorescent pattern in human cerebral cortex sections. A tissue microarray containing triplicate 600 μm diameter cores of post-mortem human cerebral cortex samples from control individuals of different ages were stained for anti-H3K9ac and anti-MeCP2 and the nuclear counterstain DAPI. Use of tissue microarrays controls for staining conditions and allows for easier comparisons to be made between age groups, as described previously (7, 34).
Figure 1 shows representative images of H3K9ac staining patterns in normal human brain samples of different ages. H3K9ac staining revealed a bright dense nuclear punctate pattern in early infant human cortex (Fig. 1, 20 d) with a relatively uniform staining pattern for all cells. In later infant samples (96 d to 2 y), a transitional change was observed in the H3K9ac staining pattern as the cells of the cerebral cortex differentiated. Glial cells (G), recognizable by a small heterochromatic nuclei (bright blue) and low MECP2 (low red), had high H3K9ac staining (bright green). For the neuronal nuclei, three distinct H3K9ac and MECP2 staining patterns emerged. In the 96 d sample, neuronal nuclei with low MECP2 showed a bright dense punctate H3K9ac staining (yellow closed arrows), similar to early infant brain. For other neuronal nuclei in 96 d to 2 y samples, bright MECP2 staining also coincided with a bright dense punctate H3K9ac staining pattern (white closed arrows). In contrast, a population of mature neuronal nuclei with high MECP2 expression and prominent nucleoli emerged in older brain samples (2 y and 29 y) that showed a lower and more diffuse nuclear H3K9ac pattern (open arrowheads). With continued neuronal maturation through juvenile and adult ages, H3K9ac staining continued to become more diffuse and decreased in staining intensity in large euchromatic neuronal nuclei with prominent nucleoli.
Figure 1. Histone H3K9 acetylation patterns in human cortical neurons correlate with elevated MeCP2 expression.
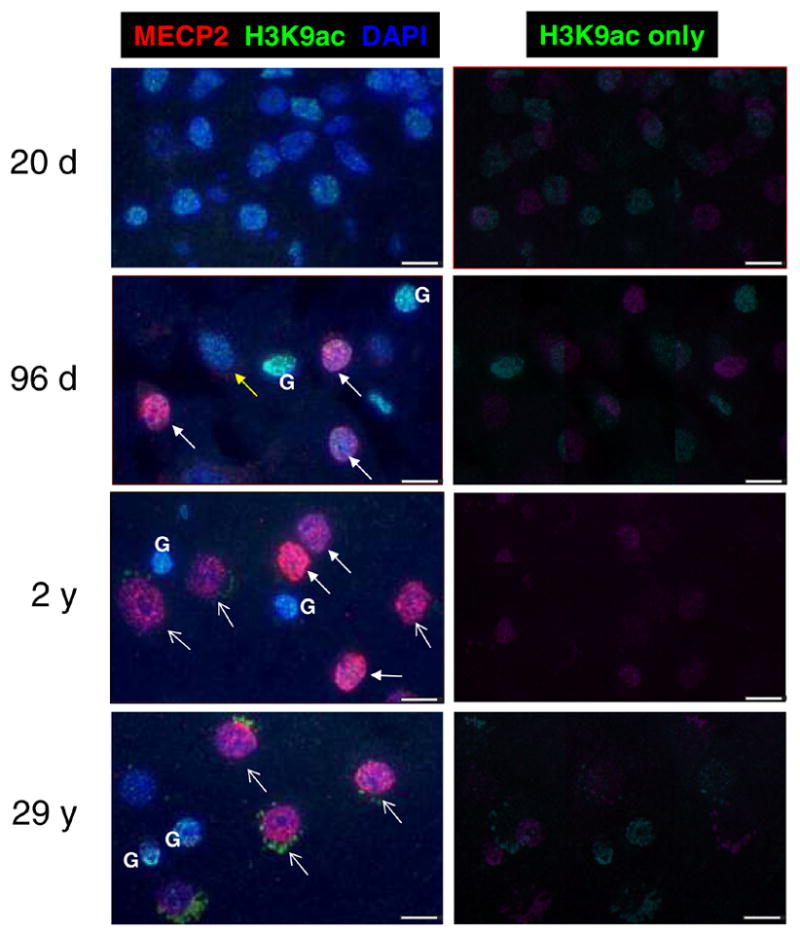
Representative images from normal control human post-mortem frontal cortex (Brodman area 9) samples from different aged individuals show heterogeneous developmental changes in H3K9ac patterns. Sections were stained with anti-MeCP2 (red fluorescence), anti-H3K9ac (green fluorescence) and nuclei counterstained with DAPI (blue fluorescence). Left panels show three-color merged images, while right panels show H3K9ac green channel only. Early infant brain showed low MECP2 expression and bright H3K9ac nuclear punctate staining in all nuclei. In contrast, later infant brain (96 d) samples showed a mixture of immature neuronal nuclei (low MECP2, bright punctate H3K9ac, yellow arrows) and “transitioning” nuclei (high MECP2, bright punctate H3K9ac, white closed arrows). A 2 y brain sample showed both “transitioning” neuronal nuclei and mature neuronal nuclei (high MECP2, low H3K9ac staining, prominent nucleoli, white open arrows), while adult brain (29 y) showed a majority of mature neuronal nuclei. Glial cell nuclei (G) were identifiable in infant-adult samples as small heterochromatic nuclei with low MECP2 and high H3K9ac. Cytoplasmic green fluorescence in adult brain is due to lipofuscin autofluorescence rather than specific H3K9ac staining, as this autofluorescent non-nuclear staining is observed using a nonspecific antibody control (data not shown). All images were captured and modified using identical settings. Scale bars represent 10 microns.
H3K9ac and MeCP2 define two distinct neuronal populations in postnatal mouse brain developmant by laser scanning cytometry
In order to obtain more precise developmental time points and to determine if the changes in H3K9ac staining patterns were also observed in mouse, a mouse tissue micoarray containing wild-type time points ranging from embryonic day 19 (E19) through postnatal day 70 (P70) was stained with antibodies for H3K9ac (or global H3ac used by a previous study (5)), MeCP2, histone H1 (control antibody), and a propidium iodide counterstain. Laser scanning cytometry (LSC) was used to quantitate the levels of histone modifications throughout mouse cerebral cortex development, as this approach has been previously used to show developmental changes in MeCP2 expression (33). LSC detects fluorescence in multiple channels for individual cells within tissue, similar to flow cytometry used for suspension cells (7). While LSC results are generally comparable to immunoblot, the ability of LSC to detect individual cell populations and protein localization changes provides a major advantage over immunoblot analysis (33, 36).
Figure 2A shows scattergrams depicting distinct cell populations and the developmental relationship between MeCP2 and H3K9ac fluorescence. In mouse embryonic brain, moderate H3K9ac levels with low MeCP2 was observed in the entire population. In early postnatal brain (P1–P7), increased H3K9ac was observed in the entire population of cells together with low MeCP2 expression. In P28–P35 cortex, however, a lack of correlation between MeCP2 and histone H3K9ac was observed in a fall-out population of mature neuronal nuclei (R2, black ovals), while another population of “transitioning” neurons showed a positive correlation between MeCP2 and H3K9ac fluorescence (R1, grey ovals). Both populations were still present in P49–P70 cortex, but a subtle shift towards an increased number of cells with the mature neuronal nuclear phenotype was observed. Figure 2B shows a similar developmental pattern when the same analysis was performed with a global anti-H3ac antibody, except that the separation between immature, transitioning, and mature neurons was less apparent with this antibody. These results demonstrate that increased MeCP2 expression within individual neuronal nuclei coincides with reduced histone H3K9ac and H3ac staining, as was suggested by the immunofluorescent staining patterns in human brain (Fig. 1). The additional population of nuclei staining high for both MeCP2 and H3K9ac in young mouse and infant human brain (Fig. 2A, R1 and Fig. 1, closed white arrows) also suggests that developmentally acquired MeCP2 expression actually precedes the changes in histone H3K9ac localization patterns.
Figure 2. Developmental changes in transitioning and mature neuronal nuclei identifiable by combined immunofluorescence for MeCP2 and H3K9ac and laser scanning cytometry.
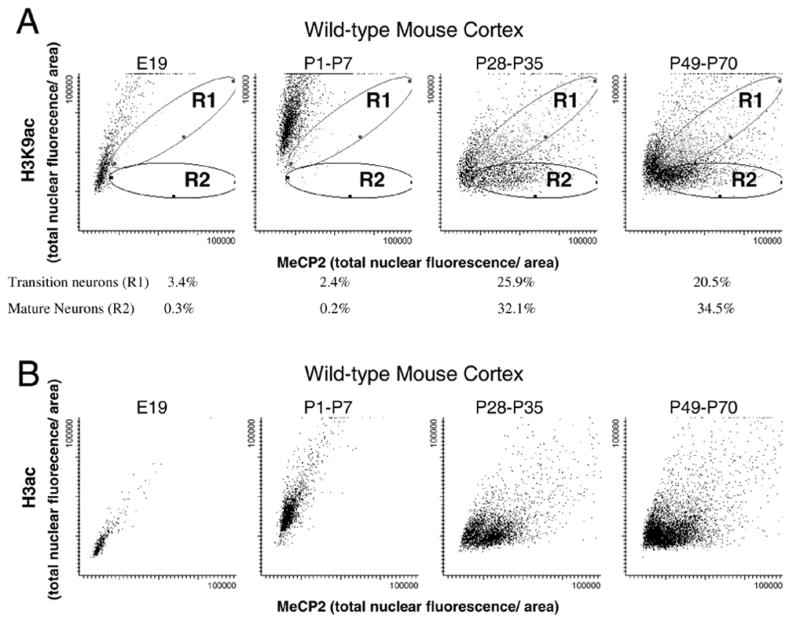
A mouse tissue microarray containing triplicate samples of cerebral cortex from different developmental stages was stained with anti-MeCP2, anti-H3K9ac (A) or anti-H3ac (B), counterstained with propidium iodide and analyzed by laser scanning cytometry (LSC) to quantitate different cell populations. LSC is similar to flow cytometry in its ability to separate distinct cell populations based on fluorescent markers. Scattergrams represent individually contoured nuclei plotted according to MeCP2 fluorescence (x-axis) and H3K9ac fluorescence (y-axis) for four developmental ranges. A) Distinct populations emerging in the postnatal brain were observed on the H3K9ac versus MeCP2 scattergrams and defined by individually gated regions. Region 1 (R1, grey ovals) defines the “transitioning” neurons, showing high MeCP2 and high H3K9ac (similar to closed arrows in Fig. 1). Region 2 (R2, black ovals) defines the “mature” neurons, showing high MeCP2 fluorescence but low H3K9ac fluorescence (similar to open arrows in Fig. 1). The percentage of cells in each of these gated populations is shown for each developmental stage. B) LSC analysis of the same mouse tissue microarray stained with anti-H3ac and anti-MeCP2 showed a similar pattern of developmental population changes, but the separation between immature, transitioning, and mature neurons was less apparent with the more global H3ac antibody.
Mecp2-deficient mouse cortex samples show increased H3K9ac, but decreased H3K9me3
In order to determine if MeCP2 was necessary for the developmental changes observed in histone H3K9ac patterns in mammalian brain, human and mouse samples with MECP2 mutation or deficiency were examined. The mouse tissue microarray contained Mecp2tm1Bird/y samples in addition to age-matched wild-type males. All samples were analyzed qualitatively, by fluorescent microscopy, and quantitatively, by LSC, and compared back to their age-matched controls. As expected based on prior immunoblot analyses (5), Mecp2 deficient adult mouse cerebral cortex (P7–10wks) showed significantly higher levels of H3K9ac (P < 0.001) compared to their wild-type littermates by LSC (Fig. 3). By fluorescent microscopy, adult Mecp2 deficient mice showed a greater number of neuronal nuclei showing bright punctate staining for H3K9ac compared to wild-type (Fig. 4A, arrows).
Figure 3. Histone H3K9ac fluorescence levels are aberrantly high in Mecp2-deficient adult mouse brain, while H3K9me3 levels are aberrantly low.
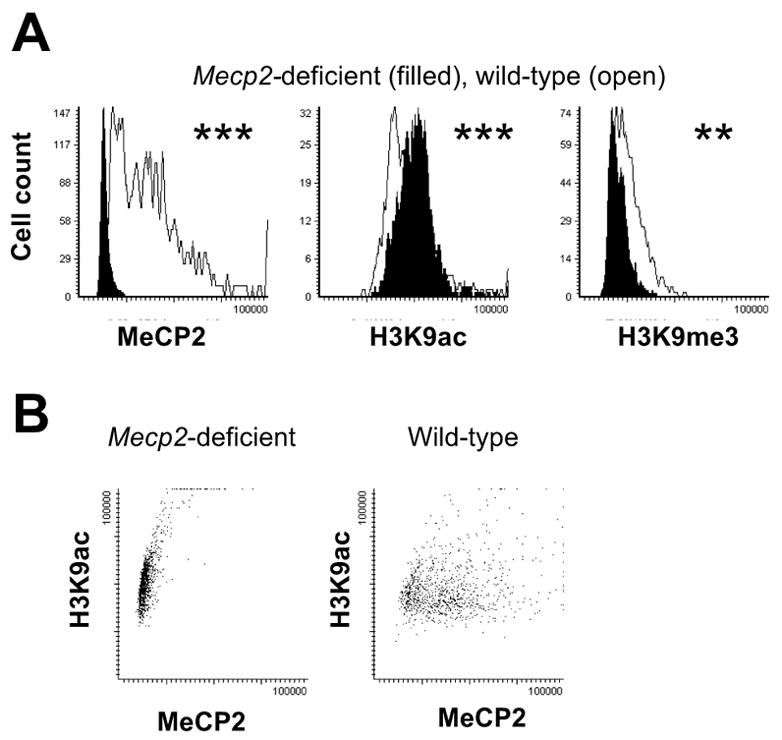
Laser scanning cytometric analysis of anti-MeCP2, anti-H3K9ac, and anti-H3K9me3 immunofluorescence for adult (P70) wild-type male (open histograms) or Mecp2−/y (filled histograms) cerebral samples. A) Mecp2−/y nuclei showed significantly lower MeCP2, significantly higher H3K9ac fluorescence, and significantly lower H3K9me3 fluorescence. Values are plotted as total nuclear fluorescence/area measurements, as in Figure 2. Representative fluorescence microscopy images are shown in
The repressive H3K9 methylation modification was also examined by immunofluorescence and LSC. H3K9me3 immunofluorescence showed a contrasting staining pattern of colocalization with DAPI-bright heterochromatin and MeCP2 staining in cerebral cortex (Fig. 4B). H3K9me3 staining revealed no change in localization with Mecp2 deficiency, but showed decreased intensity of the staining at the heterochromatic foci (Fig. 4B). Mecp2 deficient mouse brain also showed significantly lower levels of H3K9me3 than wild-type by LSC analysis (P < 0.01, Fig. 3).
RTT brain with mutant MECP2 and autism brain with MeCP2 deficiency showed increased H3K9ac punctate staining and decreased H3K9me3 fluorescence compared to controls
The human tissue microarray also included MECP2 deficient RTT and autism samples, as well as a Fragile X sample. A 2 y human male sample hemizygous for MECP2 mutation 1154Δ32 showed a greater number of nuclei with a bright punctate histone H3K9ac pattern compared to a 2 y control sample (Fig. 5). An adult female RTT sample (MECP2 R168X) showed the expected mosaic pattern of MECP2 expression due to X chromosome inactivation (7), but a bright punctate H3K9ac pattern was observed primarily in the cells expressing the MECP2 mutation (nuclei without red staining, arrows, Fig. 5). To support these descriptive findings, blind scoring for bright punctate H3K9ac staining in human cerebral cortex neurons was performed on the human tissue microarray using NeuN as a post-mitotic neuronal marker. Three replicate cores were scored for four normal controls (2–29 y), two RTT samples, and a Fragile X sample. The results of the blind scoring, shown in Figure 6, demonstrate that both RTT samples had significantly higher numbers of neurons with bright punctate staining than the age-matched controls, while a Fragile X cortex sample with no MECP2 deficiency showed no significant difference.
Figure 5. MECP2 mutant nuclei male and female human brain samples show increased H3K9ac staining in neuronal nuclei.
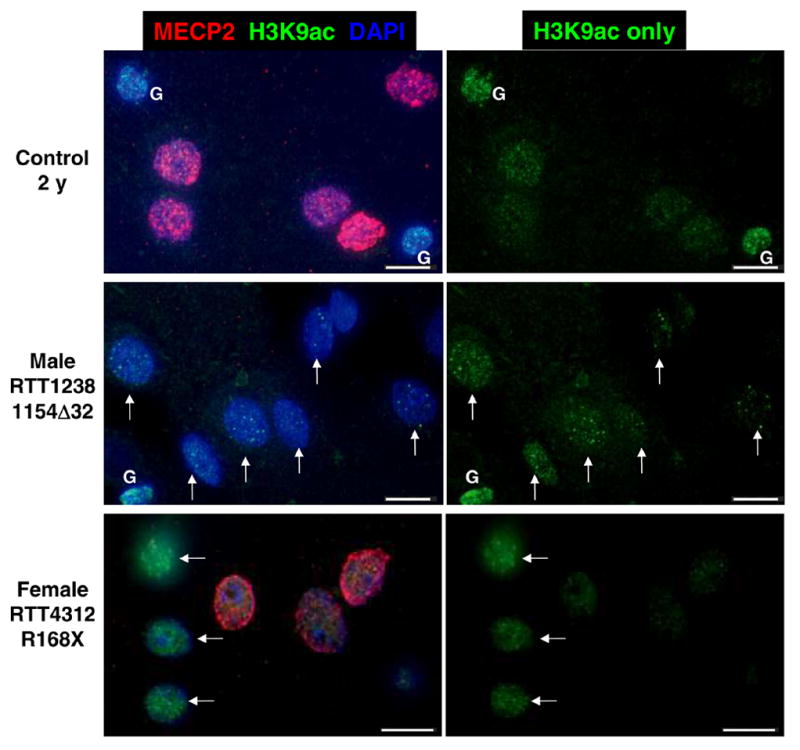
Representative images from control 2 y, MECP2 mutant male, and MECP2 mosaic mutant female RTT cerebral cortex samples. Sections were stained with anti-MeCP2 (red fluorescence), anti-H3K9ac (green fluorescence) and nuclei counterstained with DAPI (blue fluorescence). MECP2 mutant nuclei in male and female cortex are characterized by an immature bright punctate pattern of H3K9ac fluorescence. Female RTT brain is mosaic for mutant and wild-type MECP2 expression because of random X chromosome inactivation (7, 33). Mutant MECP2 expressing neurons characterized by absence of MeCP2 red fluorescence have brighter H3K9ac puncta (arrows) than neighboring wild-type expressing nuclei. Glial cells (G) are indicated. Scale bars represent 10 microns.
Figure 6. Blinded scoring of neuronal nuclei with bright H3K9ac puncta shows a significant increase in RTT brain samples compared to age-matched controls.
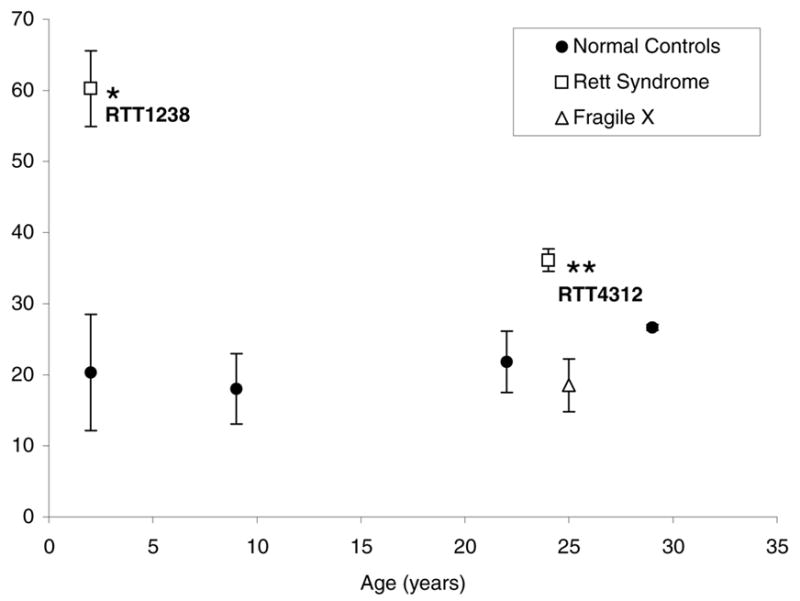
Triplicate core samples from male RTT1238, female RTT4312, and five control human brain samples of different ages were scored for neuronal nuclei (identified by NeuN staining) with bright H3K9ac puncta (similar to nuclei represented by arrows in Fig. 5) while coded for identity of the samples. Both male RTT1238 and female RTT4312 showed significantly more nuclei with an immature H3K9ac pattern compared to controls while Fragile X showed no significant difference. *P< 0.05, **P<0.01 by t-test.
LSC analysis of human brain samples also confirmed significantly increased H3K9ac and decreased H3K9me3 fluorescence in MECP2-mutant RTT brain compared to age-matched controls (Figure 7A). In addition, an autism brain sample previously shown to have significantly reduced MECP2 expression (AUT797) (34) also showed a similar significant increase in histone H3K9ac and decrease in H3K9me3 fluorescence compared to controls. Unlike RTT and autism samples, a Fragile X brain sample with normal MECP2 expression showed no significant differences in histone H3 modifications. In constrast to changes in histone H3 modifications, histone H4ac and control histone H1 showed no significant difference between samples by LSC.
Figure 7. Laser scanning cytometric analysis of cortex samples from three different neurodevelopmental disorders show that aberrantly increased H3K9ac fluorescence correlates with MECP2 mutation or deficiency.
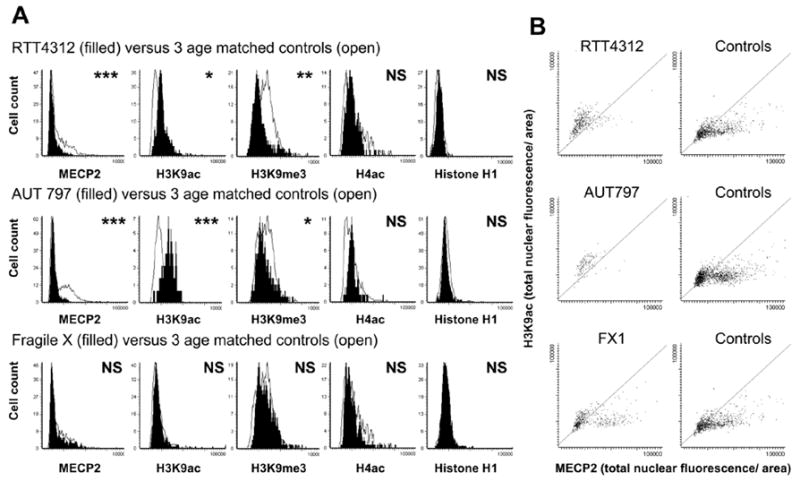
Laser scanning cytometric analysis of anti-MECP2, anti-H3K9ac, anti-H3K9me3, anti-H4ac and anti-H1 immunofluorescence for three human samples with different neurodevelopmental disorders compared to age-matched controls. All fluorescence measurements are in total nuclear fluorescence divided by area, as in Fig. 2 and 3. A) MECP2-mutant RTT4312 and MECP2-deficient AUT797 cerebral nuclei (filled histograms) showed lower MECP2, higher H3K9ac, and lower H3K9me3 fluorescence than three age-matched controls (open histograms), but no difference in histone H4ac or H1 fluorescence. In contrast, a Fragile X sample (filled histograms) with normal MECP2 expression showed no differences in histone modifications compared to controls (open histograms). *P< 0.05, ** P<0.01, ***P<0.001 by t-test. B) Scattergrams of MECP2 versus H3K9ac staining for nuclei from neurodevelopmental samples compared to age-matched controls (similar to mouse scattergrams in Fig. 2). Both RTT4312 and AUT797 lack the fallout population of mature neuronal nuclei characterized by high MECP2 and low histone H3K9ac fluorescence. In contrast the scattergram for Fragile X brain is similar to controls. Grey diagonal sections through the scattergrams are shown to better demonstrate the paucity of cells in the lower right half of the scattergram for RTT4312 and AUT797 samples compared to controls. Similar patterns were observed for each sample using an antibody recognizing H3ac (data not shown).
Figure 7B shows scattergrams of histone H3K9ac versus MECP2 fluorescence for normal control, RTT, autism and Fragile X samples, similar to those shown for mouse cortex in Figure 2A. RTT and autism samples lacked a “fall-out” population of mature neurons characterized by high MECP2 and low H3K9ac levels. In contrast, Fragile X brain nuclei showed a normal population of these mature neurons with low H3K9ac levels.
These results demonstrate that MECP2 deficiency or mutation results in an aberrant pattern of histone H3K9ac and a decrease in H3K9me3 in neuronal nuclei from two different mammalian systems. These results suggest that MECP2 mutation or deficiency results in an arrest of normal neuronal maturation to a transition stage characterized by high MECP2 expression and bright punctate histone H3K9ac patterns.
DISCUSSION
Despite the discovery of mutations in MECP2 as the genetic cause of RTT (1) and the biochemical characterization of MeCP2 as a transcriptional repressor that recruits histone deacetylase activity (17, 37), there has been limited information on how MeCP2 deficiency causes a specific defect in postnatal neuronal maturation. In this report we bring together developmental changes in the nuclear organization of a specific histone modification directly linked to gene activity, H3K9ac, with a precise role for MeCP2 in neuronal maturation.
MeCP2 is dynamically expressed during postnatal brain development, showing acquired elevated expression in individual neurons coincident with maturation stage (6, 8, 10, 38). Here we demonstrate that histone H3K9ac also shows a dynamic developmental localization pattern in maturing neurons in both mouse and human brain coincident with MeCP2 increases. Embryonic and early postnatal nuclei were characterized by high overall H3K9ac but low MeCP2 levels in all nuclei, while late infant to juvenile samples showed heterogeneous patterns. Some neuronal nuclei retained a high H3K9ac and low MeCP2 pattern, while others showed a “transition state” of high MeCP2 and bright H3K9ac punctate staining. Nuclei with reduced staining of H3K9ac had a mature neuronal phenotype characterized by high MECP2, a large euchromatic nucleus, and a single large nucleolus. This distinct population of MECP2hi/H3K9aclo neuronal nuclei increased progressively with age.
The observation that elevated MECP2 expression is coincident with bright punctate histone H3K9ac nuclei in early postnatal cortex suggested that increased MECP2 expression preceded the changes in H3K9ac distribution. A causal role for MECP2 in the H3K9ac developmental changes was demonstrated by the increase in the number of nuclei showing the “transition state” pattern of bright punctate H3K9ac staining as well as the overall increase in H3K9ac levels in Mecp2 deficient mouse brain. These results confirm a previous study by immunoblot of brain tissue from the Mecp2308/y RTT model (5), but also extend the finding to potentially explain the neuron-specific developmental arrest observed in RTT. A direct involvement of MECP2 in recruitment of HDAC to change the H3K9ac levels in maturing neurons is hypothesized based on the demonstrated association of these factors (17, 37). However, the possibility remains that MECP2 may regulate critical genes that control neuronal maturation and the observed changes in H3K9 modifications may reflect a downstream effect of gene expression differences rather than a direct effect on global histone H3 modifications.
Developmental changes in histone modifications have been previously shown in the postnatal rat corpus collosum and have been implicated in differentiation control of the oligodendrocyte lineage (39). In addition, MeCP2 has been previously implicated in large-scale chromatin reorganization during myogenic differentiation (40). To our knowledge, however, this is the first report demonstrating a role for MeCP2 in the differentiation control of large-scale histone modification changes during neuronal maturation. The developmental changes in H3K9ac patterns most likely reflect changes in gene expression that define neuronal maturational states, but global chromatin changes may also directly or indirectly control the state of cellular maturation. Interestingly, Mecp2 deficiency resulted in a change to the localization patterns of H3K9ac but not H3K9me3. Since H3K9me3 colocalizes with nuclear heterochromatin that includes completely silenced genes, this result is consistent with previous findings that MECP2/Mecp2 mutation or deficiency does not reverse the silencing of imprinted or inactive X chromosome genes (26, 36, 41). The lower level of H3K9me3 in MECP2/Mecp2 deficient brain may reflect the lack of recruitment of genes normally silenced during neuronal maturation to heterochromatic nuclear locations in the absence of MeCP2. Further investigation of candidate genes silenced by MeCP2 and their potential association and disassociation with H3K9ac puncta and H3K9me3 heterochromatin in immature and mature neurons is warranted.
A major paradox in understanding how MECP2 mutations cause RTT is explaining how a ubiquitous global transcriptional repressor is only critical for postnatal brain development. Our results begin to illuminate this puzzling area by demonstrating that MeCP2 is required for the developmental changes in histone H3K9ac patterns in the postnatal maturation of neurons. Since the level of MeCP2 expression needs to be precisely regulated for normal brain development (34, 42–44), perhaps MeCP2 is only indispensable when expressed at its highest level in maturing neurons. Postnatal elevated MeCP2 expression, potentially induced by experience-dependent activation (45), may regulate the developmental changes in H3K9 modifications both globally and in a gene-specific manner.
Abbreviations
- H3K9ac
acetylated histone H3 lysine 9
- H3K9me3
trimethylated histone H3 lysine 9
- H3ac
acetylated histone H3
- H4ac
acetylated histone H4
- HDAC
histone deacetylase
- LSC
laser scanning cytometry
- MeCP2
methyl CpG binding protein 2
- RTT
Rett syndrome
References
- 1.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong DD. Review of Rett syndrome. J Neuropathol Exp Neurol. 1997;56:843–849. doi: 10.1097/00005072-199708000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 4.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 5.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 6.Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell- specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- 7.LaSalle J, Goldstine J, Balmer D, Greco C. Quantitative localization of heterologous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- 8.Balmer D, Goldstine J, Rao YM, LaSalle JM. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J Mol Med. 2003;81:61–68. doi: 10.1007/s00109-002-0396-5. [DOI] [PubMed] [Google Scholar]
- 9.Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8:784–791. doi: 10.1006/nbdi.2001.0420. [DOI] [PubMed] [Google Scholar]
- 10.Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005;19:18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Cohen DR, Matarazzo V, Palmer AM, Tu Y, Jeon OH, Pevsner J, Ronnett GV. Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol Cell Neurosci. 2003;22:417–429. doi: 10.1016/s1044-7431(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 13.Matarazzo V, Cohen D, Palmer AM, Simpson PJ, Khokhar B, Pan SJ, Ronnett GV. The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol Cell Neurosci. 2004;27:44–58. doi: 10.1016/j.mcn.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2005 doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 15.Hendrich B, Abbott C, McQueen H, Chambers D, Cross S, Bird A. Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm Genome. 1999;10:906–912. doi: 10.1007/s003359901112. [DOI] [PubMed] [Google Scholar]
- 16.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 17.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 18.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 19.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 20.Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806–4812. doi: 10.1074/jbc.M209923200. [DOI] [PubMed] [Google Scholar]
- 21.Harikrishnan KN, Chow MZ, Baker EK, Pal S, Bassal S, Brasacchio D, Wang L, Craig JM, Jones PL, Sif S, et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet. 2005;37:254–264. doi: 10.1038/ng1516. [DOI] [PubMed] [Google Scholar]
- 22.Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC. Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J Biol Chem. 2003;278:32181–32188. doi: 10.1074/jbc.M305308200. [DOI] [PubMed] [Google Scholar]
- 23.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 24.Wan M, Zhao K, Lee SS, Francke U. MECP2 truncating mutations cause histone H4 hyperacetylation in Rett syndrome. Hum Mol Genet. 2001;10:1085–1092. doi: 10.1093/hmg/10.10.1085. [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann WE, Jarrar MH, Wang JS, Lee YJ, Reddy S, Bibat G, Naidu S. Histone modifications in Rett syndrome lymphocytes: a preliminary evaluation. Brain Dev. 2005;27:331–339. doi: 10.1016/j.braindev.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Balmer D, Arredondo J, Samaco RC, LaSalle JM. MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum Genet. 2002;110:545–552. doi: 10.1007/s00439-002-0724-4. [DOI] [PubMed] [Google Scholar]
- 27.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 28.Makedonski K, Abuhatzira L, Kaufman Y, Razin A, Shemer R. MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum Mol Genet. 2005;14:1049–1058. doi: 10.1093/hmg/ddi097. [DOI] [PubMed] [Google Scholar]
- 29.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 30.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 31.Lorincz MC, Schubeler D, Groudine M. Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized histone H3 deacetylation. Mol Cell Biol. 2001;21:7913–7922. doi: 10.1128/MCB.21.23.7913-7922.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J, Cigudosa JC, Huang TH, Esteller M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. Embo J. 2003;22:6335–6345. doi: 10.1093/emboj/cdg604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13:1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- 34.Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 35.Thatcher K, Peddada S, Yasui D, LaSalle JM. Homologous pairing of 15q11–13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet. 2005;14:785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
- 36.Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones PL, Wade PA, Wolffe AP. Purification of the MeCP2/Histone Deacetylase Complex from Xenopus laevis. Methods Mol Biol. 2001;181:297–307. doi: 10.1385/1-59259-211-2:297. [DOI] [PubMed] [Google Scholar]
- 38.Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience. 2004;123:939–949. doi: 10.1016/j.neuroscience.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 39.Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brero A, Easwaran HP, Nowak D, Grunewald I, Cremer T, Leonhardt H, Cardoso MC. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J Cell Biol. 2005;169:733–743. doi: 10.1083/jcb.200502062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gartler SM, Varadarajan KR, Luo P, Canfield TK, Traynor J, Francke U, Hansen RS. Normal histone modifications on the inactive X chromosome in ICF and Rett syndrome cells: implications for methyl-CpG binding proteins. BMC Biol. 2004;2:21. doi: 10.1186/1741-7007-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 43.Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 Region Is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]