

Abstract
Within the past decade, there has been increasing recognition that glia are far more than simply “housekeepers” for neurons. This review explores two recently recognized roles of glia (microglia and astrocytes) in: (a) creating and maintaining enhanced pain states such as neuropathic pain, and (b) compromising the efficacy of morphine and other opioids for pain control. While glia have little-to-no role in pain under basal conditions, pain is amplified when glia become activated, inducing the release of proinflammatory products, especially proinflammatory cytokines. How glia are triggered to become activated is a key issue, and appears to involve a number of neuron-to-glia signals including neuronal chemokines, neurotransmitters, and substances released by damaged, dying and dead neurons. In addition, glia become increasingly activated in response to repeated administration of opioids. Products of activated glia increase neuronal excitability via numerous mechanisms, including direct receptor-mediated actions, upregulation of excitatory amino acid receptor function, downregulation of GABA receptor function, and so on. These downstream effects of glial activation amplify pain, suppress acute opioid analgesia, contribute to the apparent loss of opioid analgesia upon repeated opioid administration (tolerance), and contribute to the development of opioid dependence. The potential implications of such glial regulation of pain and opioid actions are vast, suggestive that targeting glia and their proinflammatory products may provide a novel and effective therapy for controlling clinical pain syndromes and increasing the clinical utility of analgesic drugs.
Keywords: Interleukin-1, microglia, astrocytes, neuropathic pain, morphine, methadone, analgesia, tolerance, dependence, withdrawal
I. Overview
Within just the past decade, there has been increasing recognition that glia are far more than simply “housekeepers” for neurons, providing neurochemical precursors and energy sources to neurons, regulating extracellular ion concentrations, removing debris, and so on. It is now known that glia also importantly contribute to fever, alterations in sleep, disruption of learning and memory, as well as to neuroinflammatory/neurodegenerative conditions such as ischemia/stroke, Alzheimer’s disease, and Parkinson’s disease.
Here the focus is on two recently recognized roles of glia: (a) creating and maintaining pain facilitation, and (b) compromising the efficacy of opioids for pain control. These pain-relevant topics are of interest primarily because clinical pain syndromes occur in epidemic proportions worldwide, including chronic pain states that last for years to a lifetime of unremitting pain. In these pain states, hot, cold and hard pressure pains are greatly amplified; warm, cool and light touch are now perceived as pain. Such pain is poorly controlled by currently available therapeutics, all of which were developed to target neurons. Classically, every model used to try to understand such pain focused exclusively on neurons (Figure 1). Glia had no role, as they were not thought of in terms of neural signaling. Despite many decades of research and drug development, pain facilitation remains unresolved. The discussion to follow will build the case from literature accruing over the past 15 years that glia are key players in pain facilitation and a worthy target for drug development for clinical pain control. Regarding the second, inter-related topic of this review, glial regulation of opioid effects is a new phenomenon that has begun to develop only within the past 5 years. These findings are intriguing as they shed light on why opioids are excellent at controlling acute pain but fail to control pain facilitation. The research also reveals that glia importantly contribute to the development of opioid tolerance, whereby opioids become progressively less effective in pain control, requiring progressively more opioid to suppress pain. Lastly, the newest wrinkle in this research will be described, that being the involvement of glia in the development of morphine dependence. As a consequence of repeated opioid use, animals become dependent on opioids such that when opioid dosing is stopped, animals (including humans) enter a state of opioid withdrawal. As will be described, glia are now also implicated in opioid dependence/withdrawal.
Figure 1.
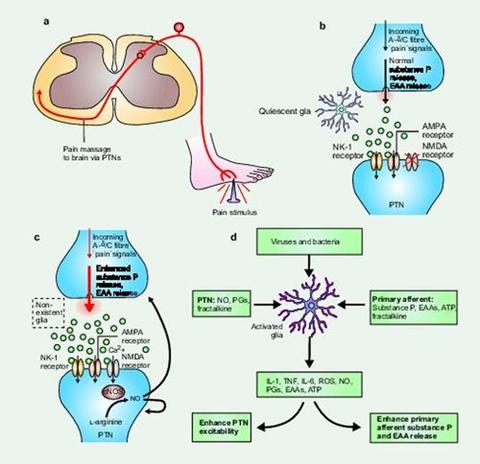
Classical and non-classical views of pain transmission and pain modulation. Panel (a). Classical pain transmission pathway. When a noxious (painful) stimulus is encountered, such as stepping on a nail as shown, peripheral “pain”-responsive A-delta and C nerve fibers are excited. These axons relay action potentials to the spinal cord dorsal horn. Here, neurotransmitters are released by the sensory neuron and these chemicals bind to and activate postsynaptic receptors on pain transmission neurons (PTNs) whose cell bodies reside in the dorsal horn. Axons of the PTNs then ascend to the brain, carrying information about the noxious event to higher centers. The synapse interconnecting the peripheral sensory neuron and the dorsal horn PTN is shown in detail in panels (b) and (c).Panel (b): Normal pain. Under basal conditions, pain is not modulated by glia. Under these circumstances, glia are quiescent, and thus not releasing pain modulatory levels of neuroexcitatory substances. Information about noxious stimuli arrives from the periphery along A-delta and C fibers, causing the release of substance P and excitatory amino acids (EAAs) in amounts appropriate to the intensity and duration of the initiating noxious stimulus. Activation of neurokinin-1 (NK-1) receptors by substance P and activation of AMPA receptors by EAAs cause transient depolarization of the PTNs, thereby generating action potentials that are relayed to brain. NMDA-linked channels are silent as they are chronically “plugged” by magnesium ions. Panel (c) Pain facilitation: classical view. In response to intense and/or prolonged barrages of incoming “pain” signals, the PTNs become sensitized and over-respond to subsequent incoming signals The intense and/or prolonged barrage depolarizes the PTNs such that the magnesium ions exit the NMDA-linked channel. The resultant influx of calcium ion activates constitutively expressed nitric oxide synthase (cNOS), causing conversion of L-arginine to nitric oxide (NO). Because it is a gas, NO rapidly diffuses out of the PTNs. This NO acts presynaptically to cause exaggerated release of substance P and EAAs. Postsynaptically, NO causes the PTNs to become hyperexcitable. Glia have not been considered to have a role in creating pain facilitation in this neuronally driven model. Panel (d): Pain facilitation: new view. Here, glial activation is conceptualized as a driving force for creating and maintaining pain facilitation. The role of glia is superimposed on the NMDA-NO-driven neuronal changes detailed in (c), so only the aspects added by including glia in the model are described here. Glia are activated (shown as hypertrophied relative to (b), as this reflects the remarkable anatomical changes that these cells undergo on activation) by three sources: bacteria and viruses which bind specific activation receptors expressed by microglia and astrocytes; substance P, EAAs, fractalkine, and ATP released by A-delta and/or C fiber presynaptic terminals (shown here) or by brain-to-spinal cord pain enhancement pathways (not shown); and NO, prostaglandins (PGs) and fractalkine released from PTNs. Following activation, microglia and astrocytes cause PTN hyperexcitability and the exaggerated release of substance P and EAAs from presynaptic terminals. These changes are created by the glial release of NO, EAAs, reactive oxygen species (ROS), PGs, proinflammatory cytokines (for example, IL1, IL6 or TNF), and nerve growth factor. Modified by Journal of Internal Medicine with permission, from Trends in Neuroscience.
Before proceeding, terminology relevant to this review will be briefly introduced. “Pain” refers to the unpleasant experience associated with actual or perceived tissue damage by a noxious (damaging) stimulus. Pain, as an experience, not only includes the perception of a simple sensory stimulus but also the cognitive analysis and emotional responses associated with that. Quantifying the pain experience in laboratory animals is arguably impossible. Given that, terminology was created that refers to the “lower order” quantifiable aspects of pain. “Nociception” (“noci” meaning “pain”) is used by the field to describe neural, physiological or behavioral responses to stimuli that humans would typically report as painful. Nociception is further broken down to reflect responding to noxious stimuli (pinch, for example) as if they are more intense than normal (hyperalgesia) versus responding to normally innocuous stimuli (light touch, for example) as if they were noxious (allodynia). For the sake of ease of communication in this general review, however, the term “pain” will refer to both animal and human responses and “pain facilitation” or “enhanced pain”, for example, will be used rather than more specialized terminology.
II. Glia
The work to be reviewed focuses on two types of glial cells, astrocytes and microglia. When these cells become activated, they (a) upregulate cell-type specific activation markers (allowing this reflection of activation to be easily visualized using immunohistochemistry) and (b) release a variety of substances (e.g., proinflammatory cytokines, chemokines, ATP, excitatory amino acids, nitric oxide, etc.) that enhance pain by increasing the excitability of nearby neurons. Microglia and astrocytes are not likely to be the only non-neuronal cells involved in pain enhancement. Endothelial cells, fibroblasts, oligodendroglia, and other cell types in both the spinal cord and overlying meninges can also produce many of the same neuroexcitatory substances as do astrocytes and microglia. However, study of the involvement of such cells has been hindered by the lack of their expression of upregulatable activation markers. As microglia and astrocytes have proven to be the cell types most amenable to study, virtually all studies to date have focused on them.
Microglia
Microglia constitute 5-12% of all cells in the CNS and 5-10% of all glia. Under basal conditions, microglia perform immune surveillance (Raivich, 2005). A shift to an activated state can occur rapidly as microglia are exquisite sensors of changes in their microenvironment. Stimuli that can activate microglia include trauma, infection, ischemia and neurodegeneration. Activation results in changes in morphology (e.g., retracted processes), proliferation, upregulated receptor expression (e.g., complement receptors, scavenger receptors), and changes in function (e.g., migration to sites of damage, phagocytosis, release of proinflammatory mediators). After resolution of challenge, microglia can either return to their basal state or may enter a “primed” state for prolonged periods of time (Perry et al., 1985). “Primed” microglia do not actively produce proinflammatory cytokines (and indeed may instead release anti-inflammatory cytokines) but now over-respond to new challenges, both in the speed and magnitude of release of proinflammatory products (Perry et al., 1985), similar in speed and magnitude to a secondary peripheral immune response. Microglial priming may prove to be important in explaining why some people greatly over-respond, in intensity and duration, to a pain-evoking event. Evidence to date from animal models suggests that prior microglial activation can leave these cells primed, such that a later pain-evoking stimulus produces both enhanced spinal proinflammatory cytokine production and enhanced pain (Spataro et al., 2006).
Astrocytes
Astrocytes constitute 40-50% of all glial cells (Aldskogius and Kozlova, 1998) and outnumber neurons (Lee et al., 2000). Astrocytes tightly enwrap the vast majority of synapses in the CNS and actively modulate neuron-to-neuron synaptic communication. The dynamic astrocyte regulation of synaptic communication has given rise to the term “tripartite synapse” to emphasize that astrocytes, in addition to the neuronal presynaptic and neuronal postsynaptic processes, are integral parts of neuronal signaling (Haydon and Carmignoto, 2006). Indeed, astrocytes are important contributors to synaptic “memory”, as prior synaptic activity leads, at later times, to greater astrocyte responses to subsequent synaptic input (Pasti et al., 2001). Beyond the synapse, astrocytes also are in intimate contact with neuronal cell bodies, dendrites, and nodes of Ranvier. They extend perivascular endfeet to contact capillaries and form a border layer in the meninges (Aldskogius and Kozlova, 1998). Under basal conditions, astrocytes provide neurons with energy sources and neurotransmitter precursors (Tsacopoulos, 2002), provide trophic support, regulate extracellular ions and neurotransmitters, and regulate neuronal survival and differentiation, neurite outgrowth, and formation of synapses (Perea and Araque, 2002). Astrocytes are activated by trauma, inflammation or infection, with progressive upregulation in glial fibrillary acidic protein (GFAP) expression, morphology and proliferation upon persistent activation (Aldskogius and Kozlova, 1998).
Microglia-Astrocyte Interactions
In vivo, astrocytes and microglia interact. Their released products can synergize, and substances released by one can activate the other. Regarding synergy, proinflammatory cytokines can synergize with each other as well as with neurotransmitters and neuromodulators, such as norepinephrine, prostaglandin E2 (PGE2), and nitric oxide (for review, see (Watkins et al., 2006)). Pain relevant examples include the synergy of TNF and IL-1 with ATP to enhance PGE2 release (Loredo and Benton, 1998); nitric oxide potentiating IL-1 induced PGE2 production and substance P release from sensory afferent terminals in spinal cord (Morioka et al., 2002); and substance P potentiating IL-1 induced release of IL-6 and PGE2 from human spinal cord astrocytes (Palma et al., 1997). Regarding cross-stimulation between glia: (a) astrocytes release substances that stimulate microglial activation, proliferation, and production of nitric oxide; and (b) microglia release substances that induce astrocyte activation, expression of adhesion molecules, functioning as antigen presenting cells, and release of glutamate, TNF, IL-1 and nitric oxide (for review, see (Watkins et al., 2006)). These microglia-astrocyte interactions are consistent with the developing pain literature that suggests that microglia are the first glial type to become activated and that their activation leads in turn to the recruitment of nearby astrocytes, such that both cell types can contribute to observed downstream alterations in pain (Ledeboer et al., 2005; Tawfik et al., 2006).
III. Glial dysregulation of pain
Under normal situations, when spinal glia are in their basal state, they have little-to-no influence on pain responsivity (Figure 1). Various drugs that suppress glial function have consistently failed to alter responsivity to heat or mechanical stimuli in normal animals (Hashizume et al., 2000; Ledeboer et al., 2005; Meller et al., 1994). While data from knockouts reveal reductions in basal pain responsivity in adult mice with disrupted IL-1 signaling, these changes appear related to developmental effects as the same effects occur when rats pups are treated only in utero with IL-1ra (Wolf et al., 2003). Thus, the weight of evidence supports that when astrocytes and microglia are not in their activated state, they do not appear to be important regulators of pain transmission. Having said this, basal cytokine levels are integral to maintaining neuronal plasticity.
When glia become activated, pain is now affected. One situation where spinal glia can become activated is in response to peripheral immune challenges that activate immune-to-brain communication (Dantzer, 2004; Maier and Watkins, 1998). Peripheral immune activated signaling to the CNS induces sickness responses. Work from our laboratory has documented that sickness responses include enhanced pain responsivity, in addition to classically recognized fever, increases in sleep, decreases in food and water intake, and generalized suppression of behavior (Dantzer, 2004; Maier and Watkins, 1998). Studies of brain-mediated sickness responses revealed that glial activation and consequent proinflammatory cytokine release are key steps in the generation of these survival-oriented changes in CNS function. These early studies predicted that sickness-induced pain enhancement would likewise be mediated by spinal glial activation and proinflammatory cytokines. This indeed turned out to be true (Watkins and Maier, 2000).
It is now known that glial regulation of pain extends well beyond enhanced pain responses that occur as a natural component of the sickness response. Both astrocytes and microglia in spinal cord are activated (as inferred from glial activation markers) in response to inflammation or damage to peripheral tissues, peripheral nerves, spinal nerves, or spinal cord (McMahon et al., 2005; Watkins and Maier, 2003). The generally accepted view has developed that microglia are activated first, followed by astrocytes (Tanga et al., 2004). While microglia were thought to fade, and astrocytes to increase, in prominence over time (Ledeboer et al., 2005; Raghavendra et al., 2003), a prolonged role for microglia has recently been proposed (Tawfik et al., 2006).
Regarding the mechanisms by which glial activation influences pain, work from our laboratory and many others have revealed that spinal glial activation and proinflammatory cytokines have been implicated in enhanced pain associated with every animal model examined to date (Table 1), with rare exception (Ledeboer et al., 2006). This conclusion is based on the facts that intrathecal (into the cerebrospinal fluid [CSF] space surrounding the spinal cord) injection of proinflammatory cytokines enhances pain (Falchi et al., 2001; Reeve et al., 2000; Tadano et al., 1999), and glial and cytokine inhibitors prevent and/or reverse such pain enhancements (McMahon et al., 2005; Watkins and Maier, 2003), as does knocking out IL-1 signaling (Honore et al., 2006; Wolf et al., 2006).
TABLE 1.
Summary of pain models with glial/cytokine involvement
| A. Glial activation spinal cord (microglia and/or astroglia): Pain models associated with upregulation of spinal cord microglial and/or astrocyte activation markers: |
|---|
| Complete Freund’s adjuvant, subcutaneous |
| Formalin, subcutaneous |
| Phospholipase A2, subcutaneous |
| Zymosan, subcutaneous |
| Sciatic nerve injury (chronic constriction injury) |
| Partial sciatic nerve ligation |
| Sciatic nerve inflammation with zymosan |
| Sciatic nerve inflammation with HIV-1 gp120 |
| Sciatic nerve inflammation with phospholipase A2 |
| Spinal nerve transection |
| Spinal nerve root injury |
| Spinal cord injury |
| Bone cancer |
| HIV-1 gp120, intrathecal |
| Chronic opioids; opioid withdrawal-induced hyperalgesia |
| B. Prevention or reversal of pain by inhibition of spinal glial activation or proinflammatory cytokine actions: | |
|---|---|
| Model | Intervention |
| Carrageenan, subcutaneous | minocycline, IL-1 knockout |
| Complete Freund’s adjuvant, subcutaneous | IL-1ra, IL-1 knockout |
| Formalin, subcutaneous | fluorocitrate, IL-1ra, minocycline; |
| IL-1 knockout | |
| Phospholipase A2, subcutaneous | fluorocitrate, IL-1ra, sTNFR |
| Zymosan, subcutaneous | fluorocitrate; |
| Sciatic nerve injury (chronic constriction injury) | IL-1ra, IL-10; IL-1 knockout |
| Sciatic nerve inflammation with zymosan | fluorocitrate, minocycline |
| IL-1ra, sTNFR, IL-6 antibody | |
| Sciatic nerve inflammation with phospholipase A2 | IL-1ra, anti-IL-6 , IL-10 |
| Sciatic nerve tetanic stimulation | fluorocitrate |
| Spinal nerve transection | propentofylline, minocycline, |
| IL-1ra, sTNFR, anti-IL-6, | |
| Spinal nerve root injury | methotrexate; IL-1 knockout |
| Spinal cord injury | IL-10, IL-1ra, minocycline |
| HIV-1 gp120, intrathecal | fluorocitrate, IL-1ra, sTNFR, |
| minocycline | |
| Dynorphin, intrathecal | IL-1ra, IL-10 |
| Fractalkine, intrathecal | minocycline, IL-1ra, anti-IL-6 |
Abbreviations: IL-1ra, interleukin-1 receptor antagonist; sTNFR, soluble TNF receptor; IL-10, interleukin-10.
Modified and updated from Ledeboer et al. (Ledeboer et al., 2006).
For complete references, see (Hains and Waxman, 2006; Honore et al., 2006; Ledeboer et al., 2006; Obata et al., 2006; Watkins et al., 2005)
Importantly, we have shown that ongoing inhibition of spinal cord proinflammatory cytokines can reverse established nerve injury-induced pain facilitation (i.e., neuropathic pain): (a) for 3+ months (Milligan et al., 2006), suggestive that other mechanisms will not reinstate pain facilitation in the absence of these cytokines, and (b) even after pain facilitation has been maintained continuously for 1-2 months (Milligan et al., 2006; Tawfik et al., 2006). Our observations provide strong support for the conclusion that glia and proinflammatory cytokines do not just induce pain facilitation but, rather, are key mediators of pain maintenance as well. This is especially important when considering the potential clinical utility of targeting proinflammatory cytokines for pain control as pain syndromes in people, such as neuropathic pain, will typically be long established and require long-term treatment. Indeed, in keeping with the recognition that the relative balance between pro- and anti-inflammatory cytokine levels may be key to the effects observed, increases in proinflammatory cytokines and/or decreases in anti-inflammatory cytokines have been found in spinal CSF samples from chronic pain patients diagnosed with complex regional pain syndrome, fibromyalgia and neuropathic pain (Alexander et al., 2005; Backonja et al., 2006).
IV. How do spinal glia “know” to become activated?
A key question is: “What is released in spinal cord, as a result of inflammation/trauma in the periphery that “tells” glia to activate?” Increasing focus is being directed at microglia as the “trigger”, as the initiator of pain facilitation. Microglia are exquisitely sensitive to signals of “not self” or “not normal”, leading them to be far more reactive in responding to CNS challenges than other candidate cell types (Kreutzberg, 1996). Similarly, spinal cord microglia are the earliest glial cell type activated in response to peripheral inflammation and injury, based both on the ability of the microglial inhibitor minocycline to prevent the onset of enhanced pain and the rapid upregulation of microglial activation markers relative to those of astrocytes (Ledeboer et al., 2005; Raghavendra et al., 2003). It appears that this initial microglial activation then recruits the activation of astrocytes (Tanga et al., 2004).
A number of putative neuron-to-glia signals have now been documented (Figure 2). These are reviewed below.
Figure 2.
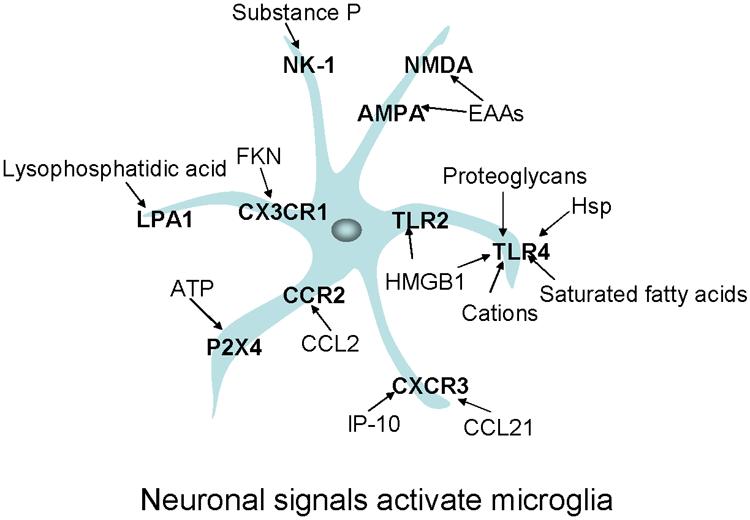
Summary of neuronal signals binding to microglial receptors leading to microglial activation and associated with pain facilitation. Neurons release these signals following specific stimulation or as a consequence of neuronal damage. As described in the main text, these receptors and signals have been associated with chronic pain states. Receptors are presented in bold. Neuronal signals bind chemokine receptors (Fractalkine [FKN] binds CX3CR1; CCL2 binds CCR2; IP-10 [interferon-inducible protein] and CCL21 [secondary lymphoid tissue chemokines] bind CXCR3); toll-like receptors (high mobility group box 1 [HMGB1] binds Toll Like Receptors 2 and 4 [TLR2 and TLR 4] and proteoglycans, heat shock proteins [HSPs], saturated fatty acids, and cations bind TLR4); and lysophosphatidic acid binds LPA1. Classical pain neurotransmitters are released by neurons and act on specific microglial receptors such as adenosine triphosphate (ATP) binding P2X4; Substance P binding NK-1; and excitatory amino acids [EAA] binding NMDA and AMPA receptors.
Neuronal chemokines
Chemokines are chemo attractant cytokines, consisting of a family of 50+ members. These chemokines are grouped into subfamilies, based on the number and positioning of amino acid(s) (X) inserted between cysteine (C) residues: CC, CXC, XC, and CXXXC (CX3C). While initially thought to be of immune origin, a number of these proteins can be produced and released by neurons as well (Stievano et al., 2004).
Fractalkine was the first chemokine discovered to be a neuron-to-glia signal. Fractalkine is also known as the chemokine ligand (L), CX3CL1. It is the only known member of this chemokine subfamily and the only known ligand of the fractalkine receptor (R), CX3CR1. This is an unusual profile for chemokines, as typically a single chemokine binds to multiple chemokine receptors and a single chemokine receptor binds to multiple chemokines (for excellent summary tables, see http://www.nature.com/bjp/journal/vgrac/ncurrent/full/0706562a.html). An additional unique feature of fractalkine is that it is tethered to the extracellular membrane of neurons. This attachment is cleaved upon strong neuronal activation, thereby forming a diffusible signaling molecule (Chapman et al., 2000). Our investigations of the distribution of fractalkine and its receptor in spinal cord revealed an intriguing pattern: fractalkine was expressed only by neurons and its receptor was expressed only by microglia (Verge et al., 2004). We observed this arrangement under both basal conditions and in response to either inflammation or trauma to the sciatic nerve (Verge et al., 2004). Intriguingly, the fractalkine receptor is upregulated in pain modulatory regions of spinal cord under neuropathic pain conditions (Lindia et al., 2005; Verge et al., 2004) and in response to ankle monoarthritis (Shan et al., 2006). Taken together, such anatomical data suggested that this chemokine might serve as a neuron-to-microglia signal, triggering microglial activation. The only known exceptions to this anatomical distribution are multiple sclerosis inflammatory lesions (Sunnemark et al., 2005) and spinal nerve trauma (Lindia et al., 2005) where novel astrocyte expression of fractalkine has been observed.
Upon intrathecal injection of fractalkine, we observed enhanced pain responsivity (Milligan et al., 2005; Milligan et al., 2004). We found that this pain enhancement by fractalkine is mediated via CX3CR1 as neutralizing antibodies against this receptor prevent pain enhancement by intrathecal fractalkine (Milligan et al., 2005). As predicted by microglial expression of CX3CR1 in spinal cord (Verge et al., 2004), our studies revealed that fractalkine enhances pain responsivity via microglial activation, as pain enhancement is blocked by the microglial inhibitor minocycline. Furthermore, fractalkine enhances pain via the release of spinal proinflammatory cytokines and nitric oxide (Milligan et al., 2005). Electrophysiological studies reveal that fractalkine causes hyper-responsivity of spinal neurons to brush and pinch, as well as increases in the numbers of neurons exhibiting prolonged afterdischarges indicative of spontaneous pain and central sensitization (Owolabi and Saab, 2006). Fractalkine binding to its receptor leads to downstream changes previously implicated in pain enhancement. For example, fractalkine binding leads to NFκB and p38 MAP kinase activation (Stievano et al., 2004) and production of proinflammatory cytokines and chemokines (Johnston et al., 2004; Stievano et al., 2004).
Importantly, endogenous fractalkine modulates pain as well. Blocking spinal fractalkine receptors delays and/or reverses neuropathic pain arising from sciatic nerve inflammation, sciatic nerve trauma, or ankle monoarthritis (Milligan et al., 2004; Shan et al., 2006), supporting the idea that endogenous fractalkine shed from neurons is importantly involved in the initiation and maintenance of neuropathic pain.
Beyond fractalkine, other neuronally derived chemokines may be neuron-to-glia signals. Two chemokine ligands of CXCR3 (CCL21/secondary lymphoid tissue chemokines; CXCL10/interferon-inducible protein of 10 kD [IP-10]) are rapidly induced in, and released from, damaged neurons (Biber et al., 2002; Rappert et al., 2004). Neurons not only produce CXCR3 ligands, but also package them in vesicles and transport them from the primary lesion site to distant presynaptic terminals. This allows for remote activation of glia, as occurs in spinal cord following peripheral nerve injury. Astrocytes and microglia each express CXCR3 mRNA and protein (Biber et al., 2002). In addition, stimulation of glia with CXCR3 agonists induce calcium and chloride transients as well as chemotaxis, indicating activation (Biber et al., 2002; Rappert et al., 2002). The importance of this communication pathway is supported by the fact that mice deficient for CXCR3 display no secondary activation of microglia in response to entorhinal cortex lesions. Whether CXCR3 agonists induce pain facilitation upon intrathecal injection has not yet been investigated.
A last neuronally derived chemokine that is intriguing with regards to pain-related neuron-to-glia signaling is CCL2, also known as monocyte chemoattractant protein-1 (MCP-1). CCL2/MCP-1 and its cognate receptor CCR2 are upregulated in damaged dorsal root ganglion neurons (White et al., 2005). While CCR2/MCP-1 is absent in normal CNS, levels rise following peripheral nerve injury as MCP-1 produced by injured neurons of the dorsal root ganglia is transported to sensory terminals in the spinal cord to form a releasable pool (Zhang and De Koninck, 2006). These sensory projections to the spinal cord appear to be the predominant source of CCL2/MCP-1 following peripheral nerve injury (Zhang and De Koninck, 2006). In addition, CCR2 is upregulated in spinal cord microglia and astrocytes following peripheral nerve injury, and these appear to be the only cell types expressing CCR2 in spinal cord (Abbadie et al., 2003; El-Hage et al., 2006). Neuronal CCR2/MCP-1 induction is followed by surrounding microglial activation, suggestive that CCR2/MCP-1 may serve as a trigger for spinal microglial activation (Zhang and De Koninck, 2006). Intrathecal injection of CCR2/MCP-1 produces pain facilitation (Tanaka et al., 2004) and intrathecal administration of neutralizing antibodies to CCR2/MCP-1 suppresses neuropathic pain (Abbadie et al., 2003). CCR2 knock out mice exhibit less activation of spinal cord astrocytes (as reflected by GFAP expression) and less activation of spinal cord microglia (as reflected by phospho-p38 MAP kinase expression) after peripheral nerve injury (Abbadie et al., 2003). In turn, CCR2 knock out mice exhibit greatly reduced central sensitization to inflammatory stimuli and are reported to fail to exhibit neuropathic pain (Abbadie et al., 2003).
Neurotransmitters
Sensory neurons relay messages of “pain” to the spinal cord using a variety of neurotransmitters, including substance P, excitatory amino acids, and ATP. As such, each may potentially be involved in glial activation. Spinal cord astrocytes express receptors for substance P and, upon stimulation with substance P, these astrocytes release PGE2 and IL-6 (Marriott et al., 1991). Spinal cord astrocytes also express metabotropic glutamate receptors, ionotropic non-NMDA receptors (AMPA and kainate) and NMDA receptors (Aicher et al., 1997; Besong et al., 2002). NMDA receptor activation leads to the activation of spinal microglia and release of both IL-1 and nitric oxide (Tikka and Koistinaho, 2001).
Of the neurotransmitters relaying “pain”, ATP has recently become the prime focus of studies exploring neuron-to-glia signaling. There is now strong evidence that the expression of a specific subset of spinal cord ATP receptors, termed P2X4, is normally low under basal conditions. Intriguingly, P2X4 receptor expression strongly upregulates in response to peripheral nerve injury, but only on microglia (Inoue, 2006). The timecourse of P2X4 upregulation mirrors the development of neuropathic pain. Suppression of P2X4 receptor signaling delays and reduces neuropathic pain (Inoue, 2006). Indeed, intrathecal injection of microglia activated by ATP in vitro proved sufficient to enhance pain (Tsuda et al., 2003). Similar to fractalkine, ATP activates microglial p38 MAP kinase and leads to the release of microglial proinflammatory cytokines (Inoue, 2006) and plasminogen, a protein which enhances NMDA receptor function.
In addition, under conditions that induce pain facilitation, neurons release neuromodulatory substances including nitric oxide and PGE2 and enhance the production of the neurotransmitter dynorphin. We have recently reported that neuronal nitric oxide enhances spinal production and release of TNF, IL-1 and IL-6 (Holguin et al., 2004), and PGE2 has been reported to be important in the initiation of both glial activation and neuropathic pain, but not in the maintenance of these effects (Takeda et al., 2005). Lastly, the pain enhancing effects of spinal dynorphin have recently been linked to selective activation of microglial p38 MAP kinase which, in turn, leads to the release of PGE2 and IL-1 (Laughlin et al., 2000; Svensson et al., 2005).
Substances released by damaged, dying and dead neurons
Substances released by neurons in response to their damage or death are likely relevant as neuron-to-glia signals, as degeneration of sensory axons occurs within the spinal cord after peripheral nerve injury, dorsal root ganglion trauma, or spinal nerve injury (Costigan et al., 1998). As the clearance of myelin and other cellular debris is a remarkably protracted process continuing for many years in the human CNS compared to the periphery (Vargas et al., 2006), this implies that substances released by injured neurons under such conditions may be able to provide ongoing stimulation to maintain equally protracted glial activation. In addition, there are reports of neurons in dorsal spinal cord dying secondary to peripheral nerve injury (Scholz et al., 2005). ATP is one major substance released by damaged and dying cells. As it is also released as a neurotransmitter in response for painful stimuli, its involvement in glial activation has already been reviewed, above. Other candidate substances, by and large, represent ligands for scavenging toll-like receptors (TLRs). The TLR family of receptors are expressed primarily by immune and immune-like cells that evolved to detect “danger signals” such as conserved motifs on bacteria and viruses, cell membrane and nuclear components released by damage, chaperone proteins (e.g., heat shock proteins; HSP) released by damage, etc. In response to nerve injury, TLR1, TLR2, and TLR4 have each been reported to be upregulated in CNS, linked to production of proinflammatory cytokines (e.g., TNF, IL-1) and chemokines (e.g., CCL2/MCP-1) (Owens et al., 2005).
In addition, lysophosphatidic acid (LPA) is a membrane phospholipid released in response to neuronal damage, which acts via the LPA(1) receptor rather than via TLRs. LPA may be an additional neuron-to-glia signal linked to pain enhancement (Inoue et al., 2004). LPA activates astrocytes, leading to the expression of IL-1, IL-6 and nerve growth factor, an additional glial product linked to pain facilitation (Watkins et al., 1997). Indeed, intrathecal LPA persistently enhances pain responses whereas intrathecal anti-sense oligodeoxynucleotides (ODN) to the LPA receptor inhibited peripheral nerve injury induced neuropathic pain (Inoue et al., 2004).
High Mobility Group Box 1 (HMGB1) is an example of a nuclear protein which, upon extracellular release by cell damage or death, is a potent inflammatory mediator (Harris and Andersson, 2004). Extracellular HMGB1 binds to and activates TLRs (Park et al., 2006). In keeping with its potential role as a neuron-to-glia signal, we have shown that HMGB1 enhances pain responsivity upon intrathecal administration (O’Connor et al., 2003).
The last candidate of this group is a TLR receptor (TLR4) rather than a ligand per se. TLR4 knockout mice and rats intrathecally administered TLR4 antisense ODN each display reduced: neuropathic pain, spinal microglial activation, and spinal proinflammatory cytokines. TLR4 is expressed only by microglia (Lehnardt et al., 2002) and is upregulated in microglia in response to spinal nerve injury correlated with enhanced pain (Tanga et al., 2004). While lipopolysaccharide (a component of bacterial cell walls) is well known as a TLR4 agonist, other candidate signals include saturated fatty acids (Hwang, 2001), ATP, heat shock proteins, and cations (Tanga et al., 2005). Thus, while the specific ligand(s) responsible for enhanced pain resulting from TLR4 activation has yet to be identified, the involvement of TLR4 remains intriguing nonetheless.
Taking this all together
A number of glial activators have been discussed above, based on studies of such mediators in isolation. It is unlikely that this accurately reflects what happens in vivo. In vivo, it makes little sense that any of these mediators act in isolation. Rather, it is far more likely that glia should detect patterns. Just as various proinflammatory substances can act in synergy to stimulate glia, so logically should putative neuron-to-glia signals. We expect that glia should be far more readily triggered to activate in response to a constellation of substances that, taken together, relay the message of peripheral nerve or tissue injury. Indeed, early evidence from our laboratory suggests this will be the case (Wieseler-Frank et al., 2004). The existence of multiple signals described above suggests that there could be patterns of signals to glia that indicate to glia the type of cell damaged, the nature of the damage, and the severity of damage.
V. How can glial activation affect neuronal activity?
It is remarkable that diverse enhanced pain states are all suppressed by inhibiting glial function (Watkins and Maier, 2003). However, this does not provide insight into what the glial products are that enhance pain. This issue is, in part, relatively simple and, in part, complicated. “Relatively simple” refers to situations in which pain enhancement can be clearly linked to the release of classical glial products, such as proinflammatory cytokines. “Relatively” simple must be kept in mind however as sensory neurons have been shown to produce and transport TNF to their presynaptic terminals in spinal cord after peripheral nerve injury (Shubayev and Myers, 2002). Whether TNF is released from these damaged axons into spinal cord is as yet unknown, but would appear reasonable to assume. Thus, the picture even for proinflammatory cytokines may not be as clear as it began. The “in part, complicated” refers to the fact that few substances are produced only or predominantly by glia. Neurons, like glia, can produce nitric oxide, ATP, prostaglandins, excitatory amino acids, and so on, blurring conclusions whether blockade of enhanced pain by antagonists/inhibitors to such mediators reflect actions on neurons or glia.
Thus, actions of proinflammatory cytokines on neurons will be described here, as it is the clearest example of how glial products can affect neuronal activity. This is not nearly as simple as merely binding of proinflammatory cytokines to known neuronal receptors (Dame and Juul, 2000; Holmes et al., 2004; Ohtori et al., 2004) so to induce rapid increases of neuronal excitability (Oka et al., 1994; Reeve et al., 2000; Samad et al., 2004). In addition, IL-1 has been shown, including in spinal dorsal horn, to enhance neuroexcitability via indirect actions; namely, via increasing calcium conductivity of neuronal NMDA receptors (Samad et al., 2004; Viviani et al., 2003). This occurs via intracellular signaling pathways leading to the phosphorylation of NMDA receptor subunits. TNF increases the conductivity of glutamatergic AMPA receptors as well (De et al., 2003) and increases spontaneous neurotransmitter release from presynaptic terminals (Grassi et al., 1994). TNF, and to a lesser extent, IL-1, upregulate the neuronal cell surface expression of both AMPA and NMDA receptors while downregulating cell surface expression of receptors for the inhibitory neurotransmitter, GABA; hence, creating an overall increase in neuronal excitatory tone (Stellwagen et al., 2005). Lastly, we have shown that IL-1 is implicated in neuropathy induced downregulation of G protein-coupled receptor kinase 2 (GRK2) expression in dorsal horn neurons, an action predicted to increase neuronal excitability by decreasing receptor desensitization (Kleibeuker et al., 2006).
Beyond these actions, proinflammatory cytokines also lead to the release of a host of neuroexcitatory substances, including more proinflammatory cytokines, nitric oxide, ATP, prostaglandins, nerve growth factors, reactive oxygen species, proinflammatory chemokines (e.g., CCL2/MCP-1, CXCL8/IL-8, CXCL10/IP-10) and BDNF (Inoue, 2006; John et al., 2005; Sperlagh et al., 2004; Watkins et al., 1999). They can also indirectly lead to elevations in extracellular glutamate levels via downregulation of glial and neuronal glutamate transporters that serve to keep extracellular glutamate levels low under normal conditions (Tawfik et al., 2006). Thus, taken together, proinflammatory cytokines exert multiple effects with the end result of neuroexcitation.
VI. Non-neuronal cells beyond glia: might they also contribute to pain enhancement?
Before leaving the topic of pain facilitation, it is worth noting that astrocytes and microglia may well not be the only non-neuronal cell types capable of enhancing pain in spinal cord. As noted above (Section II), other cell types in spinal cord can generate proinflammatory cytokines and other neuroexcitatory substances, such as fibroblasts, perivascular macrophages, dendritic cells, and endothelial cells. The potential involvement of such cells in pain facilitation has not yet been explored.
Intriguingly, immunocompetent cells exist outside the spinal cord parenchyma as well; that is, in the meninges that enwrap and protect the spinal cord, enclosing the CSF space. Meningeal tissues include many cell types capable of producing proinflammatory substances, including fibroblasts, dendritic cells, macrophages, endothelial cells, and mast cells (Wieseler-Frank et al., 2006). Indeed, we have now demonstrated that the spinal meninges elevate TNF, IL-1, IL-6 and iNOS gene expression and release TNF, IL-1 and IL-6 in response to immune activators such as HIV-1 gp120 administered intrathecally at a dose that enhances pain (Wieseler-Frank et al., 2006). As we have replicated these effects in vitro in the absence of circulating white blood cells, these proinflammatory responses are being generated by cells intrinsic to the meninges rather than by immune cells recruited from the blood (Wieseler-Frank et al., 2006). Intriguingly, similar upregulation of proinflammatory cytokines in the meninges occur following peripheral nerve injury, implying that the meninges are responsive not only to inflammatory mediators within the CSF space but also to signals released by damaged sensory afferents (Wieseler-Frank et al., 2006). Given both the density of sensory nerve fibers innervating the meninges (Strassman et al., 2004), whose only known function is to relay pain (Ray and Wolff, 1940), plus the intimate contact between the meninges and CSF space, these new findings suggest that the meninges may be an under-recognized contributor to spinal pain signaling. Consistent with this, inflammation of the arachnoid (arachnoiditis) induces back pain (Petty et al., 2000) as does meningitis (Jeha et al., 2003; Petty et al., 2000).
VII. Glial regulation of opioid analgesia, tolerance, and dependence/withdrawal
Responses of glia to chronic opioids: contribution to morphine tolerance and withdrawal-induced pain enhancement
A strong case has been made that the mechanisms underlying neuropathic pain and morphine tolerance are strikingly similar (Mayer et al., 1999). Given the strength of evidence implicating glia in pain facilitation, including neuropathic pain, it was natural to extend this question to whether glia were similarly involved in the production of morphine tolerance. As will be reviewed below, it is now clear that this is indeed the case (Watkins et al., 2005).
The first report linking glia to morphine tolerance demonstrated that chronic systemic morphine increased astrocyte activation in spinal cord (Song and Zhao, 2001). Importantly, co-administration of fluorocitrate (a glial metabolic inhibitor) with morphine significantly attenuated not only glial activation but also morphine tolerance (Song and Zhao, 2001). Hence this work associatively and causally linked the two phenomena.
Work that rapidly followed in our laboratory and others strongly supported and extended these initial observations. Chronic morphine was reported to: (a) activate microglia as well as astrocytes (Cui et al., 2006; Raghavendra et al., 2002; Tai et al., 2006), (b) upregulate TNF, IL-1 and IL-6 in spinal cord (Johnston et al., 2004; Raghavendra et al., 2002; Tai et al., 2006), (c) upregulate TNF, IL-1 and IL-6 in microglia but not neurons (Tai et al., 2006), and (d) progressively induce tolerance which was temporally correlated with increasing glial activation and proinflammatory cytokine production (Raghavendra et al., 2004). Importantly, morphine tolerance was slowed or reversed by either inhibition of spinal proinflammatory cytokines (Johnston et al., 2004; Raghavendra et al., 2002; Shavit et al., 2005) or by knocking out IL-1 signaling (Shavit et al., 2005). A link to glial p38 MAPK was reported by Cui et al. (Cui et al., 2006) who demonstrated that: (a) intrathecal administration of morphine for 7 days significantly increased the number of phospho-p38 MAPK positive cells in spinal cord, (b) this phospho-p38 MAPK expression was restricted to microglia, (c) a specific p38 inhibitor attenuated the development of tolerance to morphine analgesia. Lastly, a link has also recently been made to glial glutamate transporters. Chronic morphine regimens that induce tolerance have been demonstrated to downregulate spinal cord dorsal horn glial GLAST and GLT-1 glutamate transporters (the major transporters responsible for regulating extracellular levels of excitatory amino acids) and concomitantly upregulate extracellular excitatory amino acids (Mao et al., 2002; Tai et al., 2006). Such data suggest that tolerance may, in part, be created by an opposing increase in neuronal excitability due to increased ongoing stimulation by glutamate. Attenuation of morphine tolerance by amitriptyline was associated with upregulation of both GLAST and GLT-1 (and associated lowering of extracellular excitatory amino acids) as well as blockade of elevated proinflammatory cytokines (Tai et al., 2006). These seemingly independent events in fact may not be independent, as proinflammatory cytokines inhibit glutamate uptake by astrocytes and reduce glial GLAST and GLT-1 expression (Korn et al., 2005; Okada et al., 2005). Thus, it may be that morphine stimulates glial proinflammatory cytokines that, downstream, downregulate glial glutamate transporters, thereby enhancing neuronal excitability. This would be in addition to the various neuroexcitatory effects reviewed above (see Section V).
The work of Raghavendra et al. (Raghavendra et al., 2002; Raghavendra et al., 2004) and our laboratory (Johnston et al., 2004) also revealed a role of glia in pain enhancement that naturally occurs upon cessation of chronically administered opioids; that is, opioid withdrawal induced pain facilitation. These studies reported that withdrawal-induced pain enhancement is blocked by: (a) drugs or IL-10 (an anti-inflammatory cytokine) that block glial proinflammatory cytokine production, or (b) IL-1 receptor antagonist (Johnston et al., 2004; Raghavendra et al., 2002; Raghavendra et al., 2004).
Importantly, a link back to neuropathic pain has also been made. What is known from the clinical and laboratory animal studies is that morphine loses its efficacy in neuropathic pain patients and rats. That is, a state akin to tolerance prevails, despite the lack of prior opioid exposure; hence the patients experience naive tolerance rendering opioids ineffective. Raghavendra et al. (Raghavendra et al., 2002) demonstrated that glial activation and glial proinflammatory cytokine production by morphine was exaggerated in rats with neuropathic pain. Indeed, spinal inhibition of proinflammatory cytokines abolished the morphine resistance in neuropathic animals. That is, in the presence of proinflammatory cytokine inhibition, the analgesic efficacy of even acute morphine was restored in rats with neuropathic pain (Raghavendra et al., 2002) and therefore the detrimental effects of naive tolerance were ameliorated.
Responses of glia to acute opioids: opposition of acute opioid analgesia
While the research relating glia to morphine action began with a focus on morphine tolerance, it soon became apparent that glia modulate the acute analgesic effects of morphine as well (Watkins et al., 2005). We and others have shown that blockade of spinal proinflammatory cytokines increases the magnitude and duration of acute analgesia to morphine (Johnston et al., 2004; Shavit et al., 2005) and methadone (Hutchinson et al., 2006). Similarly, administration of a neutral dose of IL-1 (i.e., a low dose exerting no detectable effects on pain responsivity on its own) abolished, whereas knocking out IL-1 signaling potentiated and prolonged, morphine analgesia (Shavit et al., 2005). Intriguingly, administration of IL-1 receptor antagonist immediately upon apparent cessation of morphine analgesia rapidly “reinstates” analgesia suggesting that morphine-induced glial release of IL-1 “masks” ongoing morphine analgesia by exerting an opposing effect (Coats et al., 2006; Hutchinson et al., 2006; Shavit et al., 2005). Thus, IL-1 and potentially other proinflammatory cytokines, oppose the ability of acute morphine to suppress pain.
A parallel conclusion was reached using a very different approach, namely using sickness-induced pain facilitation to activate spinal cord glia prior to morphine challenge. Johnston et al. (Johnston and Westbrook, 2003;2005) demonstrated that spinal glial activation in response to a systemic injection of LPS on day 1 prevented systemic morphine analgesia on day 2, when no sickness induced pain facilitation remained. This effect was prevented by spinal administration of the glial inhibitor fluorocitrate prior to LPS (Johnston and Westbrook, 2005). The reverse case may prove true as well; that is, prior morphine may enhance proinflammatory cytokine production to a later dose of LPS. Data from microglial cell cultures clearly indicate that priming glia with prior 24 hr co-culture with morphine led to significantly increased release of TNF and superoxide upon LPS challenge (Chao et al., 1994).
While speculative, the above discussion suggests a solution to what has come to be called: “paradoxical” opioid-induced pain enhancement (King et al., 2005; Mao, 2002). The observation in humans as well as laboratory animals is that repetitive opioid exposure leads to the development of abnormal sensitivity to pain. Initial human and laboratory animal reports used repeated injections of opioid and hence it was argued that the development of “paradoxical” opioid-induced pain enhancement was the result of “mini-withdrawals” occurring between subsequent injections. However, that argument was put to rest in animal studies employing osmotic minipumps, for example, so to ensure invariant dosage of opioid across time. Even here, “paradoxical” opioid-induced pain enhancement began within a few hours of opioid initiation and developed over time (Mao, 2002; Vanderah et al., 2000). As reviewed by Mao, “collectively, these data indicate an important concept that prolonged opioid treatment not only results in a loss of opioid antinociceptive efficacy but also leads to activation of a pronociceptive system manifested as reduction of nociceptive thresholds.” To date, evidence points to involvement of dynorphin release and glutamatergic NMDA receptors (Mao, 2002). Given: (a) the progressive activation of glia with repeated opioid exposure (including with constant opioid exposure using osmotic minipumps), (b) the evidence that dynorphin and NMDA activate glia and induce the release of proinflammatory cytokines (Laughlin et al., 2000), exploring whether glial activation will solve the “paradox” of opioid-induced abnormal pain sensitivity would seem warranted for both theoretical and practical clinical reasons.
Responses of glia to repeated opioids: contribution to opioid dependence/withdrawal
Studies of glial involvement in morphine analgesic tolerance made one additional intriguing observation. That is, upon cessation of morphine, rats exhibited withdrawal-induced pain enhancement indicating that morphine dependence, as well as tolerance, had been established. This was not surprising, as dependence develops upon repetitive treatment with many drugs, including opioids. Dependence requires continued re-exposure to opioids to avoid a withdrawal syndrome that would otherwise occur when opioid dosing ceases. One sign of opioid withdrawal in rats, as well as people, is pain enhancement. What was important in the studies of Raghavendra et al. (Raghavendra et al., 2002; Raghavendra et al., 2004) and our laboratory (Johnston et al., 2004) was that rats that had either received glial modulatory drugs or intrathecal IL-10 to suppress proinflammatory cytokine production, or intrathecal IL-1 receptor antagonist throughout the tolerance/dependence regimen, showed no such withdrawal induced pain enhancement. Thus these data were the first to suggest that glia may be involved in opioid dependence/withdrawal in addition to simply tolerance to the pain suppressive actions of opioids.
To explore glial regulation of dependence/withdrawal, we gave rats repeated systemic administration of morphine to establish morphine dependence. Each morphine dose was co-administered with AV411, a blood-brain barrier permeable glial activation inhibitor (Ledeboer et al., 2006). After the last drug dose, withdrawal was precipitated by administering the opioid antagonist naloxone. Rats not receiving AV411 showed robust withdrawal signs over time (e.g., “wet dog” shakes, excessive grooming/penis licking, etc.) whereas AV411-treated rats did not (Lewis et al., 2006). Correlated with this, we found that this systemic morphine regimen upregulated astrocytic and microglial activation markers throughout the brain and spinal cord, whereas AV411 maintained glial activation at near basal levels in morphine-treated rats (Lewis et al., 2006). These data clearly suggest that glia, in addition to regulating pain facilitation, opioid analgesia and opioid tolerance, now must be considered as contributing to the phenomenon of morphine dependence/withdrawal as well. We would predict that inhibiting glial activation in response to opioids would allow opioids to be more effectively used for pain management. As fear of opioid dependence/withdrawal is cited as a major reason for underprescribing opioids by physicians, under-medicating with opioid by nurses, and avoidance of opioids by patients (Portenoy, 1994), preventing glia from driving dependence/withdrawal would be predicted to have a major positive impact on healthcare.
Non-classical effects of opioids in activating glia
A point worth noting here is that it has been assumed that morphine activates glia via classical opioid receptors, in every journal publication cited above, including our own. We have recently begun questioning whether this is in fact true for opioid-induced glial activation. Our strategy has been based on Hutchinson and colleagues’ studies demonstrating that opioids could markedly affect the function of peripheral immune cells via non-classical opioid receptors (Hutchinson and Somogyi, 2004; 2004;2005). The key point here is that classical opioid receptors, which are the only known opioid receptors on neurons, are stereoselective. That is, [-]-isomers are active (e.g., they bind the receptor and activate it) whereas [+]-isomers have greatly reduced affinity and are not. Thus [-]-methadone produces pain suppression while [+]-methadone is without effect. What is intriguing in the studies of Hutchinson et al. is that both the [+] and the [-]-isomers of opioids were equally effective in altering the function of peripheral immune cells both in vitro and in vivo, clearly documenting that a receptor other than the classical opioid receptor must, by definition, be responsible for such effects (Hutchinson and Somogyi, 2004; 2004; 2005). A structure activity relationship was also conducted using over 20 different opioid compounds & it demonstrated that the effect on peripheral immune cells did not occur in a manner that resembled classical opioid characteristics.
Extending these observations to glia, we compared the effects of chronic intrathecal [-]-morphine and [-]-methadone (both active at classical opioid receptors) to that of [+]-methadone (inactive at classical opioid receptors) and saline. The outcome measure was dorsal spinal cord IL-1 gene expression. The experimental outcome was that “inactive” [+]-methadone was equally effective as [-]-morphine and [-]-methadone in increasing IL-1 mRNA compared to saline (Coats et al., 2006). This suggests that different receptors mediate the (classical) pain suppressive effects of morphine than its (non-classical) pain enhancing effects. If this proves to be true, the clinical implications are enormous. This is because one should then be able to separate these phenomena and in doing so enhance the ability of morphine to suppress pain, reduce the development of morphine tolerance, and reduce the development of opioid dependence/withdrawal. This predicts that such clinically relevant outcomes should be attainable either by (a) modifying the physical structure of opioids to not obscure the classical opioid binding site while preventing opioid binding to the (as yet unknown) non-stereoselective receptor or (b) co-administering an [+]-opioid antagonist which blocks glial activation while leaving neuronal activation by opioids unaltered. To date we have explored the second option with exciting success. We have found that, indeed, [+]-naloxone potentiates the magnitude and duration of opioid analgesia. In addition, upon chronic co-administration with morphine in a morphine dependence paradigm, [+]-naloxone delays the development of morphine tolerance, and decreases [-]-naloxone-precipitated withdrawal behaviors (Coats et al., 2006). Thus early indications are that this may well be a clinically relevant strategy for increasing opioid analgesia while decreasing the negative consequences of repeated opioids.
VIII. How can glial activation affect neuronal responses to opioids?
This issue is obviously related to the parallel discussion in Section V, above. As we have shown that proinflammatory cytokines are also released in response to morphine (Johnston et al., 2004) the reader is referred back to the prior section for review of issues that are equally pertinent here. Beyond what has already been discussed, there are additional mechanisms that may come into play when exploring how glial activation may suppress responsivity to morphine.
The first is that opioids can induce the release of chemokines, as well as cytokines. For example, morphine induces the release of CCL1/MCP-1, CCL5/RANTES and CCL12/SDF-1 (El-Hage et al., 2006). Such chemokines release in spinal cord would be predicted to enhance pain (Abbadie, 2005).
An intriguing line of investigation has emerged indicating that chemokines can alter the actions of opioids via a mechanism quite distinct from those discussed for proinflammatory cytokines, above. This mechanism involves inactivation of opioid receptor function via heterologous desensitization. Chemokines, via binding to their cognate receptors (at least CCR1, CCR5 and CXCR4) on a cell can lead, via intracellular signaling cascades, to the desensitization/inactivation of opioid receptors expressed on the same cell (Adler et al., 2006; Zhang et al., 2004). Some forms of chemokine/opioid heterologous desensitization can be bidirectional, where opioids can desensitize chemokines receptors as well as the reverse. But there appear to be a range of possibilities including unidirectional, where only chemokines desensitize opioid receptors and not the reverse (Adler et al., 2006). While multiple chemokines can desensitize opioid receptors, it is not universal as CCL2/MCP-1 has no such actions (Adler et al., 2006).
While first discovered in studies of chemokines/opioid interactions on peripheral immune cells, the concept has proven valid for chemokines/opioid interactions on neurons as well (Adler et al., 2006). When rats were administered CXC12/SDF1 or CCL5/RANTES into the periaqueductal gray (a major site where morphine produces pain suppression) 30 minutes prior to morphine or DAMGO (a mu-opioid receptor agonist), analgesia was suppressed (Szabo et al., 2001). As AMD3100, a CXCL12 receptor antagonist, abolished the desensitization, allowing for full opioid analgesia, this confirmed that the effects observed were mediated through that chemokine receptor (Adler et al., 2006). Such effects are not restricted to mu opioid receptors but rather extends equally to delta and kappa opioid receptors as well (Adler et al., 2006).
IX. Implications for clinical pain control
The potential implications of glial involvement in the dysregulation of pain and opioid actions are vast. Regarding pain, the fundamental, inescapable fact is that present treatments for chronic pain fail. The drugs currently used to treat clinical pain problems were all developed to target neurons, prior to the discovery that glia regulate pain. Present knowledge of glial regulation of pain derives from many different laboratories using many different animal and in vitro models, and equally as many different endpoints. Such a broad consensus cannot be ignored and demands that novel therapies be examined in clinical trials to determine whether glially driven pain occurs in humans as it does in laboratory animals (Watkins and Maier, 2003). While it is difficult to change the status quo, when the status quo leaves so many pain patients in misery, it is time to seek change.
Beyond pain facilitation are the new data reviewed here, showing that glia compromise the efficacy of opioids. How broadly this impacts analgesic drugs is currently unknown, but clearly extends beyond morphine as we have observed the same effects with methadone as well (Coats et al., 2006). The fact that non-classical opioid receptors appear to mediate the glial activating effects of opioids suggests that the clinical efficacy of morphine and methadone, at minimum, can be improved in magnitude and duration by separating the glial activating effects of opioids from their pain suppressive ones. The data suggest that chemical modification of opioids may be able to retain the portions of the molecules that bind to neuronal receptors, while avoiding binding to the (as yet unknown) non-stereoselective receptor(s) that activate glia. Alternatively, one can envision the day when a glial activation inhibitor would be co-administered with every dose of opioid so as to enhance analgesia, delay the development of tolerance, and delay the development of opioid dependence/withdrawal.
Such interventions are needed, not only because clinical pain syndromes are so poorly controlled by drugs such as opioids, but also because present treatments of opioid dependence/withdrawal fail as well, with extremely high rates of return to drug use (60-80%) (Ball and Ross, 1991; Charles et al., 2003). Dependence is an expected but unwanted consequence of opioid use, and withdrawal from opioid use is highly aversive both physically and motivationally (Savage, 1999). Dependence can occur rapidly as withdrawal syndromes can occur in humans after even brief opioid use (∼2 days) (Sees and Clark, 1993). This compels humans and other organisms to seek or use opioid for relief or avoidance of withdrawal symptoms (Savage, 1999). The aversive physical and motivational changes that comprise the withdrawal syndrome are significant factors deterring drug-dependent humans from going through rehabilitation (Stewart et al., 1984). If preventing glial activation can help avoid the development of opioid dependence/withdrawal or could ease withdrawal from ongoing opioid dependencies, then one would predict that opioid use for pain control would be far safer for prolonged pain treatments.
FIGURE 3.
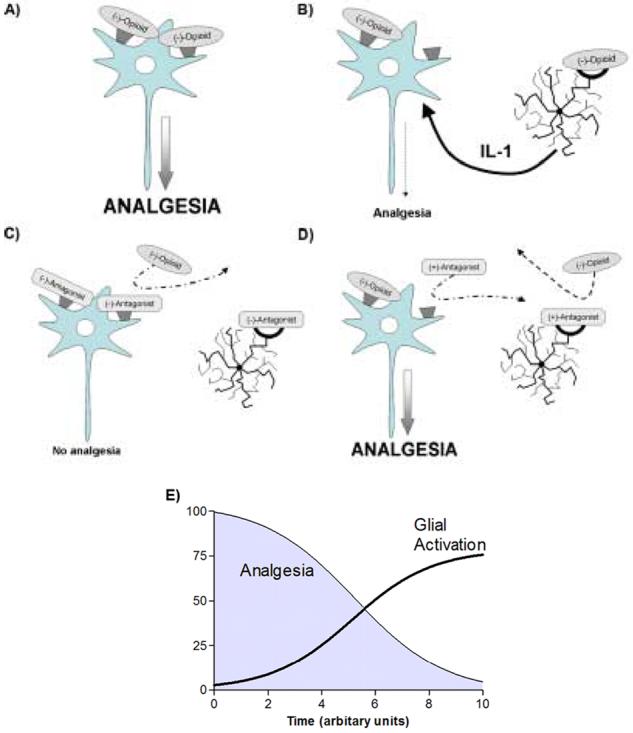
A: The classical view of opioid analgesia proposes (-)-opioid agonist isomers stereoselectively bind to classical opioid receptors producing an inhibitory influence on nociceptive signal transmission.
B: A growing body of literature suggests that the classical view of analgesia ignores an important nociceptive modulatory influence driven by opioid induced glial activation resulting from opioid agonists binding to glial opioid binding receptors which in turn increase proinflammatory cytokine (such as interleukin-1) expression & release leading to a decrease in opioid efficacy at the neuronal component.
C: (-)-Opioid antagonists bind to both the neuronal & glial components resulting in blockade of any potential opioid analgesia & glial activation.
D: Due to the stereoselectivity of neuronal opioid receptors only the (-)-isomer of opioid agonists & antagonists are able to bind. Therefore, when a combination of an opioid (+)-antagonist & an opioid (-)-agonist are introduced to this system, the (+)-antagonist is unable to bind to the stereoselective neuronal opioid receptor, but is able to block the non-stereoselective glial site. The opioid (-)-agonist can act freely at the neuronal opioid receptor, but is unable to bind to the glial site due to its blockade by the (+)-antagonist. Therefore, this situation produces opioid receptor mediated analgesia without the opposing force of opioid induced glial activation, thereby potentiating opioid analgesia.
E: Based on the above hypothesized series of events this schematic diagram demonstrates how analgesia decreases over time as a direct result of increased glial activation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Abbadie C. Chemokines, chemokine receptors and pain. Trends Immunol. 2005;26:529–34. doi: 10.1016/j.it.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–52. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler MW, Geller EB, Chen X, Rogers TJ. Viewing chemokines as a third major system of communication in the brain. Aaps J. 2006;7:E865–70. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicher SA, Sharma S, Cheng PY, Pickel VM. The N-methyl-D-aspartate (NMDA) receptor is postsynaptic to substance P-containing axon terminals in the rat superficial dorsal horn. Brain Res. 1997;772:71–81. doi: 10.1016/s0006-8993(97)00637-9. [DOI] [PubMed] [Google Scholar]
- Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interations following axon injury. Prog. Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain. 2005;116:213–9. doi: 10.1016/j.pain.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Backonja M, Muller D, Coe C. Neuropathic pain and levels of cytokines and IL-10 in the blood and cerebrospinal fluid; Proc. of the 11th World Congress on Pain; 2006.pp. 1647–P150. [Google Scholar]
- Ball JC, Ross A. The Effectiveness of Methadone Maintenance Treatment: Patients, Programs, Services & Outcomes. Springer-Verlag; J. C. Ball and A. Ross. New York: 1991. Follow-up study of 105 patients who left treatment. [Google Scholar]
- Besong G, Battaglia G, D’Onofrio M, Di Marco R, Ngomba RT, Storto M, Castiglione M, Mangano K, Busceti CL, Nicoletti FR, Bacon K, Tusche M, Valenti O, Conn PJ, Bruno V, Nicoletti F. Activation of group III metabotropic glutamate receptors inhibits the production of RANTES in glial cell cultures. J Neurosci. 2002;22:5403–11. doi: 10.1523/JNEUROSCI.22-13-05403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Dijkstra I, Trebst C, De Groot CJ, Ransohoff RM, Boddeke HW. Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience. 2002;112:487–97. doi: 10.1016/s0306-4522(02)00114-8. [DOI] [PubMed] [Google Scholar]
- Chao CC, Gekker G, Sheng WS, Hu S, Tsang M, Peterson PK. Priming effect of morphine on the production of tumor necrosis factor-alpha by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther. 1994;269:198–203. [PubMed] [Google Scholar]
- Chapman G, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJLM. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 2000;20:RC87–1. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles BK, Day JE, Rollins DE, Andrenyak D, Ling W, Wilkins DG. Opiate recidivism in a drug-treatment program: comparison of hair and urine data. J Anal Toxicol. 2003;27:412–28. doi: 10.1093/jat/27.7.412. [DOI] [PubMed] [Google Scholar]
- Coats BD, Hutchinson MR, Watkins LR. Opioid inactive (+)-naloxone potentiates morphine analgesia: further evidence that opioid analgesia is opposed by opioid induced glial activation. Proc. Soc. Neurosci. 2006 in press. [Google Scholar]
- Costigan M, Mannion RJ, Kendall G, Lewis SE, Campagna JA, Coggeshall RE, Meridith-Middleton J, Tate S, Woolf CJ. Heat shock protein 27: developmental regulation and expression after peripheral nerve injury. J Neurosci. 1998;18:5891–900. doi: 10.1523/JNEUROSCI.18-15-05891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–43. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Dame JB, Juul SE. The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev. 2000;58:25–39. doi: 10.1016/s0378-3782(00)00064-5. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur. J. Pharmacol. 2004;500:399–411. doi: 10.1016/j.ejphar.2004.07.040. [DOI] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–142. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Ambati J, Bruce-Keller AJ, Knapp PE, Hauser KF. CCR2 mediates increases in glial activation caused by exposure to HIV-1 Tat and opiates. J Neuroimmunol. 2006 doi: 10.1016/j.jneuroim.2006.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchi M, Ferrara F, Gharib C, Dib B. Hyperalgesic effect of intrathecally administered interleukin-1 in rats. Drugs Exp Clin Res. 2001;27:97–101. [PubMed] [Google Scholar]
- Grassi F, Mileo AM, Monaco L, Punturieri A, Santoni A, Eusebi F. TNF-alpha increases the frequency of spontaneous miniature synaptic currents in cultured rat hippocampal neurons. Brain Res. 1994;659:226–30. doi: 10.1016/0006-8993(94)90883-4. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–17. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris HE, Andersson U. The nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 2004;34:1503–1512. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- Hashizume H, Rutkowski MD, Weinstein JN, DeLeo JA. Central administration of methotrexate reduces mechanical allodynia in an animal model of radiculopathy/sciatica. Pain. 2000;87:159–69. doi: 10.1016/S0304-3959(00)00281-5. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–31. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Holguin A, Biedenkapp J, Campisi J, Wieseler-Frank J, O’Connor KA, Milligan ED, Maksimova E, Bravmann C, Hansen MK, Martin D, Fleshner M, Maier SF, Watkins LR. HIV-1 gp120 stimulates proinflammatory cytokine-mediated pathological pain via activation of nitric oxide synthase-I (nNOS) Pain. 2004;110:517–530. doi: 10.1016/j.pain.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Holmes GM, Hebert SL, Rogers RC, Hermann GE. Immunocytochemical localization of TNF type 1 and type 2 receptors in the rat spinal cord. Brain Res. 2004;1025:210–9. doi: 10.1016/j.brainres.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Honore P, Wade CL, Zhong C, Harris RR, Wu C, Ghayur T, Iwakura Y, Decker MW, Faltynek C, Sullivan J, Jarvis MF. Interleukin-1 alphabeta gene-deficient mice show reduced nociceptive sensitivity in models of inflammatory and neuropathic pain but not post-operative pain. Behav Brain Res. 2006;167:355–64. doi: 10.1016/j.bbr.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Maier SF, Watkins LR. Spinal proinflammatory cytokines oppose acute opioid analgesia following intrathecal and subcutaneous opioid administration. Proc. Soc. Neurosci. 2006 in pres. [Google Scholar]
- Hutchinson MR, Milligan ED, Jekich BM, Coats BD, Lewis S, Maier SF, Watkins LR. Elevations of spinal proinflammatory cytokines are mediated via non-classical opioid mechanisms. Proc. Soc. for NeuroImmune Pharmacology. 2006 [Google Scholar]
- Hutchinson MR, Somogyi AA. Relationship between 4,5-epoxymorphinan structure and in vitro modulation of cell proliferation. Eur J Pharmacol. 2004;494:251–62. doi: 10.1016/j.ejphar.2004.04.049. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Somogyi AA. (S)-(+)-methadone is more immunosuppressive than the potent analgesic (R)-(--)-methadone. Int Immunopharmacol. 2004;4:1525–30. doi: 10.1016/j.intimp.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Somogyi AA. Characterisation of the in vitro modulation of splenocyte proliferation by non-4,5-epoxymorphinan opioids. Int Immunopharmacol. 2005;5:1713–22. doi: 10.1016/j.intimp.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Hwang D. Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways. Faseb J. 2001;15:2556–64. doi: 10.1096/fj.01-0432com. [DOI] [PubMed] [Google Scholar]
- Inoue K. The function of microglia through purinergic receptors: Neuropathic pain and cytokine release. Pharmacol Ther. 2006;109:210–26. doi: 10.1016/j.pharmthera.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–8. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Jeha LE, Sila CA, Lederman RJ, Prayson RA, Isada CM, Gordon SM. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61:55–9. doi: 10.1212/01.wnl.0000073617.08185.0a. [DOI] [PubMed] [Google Scholar]
- John GR, Lee SC, Song X, Rivieccio M, Brosnan CF. IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia. 2005;49:161–76. doi: 10.1002/glia.20109. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–65. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Westbrook RF. Acute and conditioned sickness reduces morphine analgesia. Behav Brain Res. 2003;142:89–97. doi: 10.1016/s0166-4328(02)00398-4. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Westbrook RF. Inhibition of morphine analgesia by LPS: role of opioid and NMDA receptors and spinal glia. Behav Brain Res. 2005;156:75–83. doi: 10.1016/j.bbr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- Kleibeuker W, Ledeboer A, Watkins LR, Maier SF, Zijlstra J, Heijnen CJ, Kavelaars A. Neuronal G protein-coupled receptor kinase 2 is downregulated in the rat spinal during chronic constriction injury. J. Neurochemistry in review. 2006 [Google Scholar]
- Korn T, Magnus T, Jung S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: a mechanism mediated by tumor necrosis factor-alpha. Faseb J. 2005;19:1878–80. doi: 10.1096/fj.05-3748fje. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Laughlin TM, Bethea JR, Yezierski RP, Wilcox GL. Cytokine involvement in dynorphin-induced allodynia. Pain. 2000;84:159–67. doi: 10.1016/s0304-3959(99)00195-5. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Liu T, Shumilla JA, Mahoney JH, Vargas JA, Sultzbaugh L, Sanftner LM, Watkins LR, Johnson KW. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Pain in review. 2006 doi: 10.1017/S1740925X0700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Mahoney JH, Milligan ED, Martin D, Maier SF, Watkins LR. Spinal cord glia and interleukin-1 do not appear to mediate persistent allodynia induced by intramuscular acidic saline in rats. J. Pain. 2006 doi: 10.1016/j.jpain.2006.04.001. in press. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS. Gliogenesis in the central nervous system. Glia. 2000;30:105–121. doi: 10.1002/(sici)1098-1136(200004)30:2<105::aid-glia1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–86. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Coats BD, Brzeski AL, Maier SF, Watkins LR, Johnson KW. AV411, a blood brain barrier permeable glial activation inhibitor, reduces morphine withdrawal behaviors in rats. Proc. Soc. Neurosci. 2006 in press. [Google Scholar]
- Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 2005;6:434–8. doi: 10.1016/j.jpain.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Loredo GA, Benton HP. ATP and UTP activate calcium-moblizing P2U-like receptors and act synergistically with interleukin-1 to stimulate prostaglandin E2 release from human rheumatoid synovial cells. Arthritis Rheum. 1998;41:246–255. doi: 10.1002/1529-0131(199802)41:2<246::AID-ART8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev. 1998;105:83–107. doi: 10.1037/0033-295x.105.1.83. [DOI] [PubMed] [Google Scholar]
- Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100:213–7. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci. 2002;22:8312–23. doi: 10.1523/JNEUROSCI.22-18-08312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott DR, Wilkin GP, Wood JN. Substance P-induced release of prostaglandins from astrocytes: regional specialisation and correlation with phosphoinositol metabolism. J Neurochem. 1991;56:259–65. doi: 10.1111/j.1471-4159.1991.tb02590.x. [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A. 1999;96:7731–6. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192:444–62. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dyskstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, Martin D, Maier SF, Watkins LR. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci. 2005;22:2775–82. doi: 10.1111/j.1460-9568.2005.04470.x. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Langer SJ, Hughes TS, Jekich BM, Frank MG, Mahoney JH, Levkoff LH, Maier SF, Cruz PE, Flotte TR, Johnson KW, Mahoney MM, Chavez RA, Leinwand LA, Watkins LR. Repeated intrathecal injections of plasmid DNA encoding interleukin-10 produce prolonged reversal of neuropathic pain. Pain. 2006 doi: 10.1016/j.pain.2006.07.009. in press. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Morioka N, Inoue A, Hanada T, Kumagai K, Takeda K, Ikoma K, Hide I, Tamura Y, Shiomi H, Dohi T, Nakata Y. Nitric oxide synergistically potentiates interleukin-1 beta-induced increase of cyclooxygenase-2 mRNA levels, resulting in the facilitation of substance P release from primary afferent neurons: involvement of cGMP-independent mechanisms. Neuropharmacology. 2002;43:868–76. doi: 10.1016/s0028-3908(02)00143-0. [DOI] [PubMed] [Google Scholar]
- O’Connor KA, Hansen MK, Rachal Pugh C, Deak MM, Biedenkapp JC, Milligan ED, Johnson JD, Wang H, Maier SF, Tracey KJ, Watkins LR. Further characterization of high mobility group box 1 (HMGB1) as a proinflammatory cytokine: central nervous system effects. Cytokine. 2003;24:254–65. doi: 10.1016/j.cyto.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. 2006. Spinal glial activation contributes to post-operative mechanical hypersensitivity in the rat. in press. [DOI] [PubMed]
- Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine. 2004;29:1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- Oka T, Aou S, Hori T. Intracerebroventricular injection of interleukin-1β enhances nociceptive neuronal responses of the trigeminal nucleus caudalis in rats. Brain Research. 1994;656:236–244. doi: 10.1016/0006-8993(94)91466-4. [DOI] [PubMed] [Google Scholar]
- Okada K, Yamashita U, Tsujis S. Modulation of Na+-dependent glutamate transporter of murine astocytes by inflammatory mediators. J. UOEH. 2005;27:161–170. doi: 10.7888/juoeh.27.161. [DOI] [PubMed] [Google Scholar]
- Owens T, Babcock AA, Millward JM, Toft-Hansen H. Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Brain Res Rev. 2005;48:178–84. doi: 10.1016/j.brainresrev.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Owolabi SA, Saab CY. Fractalkine and minocycline alter neuronal acivity in the spinal cord dorsal horn. FEBS Letters. 2006 doi: 10.1016/j.febslet.2006.06.087. in press. [DOI] [PubMed] [Google Scholar]
- Palma C, Minghetti L, Atolfi M, Ambrosini E, Silberstein FC, manzini S, Levi G, Aloisi F. Functional characterization of substance P receptors on cultured human spinal cord astrocytes: synergism of substance P with cytokines in inducing interleukin-6 and prostaglandin E2 production. Glia. 1997;21:183–193. doi: 10.1002/(sici)1098-1136(199710)21:2<183::aid-glia2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- Pasti L, Zonata M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J. Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Communication between astrocytes and neurons: a complex language. J. Physiol. 2002;96:199–207. doi: 10.1016/s0928-4257(02)00007-4. [DOI] [PubMed] [Google Scholar]
- Perry VH, Hume DA, Gordon S. Immunohistochemical localisation of macrophages and microglia in the adult and developing mouse brain. Neurosci. 1985;15:313–326. doi: 10.1016/0306-4522(85)90215-5. [DOI] [PubMed] [Google Scholar]
- Petty PG, Hudgson P, Hare WS. Symptomatic lumbar spinal arachnoiditis: fact or fallacy? J Clin Neurosci. 2000;7:395–9. doi: 10.1054/jocn.1999.0223. [DOI] [PubMed] [Google Scholar]
- Portenoy RK. Tolerance to opioid analgesics: clinical aspects. Cancer Surv. 1994;21:49–65. [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J. Pharmacol. Exp. Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–34. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–3. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500–9. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HW, Kettenmann H. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol. 2002;168:3221–6. doi: 10.4049/jimmunol.168.7.3221. [DOI] [PubMed] [Google Scholar]
- Ray BS, Wolff HG. Experimental studies on headache: pain-sensitive structures of the head and their significance in headache. Archives of Surgery. 1940;41:813–856. [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur. J. Pain. 2000;4:247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- Samad TA, Wang H, Broom DC, Woolf CJ. Central neuroimmune interactions after peripheral inflammation: interleukin-1b potentiates synaptic transmission in the spinal cord. Proc. Soc. Neurosci. 2004;34:511–7. [Google Scholar]
- Savage SR. Opioid therapy of chronic pain: assessment of consequences. Acta Anaesthesiol Scand. 1999;43:909–17. doi: 10.1034/j.1399-6576.1999.430908.x. [DOI] [PubMed] [Google Scholar]
- Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J Neurosci. 2005;25:7317–23. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sees KL, Clark HW. Opioid use in the treatment of chronic pain: assessment of addiction. J Pain Symptom Manage. 1993;8:257–64. doi: 10.1016/0885-3924(93)90154-n. [DOI] [PubMed] [Google Scholar]
- Shan S, Hong C, Mei H, Ting-Ting L, Hai-Li P, Yu-Qiu Z. 2006. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glia.
- Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–9. doi: 10.1016/j.pain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR. Anterograde TNF alpha transport from rat dorsal root ganglion to spinal cord and injured sciatic nerve. Neurosci Lett. 2002;320:99–101. doi: 10.1016/s0304-3940(02)00010-1. [DOI] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–6. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Spataro LE, Young J, Wieseler-Frank J, Milligan ED, Jekich BM, Springer DB, Maier S, Fleshner M, Watkins LR. Abdominary surgery primes interleukin-1 and pain responses to low-dose lipopolysaccharide (LPS) and cyclophospamide (CP) Proc. Soc. Neurosci. 2006 in press. [Google Scholar]
- Sperlagh B, Baranyi M, Hasko G, Vizi ES. Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J. Neuroimmunol. 2004;15:33–39. doi: 10.1016/j.jneuroim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J, de Wit H, Eikelboom R. Role of unconditioned and conditioned drug effects in the self-administration of opiates and stimulants. Psychol Rev. 1984;91:251–68. [PubMed] [Google Scholar]
- Stievano L, Piovan E, Amadori A. C and CX3C chemokines: cell sources and physiopathological implications. Crit Rev Immunol. 2004;24:205–28. doi: 10.1615/critrevimmunol.v24.i3.40. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Weissner W, Williams M, Ali S, Levy D. Axon diameters and intradural trajectories of the dural innervation in the rat. J Comp Neurol. 2004;473:364–76. doi: 10.1002/cne.20106. [DOI] [PubMed] [Google Scholar]
- Sunnemark D, Eltayeb S, Nilsson M, Wallstrom E, Lassmann H, Olsson T, Berg AL, Ericsson-Dahlstrand A. CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflammation. 2005;2:17. doi: 10.1186/1742-2094-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Powell HC, Lai J, Porreca F, Yaksh TL. Prostaglandin E2 release evoked by intrathecal dynorphin is dependent on spinal p38 mitogen activated protein kinase. Neuropeptides. 2005;39:485–94. doi: 10.1016/j.npep.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen X-H, Xin L, Adler MW, Howard OMZ, Oppenheim JJ, Rogers TJ. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc. Natl. Acad. Sci. 2001;99:01276–10281. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadano T, Namioka M, Nakagawasai O, Tan-No K, Matsushima K, Endo Y, Kisara K. Induction of nociceptive responses by intrathecal injection of interleukin-1 in mice. Life Sci. 1999;65:255–61. doi: 10.1016/s0024-3205(99)00244-1. [DOI] [PubMed] [Google Scholar]
- Tai YH, Wang YH, Wang JJ, Tao PL, Tung CS, Wong CS. Amitriptyline suppresses neuroinflammation and up-regulates glutamate transporters in morphine-tolerant rats. Pain. 2006 doi: 10.1016/j.pain.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Takeda K, Sawamura S, Tamai H, Sekiyama H, Hanaoka K. Role for cyclooxygenase 2 in the development and maintenance of neuropathic pain and spinal glial activation. Anesthesiology. 2005;103:837–44. doi: 10.1097/00000542-200510000-00023. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–9. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–61. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Tawfik VL, Lacroix-Fralish ML, Bercury KK, Nutile-McMenemy N, Harris BT, Deleo JA. Induction of astrocyte differentiation by propentofylline increases glutamate transporter expression in vitro: Heterogeneity of the quiescent phenotype. Glia. 2006 doi: 10.1002/glia.20365. [DOI] [PubMed] [Google Scholar]
- Tawfik VL, Nutile-McMenemy N, LaCroix-Fralish ML, DeLeo JA. Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain, Behavior & Immunity. 2006 doi: 10.1016/j.bbi.2006.07.001. in press. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–33. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Tsacopoulos M. Metabolic signaling between neurons and glial cells: a short review. J. Physiol. 2002;96:283–288. doi: 10.1016/s0928-4257(02)00017-7. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–83. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan TP, Jr., Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–9. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Singh SJ, Barres BA. Glia in Health and Disease. Cold Spring Harbor Laboratories, Cold Spring Harbor Laboratories; 2006. Why is Wallerian degeneration so slow in the CNS? [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20:1150–60. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hansen MK, Nguyen KT, Lee JE, Maier SF. Dynamic regulation of the proinflammatory cytokine, interleukin-1 beta: Molecular biology for non-molecular biologists. Life Sciences. 1999;65:449–481. doi: 10.1016/s0024-3205(99)00095-8. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–9. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 2000;51:29–57. doi: 10.1146/annurev.psych.51.1.29. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat. Rev. Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–35. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wieseler-Frank J, Milligan ED, Johnston I, Maier SF. Handbook of Clinical Neurology. F. Cervero. New York: 2006. Contribution of glia to pain processing in health and disease. in press. [DOI] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102:14092–7. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieseler-Frank J, Jekich BM, Bland S, Maier SF, Watkins LR. A novel immune- to CNS communication pathway: cells of the meninges surrounding the spinal cord CSF space produce proinflammatory cytokines in response to an inflammatory stimulus. Brain, Behavior & Immunity. 2006 doi: 10.1016/j.bbi.2006.07.004. in press. [DOI] [PubMed] [Google Scholar]
- Wieseler-Frank J, Jekich BM, Mahoney JH, Lewis S, Coats BD, Maier SF, Watkins LR. Spinal cord meninges: a player in exaggerated pain states? Proc. Soc. Neurosci. 2006 in press. [Google Scholar]
- Wieseler-Frank J, Kwiecien O, Jekich B, Maier SF, Watkins LR. Putative neuron-to-glia signals synergize to enhance interleukin-1 production by rat dorsal spinal cord glial cells in vitr. Proc. Soc. Neurosci. 2004:34. [Google Scholar]
- Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain. 2006;120:315–24. doi: 10.1016/j.pain.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Wolf G, Yirmiya R, Goshen I, Iverfeldt K, Holmlund L, Takeda K, Shavit Y. Impairment of interleukin-1 (IL-1) signaling reduces basal pain sensitivity in mice: genetic, pharmacological and developmental aspects. Pain. 2003;104:471–80. doi: 10.1016/S0304-3959(03)00067-8. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang N, Rogers TJ, Caterina M, Oppenheim JJ. Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize mu-opioid receptors on dorsal root ganglia neurons. J Immunol. 2004;173:594–9. doi: 10.4049/jimmunol.173.1.594. [DOI] [PubMed] [Google Scholar]