

Abstract
Current evidence suggests that demyelination may be a necessary but not a sufficient condition for neurologic deficits associated with multiple sclerosis. Axon injury that occurs within the permissive environment of the demyelinated lesion is better correlated with functional deficits, but the mechanisms and cellular effectors of this injury are largely unknown. In an effort to identify potential axon injury mediators we examined demyelination, motor function, and the number of spinal axons in perforin-deficient mice. Perforin is a critical molecular mediator of cytotoxic immunological injury and we hypothesized that genetic deletion of perforin expression would protect demyelinated axons. Indeed, we found that while perforin-deficient mice had considerable spinal cord demyelination 180 days after infection with Theiler’s murine encephalomyelitis virus, such mice exhibited functional and axonal preservation comparable to non-demyelinated perforin-competent controls. We conclude that perforin-dependent effector cells such as cytotoxic T cells, γδ T cells, and natural killer cells may play a role in axon damage that is dependent upon but separable from demyelination.
Keywords: Multiple Sclerosis, Neuroimmunology, Cytotoxic Lymphocyte, Axon, Major Histocompatibility Complex class I
Introduction
Demyelination of the central nervous system (CNS) is a pathological hallmark of multiple sclerosis (MS). The majority of efforts seeking to ameliorate the functional deficits associated with MS focus upon resolution or repair of the demyelinating insult. Such a therapeutic focus presupposes that demyelination is the primary basis for the functional deficit. Indeed, early studies demonstrated that CNS demyelination resulted in the slowing of axon conduction velocity as well as complete blockade of axon conduction (McDonald and Sears, 1969), and such observations were generally considered sufficient to explain the majority of neurological dysfunction in MS patients. However, several clinical and basic science observations challenge this hypothesis. Magnetic resonance imaging studies have demonstrated only a weak correlation between lesion load and clinical deficit in MS patients (Stevens et al., 1986), even though pathologically such lesions were indeed demyelinated (Bruck et al., 1997) and frequently involved areas of CNS expected to result in neurologic deficits. Autopsy studies have likewise demonstrated diagnostically-relevant demyelination in individuals who exhibited normal neurologic function throughout their life (Mews et al., 1998). These observations suggest that demyelination alone is insufficient to cause permanent neurologic deficit.
Animal experiments have also questioned the direct relationship between functional deficit and demyelination. Using a viral model of MS, we showed that β2-microglobulin-deficient experienced profound demyelination of the spinal cord but exhibited normal motor function (Rivera-Quinones et al., 1998), implicating surface expression of major histocompatibility complex (MHC) class I molecules in axon injury. Likewise, while retrograde labeling experiments demonstrated a failure of axonal transport in mice with demyelination and functional deficits (Ure and Rodriguez, 2000), we found preservation of axonal transport in demyelinated mice lacking β2-microglobulin (Ure and Rodriguez, 2002). In addition, specific peptide-mediated depletion of CD8+ class I-restricted cytotoxic T cells (CTLs) in animals infected with Theiler’s murine encephalomyelitis virus (TMEV) resulted in preservation of neurologic function (Johnson et al., 2001), suggesting a direct relationship between T cell-mediated killing and axon dropout. In support of this hypothesis, CD8+ T cells have been shown to injure neurons and transect axons in vitro (Medana et al., 2001). With regard to human MS, recent pathology studies indicate that CD8+ T cells may be the most common subset of T cells in the MS brain (Babbe et al., 2000) and autopsy studies demonstrate that CD8+ T cells are frequently associated with axonal injury in MS (Bitsch et al., 2000). Moreover, demyelination leads to the upregulation of MHC class I on axons and neurons within human MS lesions (Hoftberger et al., 2004) and imaging studies indicate that axon loss rather than demyelination is the primary substrate for disability (Narayanan et al., 1997; Truyen et al., 1996; van Waesberghe et al., 1999). All together, these findings suggest that CD8+ T cell recognition of MHC class I on axons may directly mediate axonal injury and neurologic deficit in MS. CTL-induced cytotoxicity of infected targets is mediated predominantly by the release of cytolytic granules that contain the pore-forming protein perforin (PFP), the proteoglycan serglycin, and a family of serine proteases known as the granzymes. Following T cell receptor recognition of the appropriate peptide:MHC class I complex on target cells and the formation of the immune synapse, a granzyme:serglycin complex and perforin are exocytosed by the cytotoxic T cell (Ashton-Rickardt, 2005; Griffiths, 2003; Lieberman, 2003; Voskoboinik and Trapani, 2006). Once endocytosed by the target cell, perforin appears to facilitate the transfer of granzymes from endosomes to the cytosol, initiating a cascade of signals that results in cell death (Froelich et al., 2004; Metkar et al., 2002; Raja et al., 2003; Raja et al., 2002). It was previously assumed that neurons escape immune surveillance and cytotoxic attack due to a lack of MHC class I expression. However, following TMEV infection of the CNS, MHC class I is rapidly induced in most CNS cells including neurons (Altintas et al., 1993; Lindsley et al., 1992), and this induction may depend upon soluble factors such as IFN-α/β (Njenga et al., 1997b). Likewise, in vitro, electrically silent neurons exhibit increased surface expression of MHC class I, thus predisposing them to cytotoxic attack (Neumann et al., 1995). Demyelination, by altering the conduction properties of axons within the lesion, may induce the upregulation of surface MHC class I molecules on denuded axons, thereby making them targets for CTL-mediated injury (Hoftberger et al., 2004).
Based on these observations, our central hypothesis is that demyelination creates a permissive environment for perforin-mediated immunological attack of axons within the CNS (Howe and Rodriguez, 2005). In an effort to separate axon injury from demyelination, we studied C57BL/6J mice deficient in perforin expression (PFP-/-) (Kagi et al., 1994). We found that at 180 days after infection with TMEV perforin-deficient mice but not perforin-competent wildtype mice exhibited spinal cord demyelination comparable to that measured in chronically-infected C57BL/6J mice with a genetic deletion in CD4 (CD4-/-) (Murray et al., 1998b). However, despite significant demyelination, perforin-deficient mice exhibited preservation of motor function and preservation of large caliber axons in the spinal cord as compared to demyelinated CD4-/- mice. In fact, perforin-deficient mice were indistinguishable from perforin-competent wildtype mice with regard to motor function and the number of large axons in the spinal cord. This finding provides direct evidence that perforin, a molecule that mediates CTL killing of targets, is required for neurologic dysfunction and axon dropout during chronic demyelinating injury of the spinal cord.
Materials and Methods
Virus
The Daniel’s strain of TMEV was used for all experiments (Lipton, 1975). At four to six weeks of age, mice were intracerebrally infected with 2 × 105 PFU of TMEV in a total volume of 10 μL.
Animals
Mice were obtained from Jackson Laboratories (Bar Harbor, Maine). C57BL/6-Prf1tm1Sdz/J mice have a targeted disruption of the PFP gene and are homozygous for MHC class I H-2b. C57BL/6J CD4-/- mice have a targeted disruption of the CD4 gene and are homozygous for MHC class I H-2b. These animals were chosen because they exhibit demyelination within the context of an H-2b haplotype (Murray et al., 1998b) but have a functional CD8+ T cell response (Rahemtulla et al., 1991).
Demyelination analysis
At various times after infection mice were perfused via intracardiac puncture with 50 mL of Trump’s fixative. Spinal cords and brains were removed and post-fixed for 24-48 hours in Trump’s fixative in preparation for morphologic analysis. Spinal cords were removed from spinal columns and cut into one millimeter coronal blocks, and every third block was osmicated and embedded in glycol methacrylate (Rodriguez, 1991). Two μm-thick sections were prepared and stained with a modified erichrome/cresyl violet stain (Pierce and Rodriguez, 1989). Morphological analysis was performed on 12 to 15 sections per mouse as previously described (Rodriguez, 1991). Briefly, each quadrant from every coronal section from each mouse was graded for the presence or absence of demyelination. All grading was performed without knowledge of the experimental group.
Functional analysis
The Rotamex rotarod (Columbus Instruments, Columbus, OH) was used to assess motor function. This device consists of a motor-driven rotating rod suspended 28.5 cm above a grid. Mice were trained under a constant speed protocol and tested in an accelerating rod protocol according to our established procedures (McGavern et al., 1999b). On the day of testing (180 days postinfection), the change in rotarod velocity that occurred prior to the mouse falling was recorded for each mouse, and data were expressed as the percent decrease from baseline performance measured on day 21 postinfection.
Axon analysis
Calculation of axon numbers was performed essentially as reported (McGavern et al., 1999a). Sections of spinal cord were cut at 1 micron thickness and stained for exactly 20 min with the same batch of 4% para-phenylenediamine to ensure an identical intensity of myelin labeling. Digitized sample images from the smallest thoracic spinal cord section (T6) were collected at 60× magnification from each animal according to a sampling scheme developed previously (McGavern et al., 1999a). Images were captured from regions containing a relative absence of demyelination to ensure the quantitation of only myelinated fibers. Each field measured 17675 μm2, and we collected a total of 2.21 mm2 from the PFP-/- mice, 1.38 mm2 from the wildtype mice, and 1.91 mm2 from the CD4-/- mice. Myelinated axon diameters were calculated following segmentation of the gray values (145-255) corresponding to the axoplasm from each image.
Batch algorithms were generated in Matlab (The Mathworks, Nattick, MA) to automatically calculate the diameter of each axon in the field from the segmented binary image after regions corresponding to the vasculature, cell bodies, longitudinal axons, and demyelination were excluded on the basis of circularity thresholding. Diameters less than 1 μm were excluded from the analysis to eliminate small regions that did not correspond to axons. Axon area measurements were binned for analysis: (i) 1-4 μm2 (small axons); (ii) 4-10 μm2 (medium axons); and (iii) >10 μm2 (large axons). The number of axons in each bin was normalized to the area of spinal cord analyzed to yield axons per mm2. These values were averaged across all animals per group.
Statistical analysis
All data are presented as mean values ± standard error of the mean. Motor function data, percent demyelinated quadrants, and axon numbers were analyzed by one-way ANOVA using the Holm-Sidak pairwise comparison or Dunn’s test to assess intra- and inter-group differences. Demyelination frequency was analyzed by Fisher Exact analysis. SigmaStat was utilized for all statistical analysis and for guidance regarding power and sample size.
Results and Discussion
Intracerebral infection with TMEV, a picornavirus, induces a characteristic disease in the CNS of mice (Lipton, 1975). During the first 10 to 12 days of infection, the virus replicates primarily in neurons of the hippocampus, striatum, cortex, and anterior horn of the spinal cord. Oligodendrocytes and macrophages are also infected early (Njenga et al., 1997a). In mice of resistant MHC haplotypes (H-2b, k, d) the virus is rapidly cleared from the CNS (Drescher et al., 1999) and no demyelination or viral persistence develops. However, in animals of susceptible MHC haplotype (H-2q, s, v, u, r) the virus is largely cleared from neurons but persists in glial cells (Rodriguez et al., 1983) and macrophages (Dal Canto and Lipton, 1982; Levy et al., 1992; Rossi et al., 1997) within the spinal cord white matter and the brain stem. C57BL/6J mice are of the H-2b MHC haplotype and are therefore normally resistant to persistent infection with TMEV, clearing the virus from the brain and spinal cord by 45 days postinfection (Drescher et al., 1999). These mice do not normally exhibit demyelination following TMEV infection (Drescher et al., 1999). As shown in Figure 1 and in Table 1, we did not detect a significant level of demyelination in perforin-competent C57BL/6J mice 180 days after infection (Figure 1A), but we did observe substantial demyelination in both perforin-deficient C57BL/6J mice (Figure 1B) and in CD4-deficient C57BL/6J mice (Figure 1C) (Table 1). These findings are consistent with previous reports that the absence of perforin (Murray et al., 1998a) or the absence of CD4 T cells (Murray et al., 1998b) breaks resistance to persistent infection in H-2b mice. We found that there was no statistical difference between perforin-deficient C57BL/6J mice and CD4-deficient mice with regard to either the frequency of demyelination (Table 1; P=0.478 by the Fisher Exact test) or to the percentage of demyelinated quadrants in the spinal cord (Table 1; P=0.172 by ANOVA), while both strains were significantly different from the non-demyelinated perforin-competent mice (Table 1).
Figure 1.
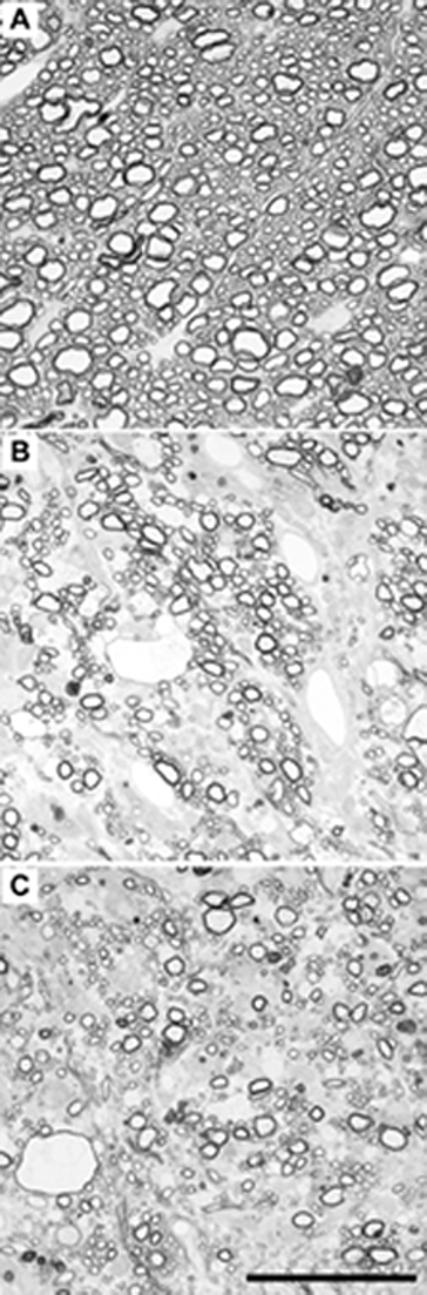
Perforin-deficient mice exhibit substantial spinal cord demyelination 180 days after infection with TMEV. A) Perforin-competent C57BL/6J mice exhibit normal spinal cord myelination. B) Perforin-deficient C57BL/6J mice have regions of profound demyelination that are comparable to the demyelination observed in chronically-infected CD4-deficient mice (C). Scale bar in (C) is 50 μm and refers to all three panels. Images are representative of at least 8 mice per group and were collected in approximately the same region of cervical spinal cord.
Table 1.
Analysis of spinal cord demyelination.
| Mouse Strain | Day* | N# | Percent of Quadrants with Demyelination | Number of Mice with Demyelination | ||
|---|---|---|---|---|---|---|
| B6 PFP+/+ | 180 | 8 | 1.2 ± 1.2 | 1/8 | ||
| B6 PFP-/- | 180 | 11 | 22.1 ± 3.5 | P<0.001@ | 10/11 | P=0.006$ |
| B6 CD4-/- | 180 | 16 | 14.5 ± 2.8 | P=0.005@; P=0.172^ | 16/16 | P<0.001$; P=0.478& |
Day after infection
Number of mice
vs B6 PFP+/+
vs B6 PFP-/-
vs B6 PFP+/+
vs B6 PFP-/-
Spinal cords were removed from infected mice and cut into one millimeter coronal blocks. Every third block was osmicated and embedded in glycol methacrylate, and two micron-thick sections were prepared and stained with a modified erichrome/cresyl violet stain. Morphological analysis was performed on 12 to 15 sections per mouse. Each quadrant from every coronal section from each mouse was graded for the presence or absence of demyelination. Values are shown as the percent of spinal cord quadrants examined with demyelination ± standard error of the mean and as number of mice per group exhibiting demyelination. All analyses were performed without knowledge of the experimental group. Demyelination quadrant data were analyzed by one-way ANOVA using the Holm-Sidak pairwise comparison. Data for number of demyelinated mice were analyzed by the Fisher Exact test.
The development of demyelination in perforin-deficient C57BL/6J mice following infection with TMEV led us to predict that these mice would exhibit functional deficits (McGavern et al., 2000). We used the rotarod test for balance, coordination, and stamina to analyze motor function in perforin-competent C57BL/6J mice, perforin-deficient C57BL/6J mice, and CD4-deficient C57BL/6J mice 180 days after infection. To our surprise, motor function in perforin-deficient mice was indistinguishable from function measured in wildtype mice (PFP-/- = 78.2 ± 7.8% of baseline, n=15; PFP+/+ = 83.2 ± 5.3% of baseline, n=9; P=0.66 by Holm-Sidak pairwise ANOVA) (Figure 2). In contrast, motor function was compromised in CD4-deficient mice (43.8 ± 11.4% of baseline, n=7; P=0.009 vs PFP-/-; P=0.007 vs PFP+/+ by Holm-Sidak pairwise ANOVA) (Figure 2). Therefore, despite the presence of demyelination in the spinal cord of perforin-deficient mice comparable to that observed in CD4-deficient mice, motor function in these animals was preserved to levels measured in perforin-competent mice that cleared the virus and did not experience demyelination. From this we conclude two things: 1) demyelination alone is not sufficient to induce motor deficits; 2) the absence of perforin within the context of demyelination leads to preservation of motor function, consistent with the hypothesis that axon injury is mediated by perforin.
Figure 2.
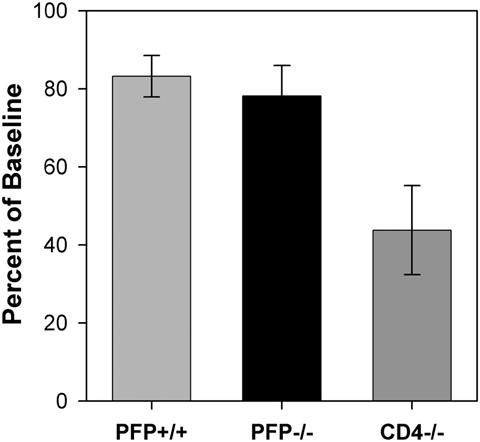
Perforin-deficient mice exhibit preservation of function despite significant demyelination. Mice from all three groups were assessed by rotarod 180 days after infection. Functional performance of the perforin-deficient C57BL/6J mice (78.2 ± 7.8% of baseline, n=15) was indistinguishable from wildtype, perforin-competent C57BL/6J mice (83.2 ± 5.3% of baseline, n=9) and was significantly better than the performance measured in demyelinated CD4-deficient mice (43.8 ± 11.4% of baseline, n=7; P<0.009 vs. PFP-/- and P<0.007 vs. PFP+/+). Thus, despite the presence of demyelination in perforin-deficient C57BL/6J mice that was comparable to that measured in CD4-deficient mice, the absence of perforin expression conferred protection of motor function.
Neurologic function depends upon transmission of signals along axons. Functional preservation in perforin-deficient mice led us to predict that these animals would also exhibit preservation of axons within the spinal cord. Indeed, the number of large caliber axons (area ≥ 10 μm2) in demyelinated perforin-deficient mice was equivalent to that measured in perforin-competent wildtype mice at 180 days postinfection, despite the fact that perforin-competent mice clear the virus soon after infection and do not exhibit demyelination (PFP-/- = 28209 ± 664 axons per mm2, n=14; PFP+/+ = 28766 ± 976 axons per mm2, n=8; P=0.616 by one-way ANOVA with Holm-Sidak pairwise analysis) (Figure 3). In marked contrast, demyelinated and functionally impaired CD4-deficient mice showed a considerable reduction in large diameter spinal axons (17129 ± 700 axons per mm2, n=12; P<0.001 vs PFP-/-; P<0.001 vs PFP+/+ by Holm-Sidak pairwise comparison) (Figure 3). Of note, the number of medium caliber axons (area = 4-10 μm2) was different between infected perforin-deficient and infected wildtype mice (135484 ± 2397 axons per mm2 vs 145916 ± 2979 axons per mm2, respectively; P=0.008 by Holm-Sidak pairwise comparison), and such axons were further reduced in infected CD4-deficient mice (127964 ± 2672 axons per mm2; P=0.04 vs PFP-/-; P<0.001 vs PFP+/+ by Holm-Sidak pairwise comparison) (Figure 3). Finally, the number of small axons (area < 4 μm2) was significantly lower in infected perforin-deficient mice (46692 ± 1461 axons per mm2) than in infected wildtype mice (62385 ± 2061 axons per mm2; P<0.001 by Kruskal-Wallis ANOVA on ranks and Dunn’s pairwise test) and was equal to the number measured in infected CD4-deficient mice (48443 ±1335 axons per mm2; P=N.S. vs. PFP-/-; P<0.001 vs PFP+/+ by Dunn’s test) (Figure 3). These findings suggest several conclusions: 1) demyelinated and functionally impaired CD4-deficient mice exhibit a loss of spinal axons across all diameters as compared to wildtype mice; 2) perforin-deficient mice also lose small and medium diameter axons when compared to perforin-competent wildtype mice and this loss is comparable to that observed in the functionally impaired CD4-deficient mice; 3) because perforin-deficient mice are not functionally impaired but have lost small and medium diameter axons we would conclude that these axons are not primarily responsible for motor function measured on the rotarod; 4) the absence of perforin confers protection of large diameter axons and preserves motor function to levels observed in non-demyelinated wildtype mice, consistent with a role for these axons in the motor function assessed by the rotarod; 5) demyelination does not predict motor dysfunction but loss of large spinal axons does; 6) large diameter demyelinated axons are injured in mice lacking CD4 T cells but not in mice lacking perforin, implicating perforin-dependent effectors such as cytotoxic T cells, NK cells, or γδ T cells in such axon injury. We conclude that the absence of perforin expression within the context of demyelination leads to preservation of motor function and the preservation of large diameter spinal axons but does not prevent the loss of small and medium diameter axons. The relationship between axonal caliber and preserved motor function is not entirely clear, but may be due to the faster conduction velocities associated with large diameter axons (Chklovskii and Stepanyants, 2003; Hoffman et al., 1987; Waxman, 1980) and the correlation between faster conduction velocity and better motor coordination (Olivier et al., 1997).
Figure 3.
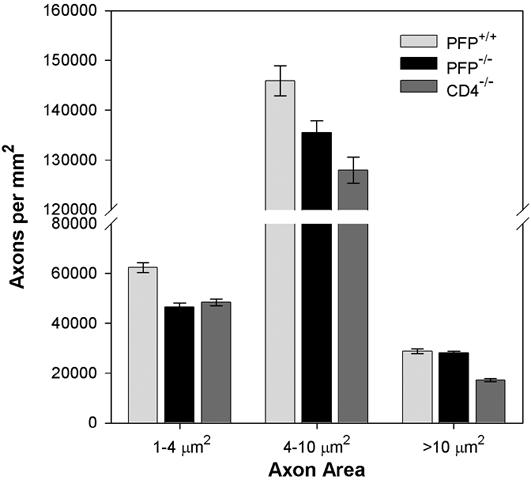
Perforin-deficient mice exhibit preservation of large diameter axons in the spinal cord despite significant demyelination. Mice from all three groups were morphologically assessed for the number of small (area = 1-4 μm2), medium (area = 4-10 μm2), and large (area ≥ 10 μm2) axons in the mid-thoracic spinal cord. Values are shown as number of axons per mm2 analyzed. Perforin-deficient and perforin-competent wildtype mice were not different with regard to the number of large axons (see text), but such axons were reduced in CD4-deficient mice. On the other hand, the number of small and medium axons was reduced in both perforin-deficient and CD4-deficient mice as compared to wildtype mice. The functional preservation observed in perforin-deficient mice relative to CD4-deficient mice suggests that protection of large axons is sufficient to maintain motor function and also suggests that perforin directly injures large spinal axons during demyelination.
An important implication of our findings is that functional deficits associated with demyelination are the result of axonal injury mediated by perforin-dependent effector cells such as cytotoxic T cells. We suggest that demyelination exposes molecular cues on the naked axon that are recognized by these cytotoxic effector cells. Whether these cues include MHC class I molecules is not addressed by our current study, but previous work indicating that mice deficient in β2-microglobulin expression (and therefore deficient in functional MHC class I expression) also exhibit preservation of axons and function despite demyelination (Rivera-Quinones et al., 1998) supports such a model. It is important to note that our proposed molecular mechanism of axon damage associated with demyelination may not be true of all strains of mice or all models of demyelination (Begolka et al., 2001; Tsunoda et al., 2002). Finally, much of the literature regarding axon injury associated with multiple sclerosis assumes that such injury is the result of a failure in oligodendrocyte-dependent growth factor-mediated support of axon survival. While such a relationship between the oligodendrocyte and the axon is certainly critical to normal CNS function, we interpret our findings as an argument against this model for axon loss following demyelination. Specifically, the fact that perforin-deficient mice had extensive demyelination but axon preservation while perforin-competent but CD4-deficient mice had both extensive demyelination and extensive axon injury suggests that the absence of oligodendrocyte trophic support cannot fully account for demyelination-dependent axon damage. We conclude that axon injury secondary to demyelination is mediated by inflammatory factors, one of which is perforin. This finding may have important implications for therapeutic intervention in multiple sclerosis, as protection of demyelinated axons via manipulation of perforin-dependent immune effector function may protect patients from functional crises triggered by recurrent demyelination.
Acknowledgements
This work was supported by grants RG3636 (CLH) and CA1011A8 (MR) from the National Multiple Sclerosis Society, by funding from Donald and Frances Herdrich (CLH), and by grants P01 NS38468 and R01 NS32129 (MR) from the National Institutes of Health. We thank Laurie Zoecklein, Mabel Pierce, Reghann LaFrance-Corey and Louisa Papke for expert technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Altintas, A, et al. Differential expression of H-2K and H-2D in the central nervous system of mice infected with Theiler’s virus. J Immunol. 1993;151:2803–12. [PubMed] [Google Scholar]
- Ashton-Rickardt, PG. The granule pathway of programmed cell death. Crit Rev Immunol. 2005;25:161–82. doi: 10.1615/critrevimmunol.v25.i3.10. [DOI] [PubMed] [Google Scholar]
- Babbe, H, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolka, WS, et al. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. J Neurovirol. 2001;7:409–20. doi: 10.1080/135502801753170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitsch, A, et al. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123(Pt 6):1174–83. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Bruck, W, et al. Inflammatory central nervous system demyelination: correlation of magnetic resonance imaging findings with lesion pathology. Ann Neurol. 1997;42:783–93. doi: 10.1002/ana.410420515. [DOI] [PubMed] [Google Scholar]
- Chklovskii, DB, Stepanyants, A. Power-law for axon diameters at branch point. BMC Neurosci. 2003;4:18. doi: 10.1186/1471-2202-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Lipton, HL. Ultrastructural immunohistochemical localization of virus in acute and chronic demyelinating Theiler’s virus infection. Am.J.Pathol. 1982;106:20–29. [PMC free article] [PubMed] [Google Scholar]
- Drescher, KM, et al. CNS cell populations are protected from virus-induced pathology by distinct arms of the immune system. Brain Pathol. 1999;9:21–31. doi: 10.1111/j.1750-3639.1999.tb00206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froelich, CJ, et al. Granzyme B-mediated apoptosis--the elephant and the blind men. Cell Death Differ. 2004;11:369–71. doi: 10.1038/sj.cdd.4401381. [DOI] [PubMed] [Google Scholar]
- Griffiths, GM. Endocytosing the death sentence. J Cell Biol. 2003;160:155–6. doi: 10.1083/jcb.200212143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, PN, et al. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci U S A. 1987;84:3472–6. doi: 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftberger, R, et al. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol. 2004;14:43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, CL, Rodriguez, M. Remyelination as neuroprotection. In: Waxman SG, editor. Multiple Sclerosis as a Neuronal Disease. Elsevier Academic Press; San Diego: 2005. pp. 389–419. [Google Scholar]
- Johnson, AJ, et al. Preservation of motor function by inhibition of CD8+ virus peptide-specific T cells in Theiler’s virus infection. Faseb J. 2001;15:2760–2. doi: 10.1096/fj.01-0373fje. [DOI] [PubMed] [Google Scholar]
- Kagi, D, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–7. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- Levy, M, et al. Theiler’s virus replication in brain macrophages cultured in vitro. J.Virol. 1992;66:3188–3193. doi: 10.1128/jvi.66.5.3188-3193.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman, J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. 2003;3:361–70. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- Lindsley, MD, et al. Coexpression of class I major histocompatibility antigen and viral RNA in central nervous system of mice infected with Theiler’s virus: a model for multiple sclerosis. Mayo Clin Proc. 1992;67:829–38. doi: 10.1016/s0025-6196(12)60820-9. [DOI] [PubMed] [Google Scholar]
- Lipton HL. Theiler’s virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect.Immun. 1975;11:1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald WI, Sears TA. Effect of demyelination on conduction in the central nervous system. Nature. 1969;221:182–3. doi: 10.1038/221182a0. [DOI] [PubMed] [Google Scholar]
- McGavern DB, et al. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. 2000;123(Pt 3):519–31. doi: 10.1093/brain/123.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, et al. Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J Neurosci Res. 1999a;58:492–504. doi: 10.1002/(sici)1097-4547(19991115)58:4<492::aid-jnr3>3.0.co;2-p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, et al. Quantitative assessment of neurologic deficits in a chronic progressive murine model of CNS demyelination. Exp Neurol. 1999b;158:171–81. doi: 10.1006/exnr.1999.7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana, I, et al. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol. 2001;159:809–15. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metkar SS, et al. Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity. 2002;16:417–28. doi: 10.1016/s1074-7613(02)00286-8. [DOI] [PubMed] [Google Scholar]
- Mews, I, et al. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult Scler. 1998;4:55–62. doi: 10.1177/135245859800400203. [DOI] [PubMed] [Google Scholar]
- Murray PD, et al. Perforin-dependent neurologic injury in a viral model of multiple sclerosis. J Neurosci. 1998a;18:7306–14. doi: 10.1523/JNEUROSCI.18-18-07306.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PD, et al. CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J Virol. 1998b;72:7320–9. doi: 10.1128/jvi.72.9.7320-7329.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, S, et al. Imaging of axonal damage in multiple sclerosis: spatial distribution of magnetic resonance imaging lesions. Ann Neurol. 1997;41:385–91. doi: 10.1002/ana.410410314. [DOI] [PubMed] [Google Scholar]
- Neumann, H, et al. Induction of MHC class I genes in neurons. Science. 1995;269:549–52. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- Njenga MK, et al. The immune system preferentially clears Theiler’s virus from the gray matter of the central nervous system. Journal of Virology. 1997a;71:8592–8601. doi: 10.1128/jvi.71.11.8592-8601.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njenga MK, et al. Interferon a/b mediates early virus-induced expresson of H-2D and H-2K in the central nervous system. Lab Invest. 1997b;77:71–84. [PubMed] [Google Scholar]
- Olivier, E, et al. An electrophysiological study of the postnatal development of the corticospinal system in the macaque monkey. J Neurosci. 1997;17:267–76. doi: 10.1523/JNEUROSCI.17-01-00267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce ML, Rodriguez, M. Erichrome stain for myelin on osmicated tissue embedded in glycol methacrylate plastic. J Histotechnol. 1989;12:35–36. [Google Scholar]
- Rahemtulla, A, et al. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 1991;353:180–4. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- Raja SM, et al. Cytotoxic granule-mediated apoptosis: unraveling the complex mechanism. Curr Opin Immunol. 2003;15:528–32. doi: 10.1016/s0952-7915(03)00111-0. [DOI] [PubMed] [Google Scholar]
- Raja SM, et al. Cytotoxic cell granule-mediated apoptosis. Characterization of the macromolecular complex of granzyme B with serglycin. J Biol Chem. 2002;277:49523–30. doi: 10.1074/jbc.M209607200. [DOI] [PubMed] [Google Scholar]
- Rivera-Quinones, C, et al. Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nat Med. 1998;4:187–93. doi: 10.1038/nm0298-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, M. Immunoglobulins stimulate central nervous system remyelination: electron microscopic and morphometric analysis of proliferating cells. Lab Invest. 1991;64:358–70. [PubMed] [Google Scholar]
- Rodriguez, M, et al. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Ann Neurol. 1983;13:426–33. doi: 10.1002/ana.410130409. [DOI] [PubMed] [Google Scholar]
- Rossi CP, et al. Role of macrophages during Theiler’s virus infection. Journal of Virology. 1997;71:3336–3340. doi: 10.1128/jvi.71.4.3336-3340.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JC, et al. Magnetic resonance imaging. Clinical correlation in 64 patients with multiple sclerosis. Arch Neurol. 1986;43:1145–8. doi: 10.1001/archneur.1986.00520110039011. [DOI] [PubMed] [Google Scholar]
- Truyen, L, et al. Accumulation of hypointense lesions (“black holes”) on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology. 1996;47:1469–76. doi: 10.1212/wnl.47.6.1469. [DOI] [PubMed] [Google Scholar]
- Tsunoda, I, et al. Induction of autoreactive CD8+ cytotoxic T cells during Theiler’s murine encephalomyelitis virus infection: implications for autoimmunity. J Virol. 2002;76:12834–44. doi: 10.1128/JVI.76.24.12834-12844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure, D, Rodriguez, M. Extensive injury of descending neurons demonstrated by retrograde labeling in a virus-induced murine model of chronic inflammatory demyelination. J Neuropathol Exp Neurol. 2000;59:664–78. doi: 10.1093/jnen/59.8.664. [DOI] [PubMed] [Google Scholar]
- Ure DR, Rodriguez, M. Preservation of neurologic function during inflammatory demyelination correlates with axon sparing in a mouse model of multiple sclerosis. Neuroscience. 2002;111:399–411. doi: 10.1016/s0306-4522(02)00012-x. [DOI] [PubMed] [Google Scholar]
- van Waesberghe JH, et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol. 1999;46:747–54. doi: 10.1002/1531-8249(199911)46:5<747::aid-ana10>3.3.co;2-w. [DOI] [PubMed] [Google Scholar]
- Voskoboinik, I, Trapani JA. Addressing the mysteries of perforin function. Immunol Cell Biol. 2006;84:66–71. doi: 10.1111/j.1440-1711.2005.01409.x. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Determinants of conduction velocity in myelinated nerve fibers. Muscle Nerve. 1980;3:141–50. doi: 10.1002/mus.880030207. [DOI] [PubMed] [Google Scholar]