

Abstract
Vaccination with Aβ1–42 and treatment with NCX-2216, a novel nitric oxide releasing flurbiprofen derivative, have each been shown separately to reduce amyloid deposition in transgenic mice and have been suggested as potential therapies for Alzheimer’s disease. In the current study we treated doubly transgenic amyloid precursor protein and presenilin-1 (APP+PS1) mice with Aβ1–42 vaccination, NCX-2216 or both drugs simultaneously for 9 months. We found that all treatments reduced amyloid deposition, both compact and diffuse, to the same extent while only vaccinated animals, with or without non-steroidal anti-inflammatory drug (NSAID) treatment, showed increased microglial activation associated with the remaining amyloid deposits. We also found that active Aβ vaccination resulted in significantly increased cerebral amyloid angiopathy and associated microhemorrhages, while NCX-2216 did not, in spite of similar reductions in parenchymal amyloid. Co-administration of NCX-2216 did not attenuate this effect of the vaccine. This is the first report showing that active immunization can result in increased vascular amyloid and microhemorrhage, as has been observed with passive immunization. Co-administration of an NSAID agent with Aβ vaccination does not substantially modify the effects of Aβ immunotherapy. The difference between these treatments with respect to vascular amyloid development may reflect the clearance-promoting actions of the vaccine as opposed to the production-modifying effects proposed for flurbiprofen.
Keywords: Microglia, Inflammation, Cerebral amyloid angiopathy, Immunotherapy Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is a progressive, neurodegenerative disease with no current disease-modifying treatment. The amyloid hypothesis of AD suggests that amyloid plaques composed of aggregated amyloid-β (Aβ) peptide are central to the progression of the disease and that preventing accumulation and/or removing existing deposits will arrest the progression of AD (Hardy and Selkoe, 2002). One approach to the removal and prevention of amyloid deposits is immunotherapy. Immunotherapy in mice was first shown by Schenk and colleagues of Elan Pharmaceuticals Inc. (Schenk et al, 1999) who showed that active immunization of PDAPP transgenic mice with Aβ1–42 resulted in the generation of anti-Aβ antibodies and also prevented amyloid accumulation in young mice and removed existing amyloid deposits in older mice. Immunization by the same protocol also prevented cognitive deficits in both doubly transgenic APP+PS1 mice (Morgan et al, 2000) and CRND8 transgenic APP mice (Janus et al, 2000). It was later shown that passive immunization of transgenic mice using anti-Aβ antibodies resulted in significant reductions in amyloid deposition (Bard et al, 2000; Wilcock et al, 2004b), but also increased incidence of microhemorrhage (Pfeifer et al, 2002; Racke et al, 2005) associated with increased cerebral amyloid angiopathy (CAA) (Wilcock et al, 2004c).
Another potential therapy for AD is the use of non-steroidal anti-inflammatory drugs (NSAIDs). Epidemiological studies have shown that chronic use of NSAIDs may delay or even prevent the onset of AD (Szekely et al, 2004; Mackenzie and Munoz, 1998). It has been suggested that the mechanism for this action is inhibition of the chronic inflammation that occurs in AD (Akiyama et al, 2000). However, recent data has suggested that some NSAIDs, especially flurbiprofen, may have a novel mechanism of action in AD which is to shift the γ-secretase cleavage from the highly amyloidogenic 42 amino acid product to an apparently innocuous 38 amino acid product (Weggen et al, 2001). We have previously shown that a novel NSAID, NCX-2216, lowers amyloid levels in doubly transgenic APP+PS1 mice (Jantzen et al, 2002). This drug consists of a nitroxybutyl ester moiety coupled to flurbiprofen via a methoxyphenyl (ferulic acid) linker. The goal of this drug is to release nitric oxide in the stomach to minimize the GI adverse events widely reported with most NSAIDs on the market (James, 1999).
The purpose of the current experiment was to compare two therapeutic approaches, vaccination and NCX-2216, which individually produce modest effects on pathology in the high amyloid producing APP+PS1 transgenic line. Theoretically, the two therapies might synergize in their amyloid-lowering effects, or alternatively negate each other through opposing influences on microglia. We administered an NCX-2216 supplemented diet to APP+PS1 mice for 9 months while also actively vaccinating some of the mice. We wished to determine a) the effects of the two treatments on amyloid removal, b) the potential for NSAIDs to interfere with the vaccine-mediated removal of Aβ by blocking microglial activation and phagocytosis of opsonized amyloid, c) whether the vascular effects of passive immunotherapy were also found with active immunization, and d) whether NSAIDs might modify these adverse effects.
Experimental Procedures
Experimental design
Doubly transgenic APP+PS1 mice were obtained by crossing Tg2576 APP transgenic mice (Hsiao et al, 1996) with line 5.1 M146L PS1 transgenic mice (Duff et al, 1996) as previously described (Holcomb et al, 1998). 10 month old APP+PS1 were assigned to one of four experimental groups. The first group received Aβ1–42 vaccination as described below along with a control diet. The second group received an NCX-2216 supplemented diet as described below with no vaccination. A third group received an NCX-2216 supplemented diet with Aβ1–42 vaccination. The final group received a control diet with no vaccinations.
Drug treatments
Human Aβ1–42 peptide (Bachem, King of Prussia, PA) was suspended in pyrogen-free Type I water at 2.2 mg ml−1, then mixed with 10X PBS to yield 1X PBS and incubated overnight at 37 °C. For the first vaccination, the antigen suspension was mixed 1:1 with Freund's complete adjuvant (Sigma-Aldrich, St Louis, MO) and 100μg of Aβ was injected subcutaneously. For the following four vaccinations, the antigen suspension was mixed 1:1 with Freund’s incomplete adjuvant (Sigma-Aldrich, St Louis, MO). For the remaining vaccinations, the antigen suspension was mixed 1:1 with mineral oil. The first four vaccinations were administered biweekly, the next two vaccinations were monthly and the remaining two vaccinations were administered bi-monthly, for a total of 8 vaccinations over 10 months.
The NCX-2216-supplemented diet and its control diet were prepared by Harlan Teklad (Madison, WI). The control diet was a specifically designed balanced fixed formula diet for rodents (Harlan Teklad 22/5 rodent diet, Madison, WI). This diet was initally supplemented with NCX-2216 at a concentration of 375ppm. Due to attrition in the groups receiving this diet over the first 6 weeks of treatment (2 NCX-2216 and 5 NCX-2216 plus vaccination), the diet was reformulated to a concentration of 187 ppm. There were no obvious gross lesions upon necropsy and no sickness behavior was observed prior to death. Mice started receiving NCX-2216 (375ppm) 3 weeks after the beginning of the study (following the second innoculation). Seven weeks later mice receiving NCX-2216 received the lower dose diet of 187 ppm and continued on this diet for a total of 9 months until the end of the study. Final sample sizes at the end of the study were 12 untreated APP mice, 7 vaccinated mice, 10 NCX-2216 treated mice and 3 mice given both treatments.
Tissue preparation and histology
Mice were sacrificed at 20 months of age, two months following the final vaccination. On the day of sacrifice, mice were weighed, overdosed with 100mg/kg of Nembutal sodium solution (Abbott laboratories, North Chicago IL), blood samples were collected, and the mice were intracardially perfused with 25ml of 0.9% sodium chloride. Brains were rapidly removed and the left half of the brain was immersion fixed for 24 hours in freshly prepared 4% paraformaldehyde in 100mM potassium phosphate buffer (pH 7.2) for histopathology. The hemi-brains were then incubated for 24 hours in 10, 20 and 30% sucrose sequentially to cyroprotect them. Horizontal sections of 25μm thickness were collected using a sliding microtome and stored at 4oC in Dulbecco’s phosphate-buffered saline with sodium azide (pH 7.2) to prevent microbial growth. A series of eight equally spaced tissue sections 600μm apart were randomly selected spanning the entire brain and stained using free-floating immunohistochemistry for total Aβ (rabbit polyclonal anti-Aβ, raised at USF; 1:10,000 dilution) and CD45 (rat monoclonal anti-CD45, 1:5000 dilution, Serotec, Raleigh, NC) as previously described (Gordon et al, 2001, Wilcock et al, 2004b). A second series of tissue sections 600μm apart were stained using 0.2% Congo red solution in NaCl-saturated 80% ethanol. Another set of sections were also mounted and stained for hemosiderin using 2% potassium ferrocyanide in 2% hydrochloric acid for 15 minutes followed by a counterstain in a 1% neutral red solution for 1 minute.
Quantification of Congo red staining, CD45 and Aβ immunohistochemistry was performed using the Image-Pro Plus (Media Cybernetics, Silver Spring, MD) software to analyze the percent area occupied by positive stain. Images from one region of the frontal cortex and three non-overlapping regions of the hippocampus were collected at 100X magnification using constant bulb temperature and exposure, with all images collected in the same session. Images representing high and low levels of staining were used to establish red, green and blue values (RGB) to distinguish positive stain using the Image-Pro Plus software. The same RGB values were used to collect percent area occupied by positive stain for each image. Average values for each animal were calculated for the frontal cortex and all regions of the hippocampus were combined to yield a single value for the hippocampus. The initial analysis of Congo red was performed to give a total value. A second analysis was performed after manually editing out all of the parenchymal amyloid deposits to yield a percent area restricted to vascular Congo red staining. To estimate the parenchymal area of Congo red, we subtracted the vascular amyloid values from the total percentage. For the hemosiderin stain, eight equally spaced sections were examined and the numbers of Prussian blue positive deposits were counted on all sections and the average number of hemosiderin deposits per section was calculated. To assess possible treatment-related differences, the values for each treatment group were analyzed by one-way analysis of variance followed by Fischer’s least square means difference comparisons using StatView (SAS Institute Inc, NC).
The blood samples were allowed to clot for two hours at 4oC. The samples were then centrifuged at 3800rpm for 15 minutes after which the serum was collected and stored at −80oC.
Antibody titer measurements
Antibody titers were measured in serum using a method to dissociate Aβ from the anti-Aβ antibodies in the serum. This method was previously described (Li et al, 2004), however, in the current study dissociation was performed at a pH of 3.5 as opposed to pH 2.5 as was previously reported. Briefly, serum was diluted 1:100 using a dissociation buffer at pH 3.5 and incubated at room temperature for 20 minutes. The sera were then centrifuged at 8,000 × g for 20 min. at room temperature using a Microcon centrifugal device (YM-10, 10,000MW cut-off, Millipore, Billerica, MA). The sample reservoir was then placed inverted into a second tube and centrifuged at 1000 × g for 3 min. The collected solution containing the antibody dissociated from the Aβ peptide was adjusted to pH 7.0. These samples were then analyzed by ELISA to determine the antibody titer. 96-well plates were coated with Aβ1–42 peptide overnight, blocked and then incubated for 1 hour at 37oC with two-fold serial dilutions of serum samples. Anti-mouse peroxidase (Sigma-Aldrich, St. Louis, MO) was added and the ELISA was developed using 3, 3′, 5, 5′-tetramethylbenzidine (Sigma-Aldrich, St. Louis, MO).
Results
Anti-Aβ antibody titers were generated in response to the Aβ1–42 vaccination protocol. Significant antibody titers were observed in APP+PS1 transgenic mice treated with Aβ vaccination alone or with Aβ vaccination and NCX-2216, with no significant differences observed between these two groups (Fig. 1). As expected, control treated APP+PS1 mice as well as mice receiving only NCX-2216 demonstrated no anti-Aβ antibody titers. These titers (approximately 8 μg/ml) are low compared to results reported by others after inoculations in active immunization protocols (Seabrook et al, 2004; Das et al, 2003). This may reflect the advanced age of the mice at sacrifice (20 mo), the variations between laboratories in the ELISA protocol used to measure the antibody titers or the 2 month interval from the last inoculation. However, we have previously shown that antibody titers decline only 20% two months after withdrawal of inoculations (Dickey et al, 2001).
Figure 1.
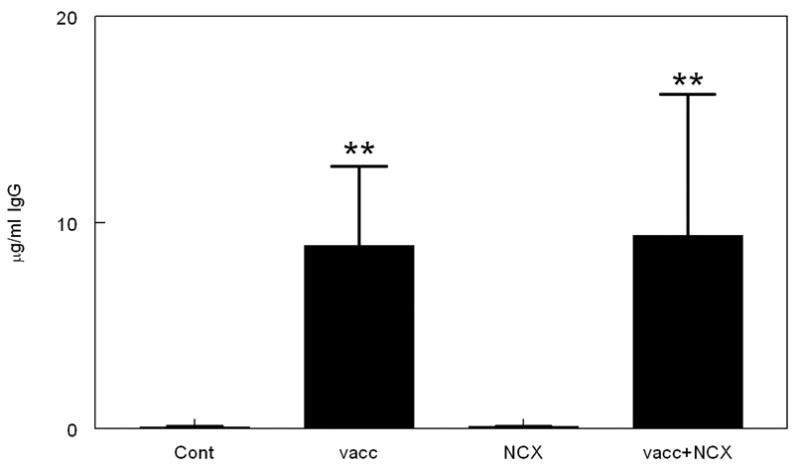
Aβ1–42 vaccination, with or without NCX-2216 treatment, produces detectable anti-Aβ antibody titers. The graph shows antibody titers for mice receiving either no treatment (Cont), Aβ1–42 vaccination (vacc), NCX-2216 diet (NCX) or both Aβ1–42 vaccination with NCX-2216 diet (vacc+NCX). Titers are shown as μg IgG per ml serum after dissociation from circulating Aβ. ** indicates P<0.01.
The pathology of APP+PS1 transgenic mice at 10 months of age, the age of the mice at the beginning of the study, has been described in detail previously (Gordon et al, 2002). In summary, at 10 months of age, these mice show 25% of the frontal cortex and 8% of the hippocampus occupied with total Aβ staining while Congo red staining shows 10% of the frontal cortex and 2% of the hippocampus occupied with positive stain. While not quantified, vascular amyloid is detectable in 10 month old APP+PS1 transgenic mice. Microglial activation is low at 10 months of age with only 0.2% of the frontal cortex occupied with positive stain.
Immunohistochemistry for Aβ showed the predicted staining pattern for the APP+PS1 transgenic mice of this age in the hippocampus (Fig. 2 Panel A). There was a high density of deposited Aβ in the CA1 region as well as in the dentate gyrus and hilus region of the hippocampus. There were fewer deposits throughout the remainder of the hippocampus. Other brain regions displayed Aβ staining similar to that shown previously for the APP+PS1 transgenic mice (Gordon et al, 2002). Following treatment with either vaccination or NCX-2216, reductions in Aβ immunohistochemistry were observed throughout the brain. In the hippocampus, significant reductions were observed throughout, particularly in the CA1 and dentate gyrus regions (Fig. 2 Panels B -vaccination and C – NCX-2216). Interestingly, despite the efficacy of each treatment alone, no additional reductions in Aβ were observed when the treatments were administered together (Fig. 2 Panel D). Analysis of the percent area occupied with positive stain showed that vaccination alone, NCX-2216 alone and vaccination plus NCX-2216 reduced Aβ deposition in both the frontal cortex and hippocampus (Fig. 3). Aβ1–42 vaccination alone showed a 35% reduction in Aβ deposition (P<0.01) in the frontal cortex (Fig. 3 Panel A) and a 25% reduction (P<0.01) in the hippocampus (Fig. 3 Panel B). NCX-2216 treatment alone showed an 18% reduction (P = 0.06) in the frontal cortex and a 20% reduction (P<0.01) in the hippocampus (Fig. 3 Panel B). Aβ1–42 vaccination administered together with NCX-2216 showed a 40% reduction (P<0.01) in the frontal cortex (Fig. 3 Panel A) and a 30% reduction (P<0.05) in the hippocampus (Fig. 3 Panel B). These reductions were not significantly different comparing the three treatment groups.
Figure 2.

Aβ immunohistochemistry is reduced in all three treatment groups. Aβ immunohistochemistry is shown in the hippocampus for APP+PS1 transgenic mice receiving no treatment (Panel A), Aβ1–42 vaccination (Panel B), NCX-2216 diet (Panel C) or both Aβ1–42 vaccination with NCX-2216 diet (Panel D). In Panel A, CA1: cornu ammonis 1, CA3: cornu ammonis 3, DG: dentate gyrus. Scale bar in Panel A = 120μm.
Figure 3.

Quantification of Aβ immunohistochemistry reveals reductions in all three treatment groups compared with control treated APP+PS1 transgenic mice. The graphs show percent area occupied with positive stain for mice receiving either no treatment (cont), Aβ1–42 vaccination (vacc), NCX-2216 diet (NCX) or both Aβ1–42 vaccination with NCX-2216 diet (vacc+NCX). Panel A shows quantification in the frontal cortex while panel B shows quantification in the hippocampus. * indicates P<0.05, ** indicates P<0.01 compared to untreated mice.
Congo red is a histochemical dye that binds compact amyloid deposits in the βpleated sheet formation. The distribution of Congo red staining in APP+PS1 transgenic mice of this age is very similar to that observed with Aβ immunohistochemistry. However, as opposed to 30% of the hippocampus being occupied with Aβ positive stain, only 2% of the hippocampus was occupied with Congo red positive stain (Fig. 4 Panel A), demonstrating the abundance of diffuse plaques in the Aβ immunohistochemical measurements. After Aβ vaccination, we observed reductions in Congophilic amyloid deposits, particularly around the hilus region of the hippocampus as well as the CA1 and dentate gyrus regions (Fig. 4 Panel B). Similar reductions in compact amyloid deposits were observed in mice treated with either NCX-2216 (Fig. 4 Panel C) or mice receiving both Aβ1–42 vaccination and NCX-2216 (Fig. 4 Panel D). Quantification of percent area occupied with Congo red positive stain revealed significant reductions in total Congo red stain in the frontal cortex and hippocampus for all three treatment groups compared with control APP+PS1 transgenic mice (Fig. 5 Panel A). These reductions were 25–35% in the frontal cortex and 30–40% in the hippocampus with no significant differences between any of the three treatment groups.
Figure 4.
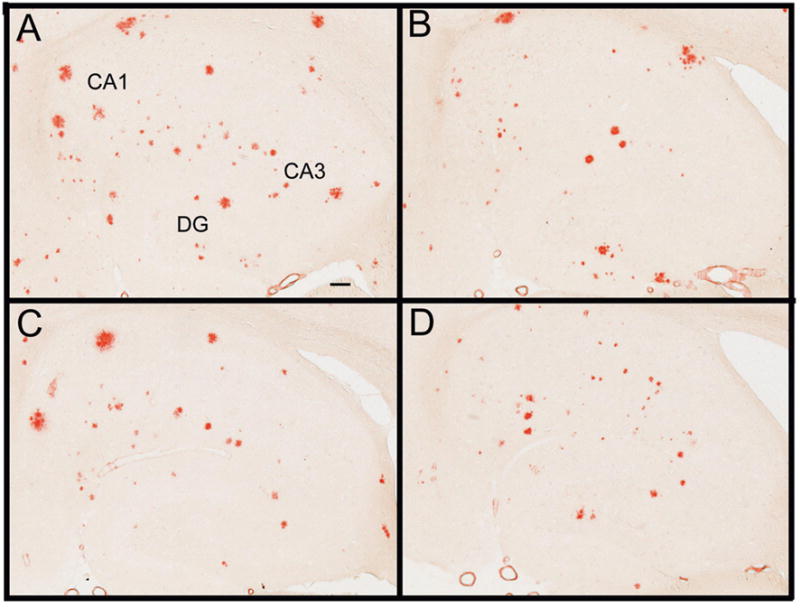
Congo red staining is reduced in all three treatment groups. Congo red staining is shown in the hippocampus for APP+PS1 transgenic mice receiving no treatment (Panel A), Aβ1–42 vaccination (Panel B), NCX-2216 diet (Panel C) or both Aβ1–42 vaccination with NCX-2216 diet (Panel D). In Panel A CA1: cornu ammonis 1, CA3: cornu ammonis 3, DG: dentate gyrus. Scale bar in Panel A = 120μm.
Figure 5.

Quantification of Congo red staining reveals reductions in total Congo red in all three treatment groups, but increased vascular Congo red following Aβ1–42 vaccination. The graphs show percent area occupied with positive stain for mice receiving either no treatment (cont), Aβ1–42 vaccination (vacc), NCX-2216 diet (NCX) or both Aβ1–42 vaccination with NCX-2216 diet (vacc+NCX). Solid bars indicate values for the frontal cortex while open bars indicate values for the hippocampus. Panel A shows quantification of total Congo red, panel B shows quantification of vascular Congo red and panel C shows calculation of parenchymal Congo red. ** indicates P<0.01 compared to control and as shown in panel B.
Interestingly, when we quantified Congo red staining in the vasculature on the same images used for total Congo red quantification, we observed increases in mice receiving Aβ vaccination with or without NCX-2216 treatment but not in mice receiving NCX-2216 alone (Fig. 5 Panel B). Treatment with Aβ1–42 vaccination alone resulted in a 2-fold increase in vascular amyloid in the frontal cortex and a 1.75-fold increase in the hippocampus (Fig. 5 Panel B). When Aβ1–42 vaccination was administered with NCX-2216 we observed a 3-fold increase in the frontal cortex and a 2.5-fold increase in the hippocampus (Fig. 5 Panel B), and these two treatment groups were not significantly different.
Vascular Congo red values were subtracted from total Congo red values to calculate parenchymal amyloid load. This showed that Aβ1–42 vaccination with or without NCX-2216 treatment was slightly more efficacious than NCX-2216 in reducing parenchymal amyloid deposits in the frontal cortex (P<0.05) and hippocampus (P=0.4).
Prussian blue staining detects hemosiderin, a degradation product of hemoglobin caused by microglia present where microhemorrhages have occurred. Microhemorrhages were detected in low numbers in control APP+PS1 mice as well as mice treated with NCX-2216. However, following vaccination with Aβ1–42 we observed a greater number of microhemorrhages as well as some larger bleeds as illustrated in Figure 6 (Panel B and D). Quantitatively, we observed a 4-fold increase in the numbers of microhemorrhages following Aβ1–42 vaccination and this increase was the same when mice were administered NCX-2216 with the vaccination (Fig. 6 Panel E). In addition, it is important to note that the microhemorrhages appeared larger following vaccination.
Figure 6.

CAA- associated microhemorrhage is increased following Aβ1–42 vaccination. Sections are stained for hemosiderin (extravenous iron) with Prussian blue and cells are counterstained with Texas red. Panels A and C show the hippocampus of a control treated APP+PS1 transgenic mouse while panels B and D show the hippocampus of an Aβ1–42 vaccinated APP+PS1 transgenic mouse. For Panels A and B, the scale bar in panel A represents 120μm. Panels C and D are a higher magnification of the area shown in the box of panels A and B, and the scale bar in panel C = 50μm. In Panel A, CA1: cornu ammonis 1, CA3: cornu ammonis 3, DG: dentate gyrus. Panel E shows quantification of Prussian blue staining and is shown as the number of positive profiles per section for mice receiving either no treatment (cont), Aβ1–42 vaccination (vacc), NCX-2216 diet (NCX) or both Aβ1–42 vaccination with NCX-2216 diet (vacc+NCX). ** indicates P<0.01 compared to the control group.
CD45 immunohistochemistry reveals microglial cells in an activated state. CD45 immunostaining counterstained with Congo red revealed activation of microglial cells around compact, Congophilic amyloid deposits in the control treated APP+PS1 transgenic mice (Fig. 7 Panel A). Treatment with Aβ1–42 vaccination alone appeared to show more microglial activation around the remaining compact amyloid deposits and vascular deposits (Fig. 7 Panel B). NCX-2216 treatment alone showed no change in the microglial activation around the remaining amyloid deposits. (Fig. 7 Panel C). Interestingly, Aβ1–42 vaccination along with NCX-2216 treatment showed the same staining pattern as vaccination alone; there appeared to be more microglial activation around the remaining amyloid deposits both in the parenchyma and the vasculature (Fig. 7 Panel D). Quantification of percent area occupied with positive CD45 immunohistochemical stain showed no significant changes in any treatment groups in the frontal cortex (Fig. 8 Panel A). In the hippocampus significant reductions were observed in microglial activation following NCX-2216 treatment (Fig. 8 Panel B), however, no reduction was observed following Aβ1–42 vaccination or when both treatments were administered together (Fig. 8 Panel B). However, these measurements reflect not only the amount of microglial activation per deposit, but also the number of deposits present. Due to the observation that compact amyloid deposits were significantly reduced with all treatments, we calculated the ratio of CD45 immunohistochemistry to total Congo red area to estimate microglial activation per amyloid deposit. We found that Aβ1–42 vaccination with or without NCX-2216 treatment increased the ratio of CD45: Congo red suggesting increased microglial activation associated with the remaining amyloid deposits in both the frontal cortex (Fig. 8 Panel C) and hippocampus (Fig. 8 Panel D). No change in the ratio was observed following treatment with NCX-2216 in either the frontal cortex (Fig. 8 Panel C) or hippocampus (Fig. 8 Panel D).
Figure 7.

CD45 immunohistochemistry is increased around remaining plaques in mice administered Aβ1–42 vaccination. CD45 immunohistochemistry counterstained with Congo red is shown in the hippocampus for APP+PS1 transgenic mice receiving no treatment (Panel A), Aβ1–42 vaccination (Panel B), NCX-2216 diet (Panel C) or both Aβ1–42 vaccination with NCX-2216 diet (Panel D). In Panel A CA1: cornu ammonis 1, CA3: cornu ammonis 3, DG: dentate gyrus. Scale bar in Panel A = 120μm.
Figure 8.
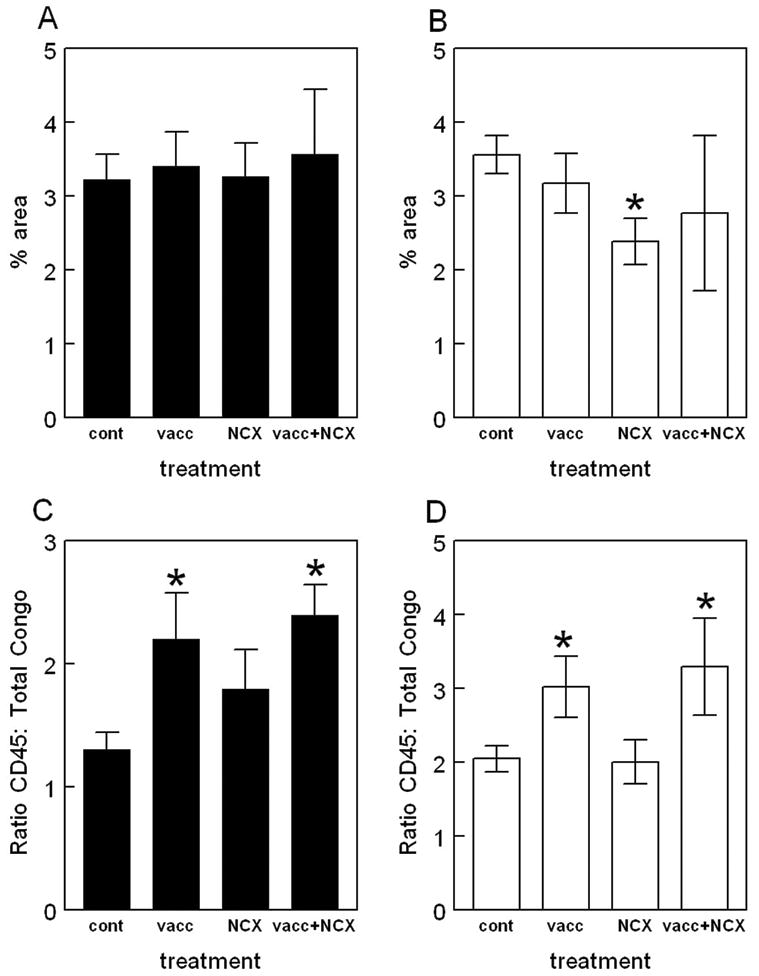
Quantification of CD45 immunohistochemistry reveals increases around remaining deposits following Aβ1–42 vaccination. The graphs show percent area occupied with positive stain for mice receiving either no treatment (cont), Aβ1–42 vaccination (vacc), NCX-2216 diet (NCX) or both Aβ1–42 vaccination with NCX-2216 diet (vacc+NCX). Panels A and B show quantification of total percent area occupied with positive stain for CD45 while panels C and D show calculated ratios of CD45 staining to Congo red. Panels A and C show quantification in the frontal cortex while panels B and D show quantification in the hippocampus. * indicates P<0.05 compared to control APP+PS1 mice.
Discussion
We have shown here that two Aβ-reducing therapies, Aβ1–42 vaccination and NCX-2216, were equally effective in reducing Aβ deposition both in the compact and diffuse states. However, administration of the two therapies together did not increase amyloid removal. Active immunization with Aβ increased levels of CAA and microhemorrhage, an effect not observed with NCX-2216 treatment. NCX-2216 did not attenuate this potentially adverse effect of Aβ vaccination.
NCX-2216 has been shown previously to reduce amyloid deposition in APP+PS1 transgenic mice. Jantzen et al (2002) showed that 5 months of administration of NCX-2216 in the diet of APP+PS1 transgenic mice significantly reduced amyloid load. In the current study we show that nine months of treatment with NCX-2216 significantly reduced amyloid load as detected by Aβ immunohistochemistry, in which the majority of stained deposits are diffuse, and Congo red, detecting compact amyloid deposits. These reductions were similar in the percent reductions to that observed previously (Jantzen et al, 2002). The mechanism by which NCX-2216 reduces amyloid deposition remains unclear. It has been shown in vitro that some NSAIDs can reduce Aβ production via a γ-secretase inhibition unrelated to cyclooxygenase inhibition (Weggen et al, 2001). It has also been shown that NCX-2216 activates peroxisome proliferator-activated receptor-γ (PPAR-γ much more effectively than the parent NSAID, flurbiprofen (Bernado et al, 2006), suggesting another mechanism for Aβ reductions since pioglitazone, a PPAR-γ agonist, has also been shown to reduce Aβ in vivo in a transgenic mouse model (Heneka et al, 2005), possibly by down-regulating β-secretase RNA expression (Sastre et al, 2006).
Aβ1–42 active vaccination has previously been shown to produce anti-Aβ antibody titers and reduce amyloid deposition in transgenic mice (Schenk et al, 1999). It has also been shown to attenuate memory loss in APP+PS1 transgenic mice (Morgan et al, 2000) and in singly transgenic APP mice (Janus et al, 2000). We have also shown that there appears to be a dependence on microglial activation for the removal of compact, Congophilic amyloid deposits following active vaccination (Wilcock et al, 2001) and following direct intracranial injection of anti-Aβ antibodies (Wilcock et al, 2003). In the current study we show that Aβ vaccination results in generation of significant anti-Aβ antibody titers, reduced amyloid deposition and increased microglial activation associated with the remaining compact amyloid deposits. It has been suggested that Aβ immunotherapy may either prevent further amyloid accumulation or remove existing deposits. The current study is inconclusive with regard to this issue. While the amyloid loads in the Aβ1–42 vaccinated mice are greater than we have previously observed in 10 month old mice (the age of the mice at the start of the current study) (Gordon et al, 2002) it is unclear whether results primarily reflect a partial prevention of amyloid deposition or whether removal and turnover of some deposits is occurring.
Interestingly, we also show that there is an apparent increase in CAA and CAA-associated microhemorrhage following vaccination. We have previously shown that this phenomenon is occurring following passive immunization with Aβ antibodies (Wilcock et al, 2004c). Other groups have also shown that treatment of aged APP transgenic mice with anti-Aβ antibodies results in an increased incidence of vascular microhemorrhage (Pfeifer et al, 2002; Racke et al, 2005). However, to date, this vascular adverse event has not been reported following active immunization. Some studies examining anti-Aβ immunotherapy using transgenic mice have examined the brains for microhemorrhage and have not detected such events. One such study used a different transgenic mouse model (J20 APP transgenic mouse) and only examined them at 10 months of age, following 6 months of vaccination (Seabrook et al, 2006). It would appear that age is a significant factor in the development of immunotherapy-associated microhemorrhage as Jucker and colleagues (2002) showed following passive immunotherapy in old and young APP transgenic mice. Only the old mice with significant CAA developed the microhemorrhages while 6 month old mice receiving the same passive immunization protocol failed to show any microhemorrhage. A study by Racke and colleagues (2005) also demonstrated that the occurrence of microhemorrhage is dependent on the antibody being used. 22-month-old PDAPP transgenic mice were passively immunized for 6 weeks with either 266 (mid-domain antibody; does not bind plaques) or 3D6 (N-terminal antibody; binds plaques) anti-Aβ antibodies and found that only the 3D6 antibody resulted in an increased incidence of CAA. The authors suggest that the results indicate plaque binding is necessary for increased microhemorrhage to occur. We have previously shown that the anti-Aβ antibodies produced following the active vaccination protocol used here are primarily N-terminal antibodies with Aβ reactivity being competed by soluble Aβ1–40, Aβ1–42 and Aβ1–16 (Dickey et al, 2001), but not by Aβ 10–20, 20–29 or 29–40. We also showed that active vaccination with this regimen produces antibodies primarily of the IgG1 and IgG2b isotypes, along with low levels of IgG2a (Dickey et al, 2001).
Active Aβ vaccination advanced to phase 2 clinical trials where dosing was terminated due to 6% of the patients developing meningoencephalitis (Orgogozo et al, 2003). Despite the trial being halted, the patients continued to be observed and some continued to produce anti-Aβ antibodies. Two autopsy reports from patients in the trial showed that those who developed meningoencephalitis had T-cell infiltration in the brain (Nicoll et al, 2003; Ferrer et al, 2004). Of the three autopsy reports published to date, all suggest that the severity of CAA is greater in vaccinated patients than the average for AD patients of the same stage (Nicoll et al, 2003; Ferrer et al, 2004; Masliah et al, 2005). One of these reports also notes the presence of “multiple cortical hemorrhages” in the brain (Ferrer et al, 2004) while a second indicates the CAA is "severe” (score of 3) typically indicating hemorrhage is present (Masliah et al, 2005).
While one might hypothesize that a lower dose of antibody may prevent the occurrence of vascular adverse events due to immunotherapy, it appears that even relatively low levels of anti-Aβ antibodies over time are sufficient to produce CAA associated microhemorrhage. Moreover, the NCX-2216 treatment alone reduced amyloid deposits to a similar degree as the vaccine, yet did not produce the increase in vascular deposits. Given the presence of increased vascular staining with both active and passive immunization, it would appear that something associated with the antibody -mediated clearance is responsible for the increase in CAA, rather than amyloid reduction per se.
Based on approximate calculations, the titers (8 μg in 1ml serum) achieved in the current active vaccination study appears to equate to a 3mg/kg dose of anti-Aβ antibodies when translating to a passive immunization protocol. This is 30% of the dose we have previously shown (using passive immunization) to result in microhemorrhage and exacerbation of CAA following three months of anti-Aβ antibody treatment (Wilcock et al, 2004c). Using a similar active vaccination regimen to the one reported here, we have previously shown that antibody titers reach their maximum levels following 7 immunizations. Following inoculation cessation, antibody titers declined only 20% after two months, with detectable titers still present 14 months after the last immunization (Dickey et al, 2001). Therefore, we would not predict that the antibody titers were substantially greater earlier in the study, although it is plausible that serum antibody titers were slightly higher immediately following the final immunization. Together with our previous studies (Wilcock et al, 2004c), the data presented here suggest that the levels of anti-Aβ antibody may not be as significant as duration of treatment and mouse age when increasing vascular amyloid with immunotherapy.
Furthermore, treatment with an NSAID along with the active Aβ vaccination did not attenuate the vascular consequences of vaccination, nor did it impair clearance. In our earlier work with intracranially applied anti-Aβ antibodies, we found that NCX-2216 partially attenuated both the activation of microglia and the clearance of compact amyloid deposits caused by the antibody (Wilcock et al, 2004a). In contrast, dexamethasone completely suppressed both effects, implying that the NCX-2216 does not maximally suppress microglia in vivo.
Administration of NCX-2216 with Aβ1–42 vaccination did not result in greater amyloid removal than either treatment alone. It is plausible that there is a limit to the extent of amyloid removal and a dynamic equilibrium exists between Aβ removal and production. We have observed what appears to be an equilibrium effect previously following intracranial LPS administration, where amyloid was removed within seven days, but returned to control levels by four weeks. In essence, the mouse accumulated the same amount of amyloid in three weeks that originally accumulated over 16 months (Herber et al, 2004). There are three proposed mechanisms for antibody-mediated Aβ removal. These are Fc-receptor mediated microglial phagocytosis (Bard et al, 2003), efflux of Aβ from the brain into the plasma (DeMattos et al, 2001), possibly via the FcRn receptor (Deane et al, 2005) and a direct, catalytic disaggregation of amyloid deposits by the antibody (Solomon et al, 1997). The proposed mechanisms of NSAID-mediated reductions in Aβ (vide supra) do not in any way overlap with the mechanisms of antibody-mediated reductions. It is possible that antibody-mediated microglial activation results in a phenotypic switch to an activated microglial cell state where NSAIDs no longer have an effect. We have previously suggested that microglia may exist in distinct activation states, one of which may actually be beneficial in AD (Morgan et al, 2005).
In summary, we have shown here that two amyloid-lowering therapies administered together do not show additive or synergistic effects in their amyloid-lowering abilities. However, importantly, we show that active immunization with Aβ1–42 increases levels of CAA and microhemorrhage. This effect is not attenuated by an NSAID such as NCX-2216. While it is unclear what impact microhemorrhages have clinically in AD, it has been shown that CAA is associated with intracerebral hemorrhages as well as ischemic infarcts (Attems, 2005). While we have previously shown that microhemorrhage as a result of immunotherapy does not impact mice cognitively (Wilcock et al, 2004c) the data presented here should increase concern with respect to ongoing clinical trials that even low levels of circulating anti-Aβ antibodies may result in vascular complications with long durations of treatment.
Acknowledgments
This work was supported by NIH grants AG 15490 and AG 18478. DGM also receives salary support from AG25711. Donna M. Wilcock was the Benjamin Scholar for Alzheimer's Research. The authors wish to thank Karen Ashe and Karen Duff for early access to the transgenic mice used in this study. We thank Ennio Ongini and Nicox, S.A. for donating the NCX-2216 for our research. We also thank Paul E. Gottschall for providing us with the polyclonal anti-Aβ antibody for immunohistochemistry.
Abbreviations
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- CA
cornu ammonis region of the hippocampus
- CAA
cerebral amyloid angiopathy
- ELISA
enzyme-linked immunosorbent assay
- NSAID
Non-steroidal anti-inflammatory drug
- PPARγ
peroxisome proliferator-activated receptor gamma
- PS1
Presenilin-1
- RGB
red, green, blue video segmentation
- RNA
ribonucleic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyama I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attems J. Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005;110:345–359. doi: 10.1007/s00401-005-1074-9. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer's disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, Seubert P, Yednock T. Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci USA. 2003;100:2023–2028. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado A, Gasparini L, Ongini E, Minghetti L. Dynamic regulation of microglial functions by the non-steroidal anti-inflammatory drug NCX 2216: Implications for chronic treatments of neurodegenerative diseases. Neurobiol Dis. 2006;22:25–32. doi: 10.1016/j.nbd.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Das P, Chapoval S, Howard V, David CS, Golde TE. Immune responses against Abeta1–42 in HLA class II transgenic mice: implications for Abeta1–42 immune-mediated therapies. Neurobiol Aging. 2003;24:969–76. doi: 10.1016/s0197-4580(03)00036-8. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzehimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, Wu Z, Holtzman DM, Zlokovic BV. IgG-assisted age-dependent clearance of Alzheimer's amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci. 2005;25:11495–11503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Morgan DG, Kudchodkar S, Weiner DB, Bai Y, Cao C, Gordon MN, Ugen KE. Duration and specificity of humoral immune responses in mice vaccinated with the Alzheimer’ Disease-associated β-amyloid 1–42 peptide. DNA Cell Biol. 2001;20:723–729. doi: 10.1089/10445490152717587. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refelo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada R, Sanchez M, Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MN, King DL, Diamond DM, Jantzen PT, Boyett KW, Hope CE, Hatcher JM, DiCarlo G, Gottschall PE, Morgan D, Arendash GW. Correlation between cognitive deficits and Abeta deposits in transgenic APP+PS1 mice. Neurobiol Aging. 2001;22:377–385. doi: 10.1016/s0197-4580(00)00249-9. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O' Callaghan JP, Morgan D. Time Course of the Development of Alzheimer-like Pathology in the Doubly Transgenic PS1 + APP Mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- James DS. The multisystem adverse effects of NSAID therapy. J Am Osteopath Assoc. 1999;99:S1–S7. doi: 10.7556/jaoa.1999.99.11.S1. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial Activation and β-Amyloid Deposit Reduction Caused by a Nitric Oxide-Releasing Nonsteroidal Anti-Inflammatory Drug in Amyloid Precursor Protein Plus Presenilin-1 Transgenic Mice. J Neurosci. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HTJ, Nixon PA, Mercken M, Bergeron C, Fraser PE, St George-Hislop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Li Q, Cao C, Chackerian B, Schiller J, Gordon M, Ugen KE, Morgan D. Overcoming antigen masking of anti-amyloid beta antibodies reveals breaking of B cell tolerance by virus-like particles in amyloid beta immunized amyloid precursor protein transgenic mice. BMC Neurosci. 2004;5:21. doi: 10.1186/1471-2202-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology. 1998;50:986–990. doi: 10.1212/wnl.50.4.986. [DOI] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon MN, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–53. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2001;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Jiang L, Thomas K, Lemere CA. Boosting with intranasal dendrimeric Abeta1-15 but not Abeta1-15 peptide leads to an effective immune response following a single injection of Abeta1-40/42 in APP-tg mice. J Neuroinflamm. 2006;3:14. doi: 10.1186/1742-2094-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabrook TJ, Bloom JK, Iglesias M, Spooner ET, Walsh DM, Lemere CA. Species-specific immune response to immunization with human versus rodent A beta peptide. Neurobiol Aging. 2004;25:1141–51. doi: 10.1016/j.neurobiolaging.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frenkel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed monoclonal antibodies. Proc Natl Acad Sci USA. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004 2004 Jul–Aug;23(4):159–69. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D. Number of A inoculations in APP + PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol. 2001;20:731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms independent of and associated with microglial activation. J Neurosci. 2003;213:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Muniredddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial Activation Facilitates Aβ Plaque Removal Following Intracranial Anti-Aβ Antibody Administration. Neurobiol Dis. 2004a;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004b;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004c;8:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]