

Abstract
The accumulation of misfolded proteins is associated with various neurodegenerative conditions. Peripheral myelin protein 22 (PMP22) is a hereditary neuropathy-linked, short-lived molecule that forms aggresomes when the proteasome is inhibited or the protein is mutated. We previously showed that the removal of pre-existing PMP22 aggregates is assisted by autophagy. Here we examined whether the accumulation of such aggregates could be suppressed by experimental induction of autophagy and/or chaperones. Enhancement of autophagy during proteasome inhibition hinders protein aggregate formation and correlates with a reduction in accumulated proteasome substrates. Conversely, simultaneous inhibition of autophagy and the proteasome augments the formation of aggregates. An increase of heat shock protein levels by geldanamycin treatment or heat shock preconditioning similarly hampers aggresome formation. The beneficial effects of autophagy and chaperones in preventing the accumulation of misfolded PMP22 are additive and provide a potential avenue for therapeutic approaches in hereditary neuropathies linked to PMP22 mutations.
Keywords: autophagosome, proteasome, neuropathy, Schwann cells, protein misfolding, aggresomes, heat shock proteins, heat shock
Introduction
In order to maintain cellular homeostasis, different quality control mechanisms evolved which prevent the accumulation of misfolded or nonfunctional polypeptides in aggregates (Goldberg et al., 2003). When the correct folding and/or trafficking of a nascent polypeptide fail, the protein is commonly targeted for degradation by the ubiquitin-proteasome system (UPS) (Goldberg et al., 2003). The proteasome is an enzymatic complex that contains a catalytic chamber inside of which unfolded polypeptides are degraded (Groll and Clausen, 2003). In addition to the proteasome, unwanted cytoplasmic proteins can be degraded within lysosomes. Macroautophagy, from here on referred to as autophagy, is a constitutive event by which cytoplasmic cargo is engulfed in double membrane-bound structures called autophagosomes that later fuse with lysosomes, ensuring degradation of the cargo (Klionsky and Emr, 2000). When the balance between synthesis/folding and degradation pathways is disrupted, the accumulation of proteins in aggregates is favored, which constitutes the pathological hallmark of several neurodegenerative conditions (Ciechanover and Brundin, 2003).
Peripheral myelin protein 22 (PMP22) is a hydrophobic, aggregation-prone, membrane protein expressed mainly in myelinating Schwann cells (SCs) (Snipes et al., 1992; Tobler et al., 2002). In non-myelinating and myelinating SCs, the majority (~80%) of the newly-synthesized PMP22 is rapidly degraded by the UPS, presumably due to misfolding (Pareek et al., 1997). Mutations in PMP22, or duplication of the PMP22 gene, are associated with hereditary demyelinating neuropathies among which, Charcot Marie Tooth type 1A (CMT1A) is the most common form (Young and Suter, 2001). Although the molecular mechanisms underlying CMT1A are not well understood, halted intracellular protein trafficking and formation of aggregates are believed to play roles (D’Urso et al., 1998; Tobler et al., 1999; Naef and Suter, 1999; Colby et al., 2000; Fortun et al., 2003; 2006). In SCs of CMT1A mouse models, the spontaneous accumulation of misfolded PMP22 in aggregates correlates with impaired proteasome activity (Fortun et al., 2005; 2006). These aggregates resemble PMP22 inclusions formed in cultured SCs upon pharmacological inhibition of the proteasome, termed aggresomes (Johnston et al., 1998; Notterpek et al., 1999).
It is still debated whether the formation of cytosolic protein aggregates, such as of PMP22, are harmful or protective in given disease paradigms. In models of CMT1A, PMP22 aggregates associate with autophagosomes and lysosomes, as well as heat shock proteins (HSPs), suggesting endogenous activation of these pathways in response to the presence of misfolded proteins (Notterpek et al., 1997; Ryan et al., 2002; Fortun et al., 2003; 2005). Cooperation of the aggresome and autophagy in the removal of aggregation-prone proteins is supported by pharmacologic studies in HeLa cells (Iwata et al., 2005). Using a similar approach, previously we showed that removal of pre-existing aggresomes formed by endogenous PMP22 is aided by autophagy (Fortun et al., 2003). A second cellular mechanism that can influence protein aggregation is the heat shock response. HSPs are molecular chaperones that prevent protein aggregation, either by aiding degradation via chaperone-mediated autophagy (CMA) or the UPS, or by promoting re-folding (Muchowski and Wacker, 2005). In proteasome-inhibited cells, overexpressed wild type (wt) and mutant PMP22s, as well as the spontaneous aggregates in neuropathic mouse nerves, recruit HSPs (Ryan et al., 2002; Fortun et al., 2003). Nonetheless, it is not known if elevated levels of chaperones are able to influence the folding efficiency of PMP22.
As a step toward the development of therapeutic approaches for PMP22-associated neuropathies, here we asked whether the induction of autophagy and/or HSPs could prevent the formation of cytosolic PMP22 aggregates. Indeed, the stimulation of autophagy or the heat shock response is able to significantly reduce the formation of aggregates, and relieve the impairment of the proteasome. These results support the further development of pharmacologic agents targeted at modulating the autophagic and chaperone systems.
Materials and Methods
Animal housing
Trembler J (TrJ) (Jackson laboratories) mouse breeding colony is housed under SPF conditions at the McKnight Brain Institute animal facility. The use of animals for these studies is approved by a University of Florida Institutional Animal Care and Use Committee. Genomic DNA was isolated from tail biopsies of mouse pups up to 8-days old and litters were genotyped by PCR (Notterpek et al., 1997). For the Western blotting experiments, sciatic nerves from 6-month old (adult) wt and heterozygous neuropathic mice were used. For the primary mouse SC cultures, sciatic nerves from genotyped postnatal day 5 (P5) littermates were used.
Cell culture and aggresome formation
Primary SC cultures were established from newborn rat or P5 mouse pups (Ryan et al., 2002). Rat SCs were grown to ~80% confluency in 10% fetal calf serum (Hyclone, Logan, UT), 5 μM forskolin (Calbiochem, La Jolla, CA) and 10 μg/ml bovine pituitary extract (Biomedical Technologies Inc, Stoughton, MA) containing Dulbecco's modified eagle's medium. SCs were treated with the proteasome inhibitor lactacystin (Lc) (10 μM) (Biomol Research Laboratories, PA), or its vehicle, dimethlysulfoxide (DMSO) (Sigma), as a control (Notterpek et al., 1999). Treatments were performed for 12 h or 16 h, as indicated in the text. The proteasome inhibitors epoxomicin (5 μM) and MG-132 (20 μM) (both from Biomol) were also used to exclude that effects were unique to lactacystin. After each treatment paradigm, the cells were immediately processed for immunostaining to visualize the aggregates, or for Western blotting to evaluate the accumulation of ubiquitinated proteins (Notterpek et al., 1999). Primary mouse SCs from wt and neuropathic nerves were used within 24 h of culture, without pharmacologic treatments.
Visualization of autophagosomes by direct fluorescence microscopy
Autophagic vacuoles were visualized by labeling with monodansylcadaverine (MDC) or GFP-LC3 (Munafo and Colombo, 2001; Kabeya et al., 2000). SCs from wt or TrJ mice grown on coverslips were labeled with 100 μM MDC (Sigma) for 1h in serum free medium (DMEM) and immediately imaged, or fixed with 4% paraformaldehyde prior to imaging. Rat or mouse SCs plated at equal densities were transfected with GFP-LC3 plasmid (Kabeya et al., 2000) using the lipofectamine method (Invitrogen, Life Technology, Carlsbad, CA). Transfection efficiency was ~36% using the GreenLantern plasmid (Invitrogen). Twenty-four hours after transfection, the relationship between spontaneous (mouse SCs) or Lc-induced (16 h, 10 μM in rat SCs) aggregates and GFP-LC3 fluorescence was evaluated by immunostaining using anti-PMP22 antibodies, as explained in the text.
Ultrastuctural studies
Rat SCs treated with or without proteasome inhibitors for 12 h or 16 h, were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.5), with 7% sucrose, at 4°C for 60 min. The specimens were then treated with 2% OsO4 and uranyl acetate, and dehydrated and embedded in Epon resin. For localization of acid phosphatase (AcP) activity, the samples were treated with cytidine 5′-monophosphate and cerium chloride after fixation (Lenk et al., 1992). Thin sections were examined on a JEOL 100CX transmission electron microscope. The volume of autophagosomes divided by the volume of cytoplasm was determined in eight independent samples of proteasome inhibited or control cells. The results are presented as percentages of cytoplasm volumes. Statistical significance between the various paradigms was determined using a Student's t-test.
Modulation of autophagy and molecular chaperones
To determine the effects of autophagy and molecular chaperones on the formation of protein aggregates, proteasome inhibition was carried out in the absence or the presence of the following modulators. For the stimulation of autophagy, SCs were incubated in media deprived of amino acids and serum (starvation medium) (Fortun et al., 2003), or treated with rapamycin (200 nM) (Calbiochem) (Ravikumar et al., 2004). To block autophagy, 3-methyladenine (3-MA) (10 mM) (Sigma) was included, without or during proteasome inhibition, in normal or starvation medium (Fortun et al., 2003). The effects of elevated HSPs were evaluated by two approaches, with and without Lc. In the first paradigm, cells were treated with geldanamycin (GA) (125 nM-2 μM) for 16 h (McLean et al., 2004). Alternatively, cells were incubated at 45°C for 20 min (heat shock; HS) followed by a chase for 0–16 h (Allen et al., 2004). For the combined treatment, cells were first exposed to a 20 min HS, followed by incubation at 37°C in fresh starvation medium, with or without Lc. Cells with aggresomes were visualized by immunostaining with monoclonal anti-PMP22 and polyclonal anti-ubiquitin antibodies (see below).
Immunofluorescence and aggresome quantification
Cultured SCs on glass coverslips were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 for 15 min at room temperature. After blocking with 10% normal goat serum, the samples were incubated with the indicated primary antibodies overnight at 4 °C. Bound antibodies were detected using Alexa Fluor 594-conjugated (red) anti-rabbit and Alexa Fluor 488-conjugated (green) anti-mouse antibodies (Molecular Probes, Eugene, OR). Samples were then mounted using the Prolong Antifade kit (Molecular Probes). Images were acquired with a SPOT digital camera (Diagnostic Instrumentals, Sterling Heights, MI) attached to a Nikon Eclipse E800 (Tokyo, Japan), or an Olympus MRC-1024 confocal microscope. The number of cells with aggresomes was quantified and expressed as a percent of total cells, as detected by nuclear staining with Hoechst #33258 (Molecular Probes, Eugene, OR). All experiments were repeated independently at least three times, and in each one, cells in eight different visual fields (0.277 μm2 actual area) were counted. The criteria for aggresome counting included: 1) immunoreactivity for ubiquitin and PMP22; 2) assembly at perinuclear region; 3) exclusion from ER and Golgi and 4) diameter larger than 1 μm. Statistical significance between the various treatment paradigms was evaluated using a Student's t-test. Images were processed for printing using Adobe Photoshop 5.0 (Adobe Systems, San Jose, CA).
Primary Antibodies
A mouse monoclonal antibody was used to detect PMP22 in the studied samples (Chemicon, Temencula, CA). To monitor autophagy, a polyclonal rabbit anti-microtubule-associated protein 1, light chain 3 (LC3) antibody, generated against an eighteen amino acid portion of the human protein [SEKTFKQRRTFEQRVEDV(C)], was utilized. Antibodies for protein chaperones included anti-calnexin, anti-heat shock protein 70 (Hsp 70), Hsp 40 and αB-crystallin (all from Stressgen, Victoria British Columbia, Canada). Monoclonal anti-actin, -tubulin (both from Sigma), -ubiquitin (Santa Cruz Biotechnology, CA) and polyclonal anti-GFP (gift from Dr. G. Shaw, University of Florida), -ubiquitin (Dako, Carpinteria, CA), cathepsin D (Cortex Biochem, San Leandro, CA) were obtained from the indicated suppliers.
Western blot analyses
Frozen sciatic nerves from 6-month old genotyped mice were crushed under liquid nitrogen, lysed in immunoprecipitation buffer [10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 1% Nonidet P-40, 0.5% deoxycholate and 150 mM NaCl] supplemented with a mixture of protease inhibitors (Complete™; Roche, Indianapolis, IN) (Fortun et al., 2003). For each lysate, at least six nerves from three mice per genotype were used. The lysates were microcentrifuged, the supernatant removed and the insoluble material incubated with 10 mM Tris-HCl, 3% SDS for 10 min at room temperature. Following a brief sonication, total protein concentrations were measured by BCA assay (Pierce, Rockford, IL). After exposure to the specified conditions, cultured rat SCs were lysed with 3% sodium dodecylsulfate (SDS) containing gel sample buffer, supplemented with protease inhibitors (Fortun et al., 2006). Protein lysates were separated on SDS gels and transferred onto nitrocellulose or PVDF membranes (for ubiquitin and LC3). Blots were blocked and incubated with the indicated primary antibodies. After incubation with anti-rabbit or anti-mouse horseradish peroxidase (HRP) conjugated secondary antibodies (Sigma, St. Louis, MO), membranes were reacted with an enhanced chemiluminescent substrate (Perkin Elmer, Boston, MA). Films were digitally imaged using a GS-710 densitometer (Bio-Rad Laboratories).
Proteasome reporter assay
To monitor ubiquitin-mediated proteolysis by the UPS, a green fluorescent protein (GFP)-based substrate was utilized (Dantuma et al., 2000). Rat SCs plated at equal densities were transfected with an UbG76V-GFP reporter construct using the lipofectamine method (Invitrogen, Life Technology, Carlsbad, CA). The attachment of a mutated uncleavable ubiquitin moiety to the normally very stable GFP results in a fusion protein (UbG76V-GFP) that is rapidly degraded by the proteasome and is hardly detected under normal conditions (Dantuma et al., 2000). The efficiency of transfection was ~43% using the GreenLantern GFP plasmid (Invitrogen) (Fortun et al., 2006). Twenty-four hours after transfection, the cells were treated with Lc (10 μM) with or without autophagy modulation, as explained in the text. Sixteen hours later, cells were imaged for GFP expression and phase contrast using a T1-SM inverted microscope equipped with a Nikon DS-L1 camera. Cellular levels of GFP were determined by Western blotting using anti-GFP and anti-tubulin (loading control) antibodies. The intensity of bands was quantified using Scion Images Software (Scion, Frederick, MD). The relative levels of UbG76V-GFP were expressed as the absorbance units of the GFP band, after correction for tubulin. The results from duplicates of three independent experiments were averaged, graphed and analyzed using a Student’s t-test.
Results
Correlation between the formation of spontaneous PMP22 aggregates and autophagy
Spontaneous protein aggregates accumulate in the cytoplasm of SCs from CMT1A-like neuropathic mice based on PMP22 point mutation (TrJ) or overexpression of the wt protein (Ryan et al., 2002; Fortun et al., 2003; 2006). Morphological studies show that these aggregates are often surrounded by lysosomes and autophagosomes, suggesting ongoing autophagy. To further examine the involvement of autophagy in neuropathic samples, primary SCs derived from P5 TrJ mouse nerves were examined (Fig. 1). When SCs from wt and TrJ neuropathic mice are incubated with MDC dye, a marker for autophagic vacuoles, brighter and more numerous vesicles are detected in TrJ, as compared to wt cells (Fig. 1A). As a biochemical measure of autophagy, next we evaluated the levels and lipidation of the autophagic marker LC3 (Fig. 1B). After cleavage of the newly-synthesized LC3, the cytosolic form (LC3 I) becomes lipidated (LC3 II) and incorporates into the limiting membrane of autophagosomes, which is detected as a change in mobility on gel electrophoresis (Tanida et al., 2004). Analysis of total (T), detergent-soluble (S), and -insoluble (I) fractions of sciatic nerve lysates from wt and TrJ mice reveals an increase in the levels of LC3 I in the neuropathic sample, as compared to wt. Furthermore, LC3 II, the lipidated form bound to the autophagosome membrane, is below detection in lysates of wt nerves, but prominent and readily visible in TrJ. Surprisingly, only a small fractions of LC3 I, but not LC3 II, are found in the detergent-insoluble fraction. It is feasible that LC3 I associates with the aggregates and hence its detergent-insolubility, whereas the binding of LC3 II to the autophagosome membrane confers solubility to the aggregate in its route to degradation.
Figure 1. Aggregates in neuropathic SCs recruit autophagosomes.
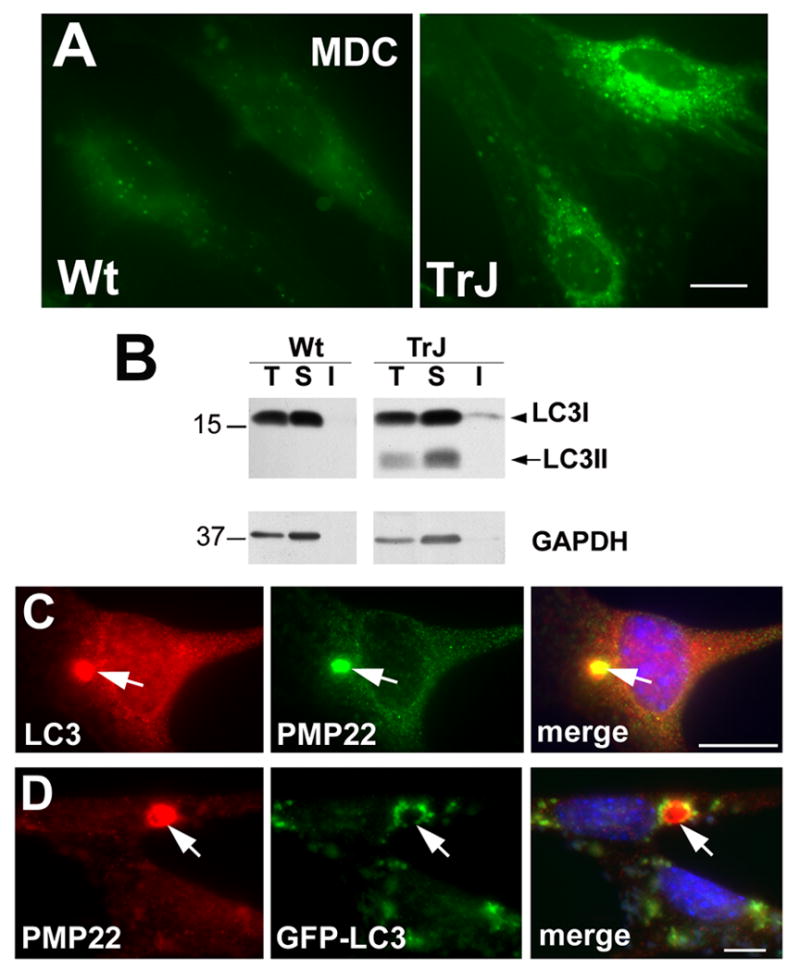
(A) Autophagosomes were visualized by MDC dye uptake in acutely cultured SCs from wt and TrJ mouse nerves. Images shown were collected at identical settings of the camera. (B) Whole lysates (T), detergent-soluble (S) and detergent-insoluble (I) fractions from wt and TrJ neuropathic mouse nerves (15 μg/lane) were blotted with anti-LC3 antibodies. The arrowhead and arrow point at the LC3 I and LC3 II forms, respectively. Molecular mass are indicated at the left, in kDa. (C) The localization of endogenous LC3 was evaluated by double labeling with anti-LC3 (red) and anti-PMP22 (green) antibodies in SCs from TrJ mice. (D) The recruitment of autophagosomes (green) to PMP22 aggregates (red) is visualized by direct GFP fluorescence in GFP-LC3 transfected TrJ cultures (D). Scale bars, 10 μm.
To examine the spatial association of autophagosomes with PMP22 aggregates, primary SCs isolated from P5 TrJ mice were costained with anti-LC3 and PMP22 antibodies (Fig. 1C). The merged image shows a perinuclear PMP22 aggregate in close association with the endogenous LC3. In agreement, cultures transfected with a GFP-LC3 plasmid to visualize autophagic vacuoles by direct fluorescence, show GFP-labeled autophagosomes in the vicinity of PMP22 aggregates (Fig. 1D). Autophagosmes were also detected near protein aggregates in nerves and SCs isolated from PMP22 overexpressor mice (Fortun et al., 2006). Together, these results indicate that compared to wt, autophagy is induced in neuropathic mouse nerves containing spontaneous aggregates of mutated (Fig. 1) or overproduced PMP22 (Fortun et al., 2006).
PMP22 aggregates formed under proteasome inhibition recruit autophagosomes
A large fraction of the newly-synthesized wt PMP22 is substrate for the UPS and forms aggregates when this pathway is inhibited (Notterpek et al., 1999). Specifically, over ninety percent of cultured SCs contain aggresomes of endogenous PMP22 after a 16 h treatment with proteasome inhibitors (Notterpek et al., 1999; Ryan et al., 2002; Fortun et al., 2003). These aggregates fulfill all established characteristics of aggresomes, including exclusion from the main organelles, recruitment of ubiquitin and microtubule-dependent accumulation (Johnston et al., 1998; Notterpek et al., 1999). Using this in vitro model, we sought to evaluate the autophagic response during de novo formation of PMP22 aggregates (Figs. 2 and 3). First, the levels of LC3 in lysates of lactacystin-treated rat SCs were examined (Fig. 2A, B). Similar to the neuropathic samples (Fig. 1), the formation of PMP22 aggresomes correlates with higher levels of LC3, both LC3 I and the lipidated LC3 II forms, as compared to DMSO treated controls (Fig. 2A). As positive controls for the activation of autophagy and for the conversion of LC3 I to LC3 II, cells treated with rapamycin (Rp) were included (Fig. 2A). Quantification of LC3 levels in five independent experiments, after correction for actin, corroborates the increase in LC3 I and II in proteasome-inhibited cells (Fig. 2B). Significantly, the ratio of LC3 II/I is increased nearly 2-fold (0.287 ± 0.221 vs. 0.429 ± 1.385), as compared to control cells (p=0.05). Similar results were obtained when using other proteasome inhibitors, such as epoxomicin or MG132 (not shown). The elevated levels of LC3 are also noticeable by immunostaining (Fig. 2C). In DMSO treated cells, LC3-like immunoreactivity is low (red), with a diffuse distribution throughout the cell (Fig. 2C, inset). After 16 h of proteasome inhibition, LC3-labeled autophagosomes (red) are detected prominently at the periphery of PMP22 aggregates (green). Partial co-localization of LC3 and PMP22 is also evident (yellow on merged image). Utilizing the LC3-GFP transfection approach as for the primary mouse SCs (Fig. 1D), green fluorescent autophagosomes are detected around the Lc-induced PMP22 aggregates (red) (Fig. 2D). A similar induction of autophagy in response to proteasome inhibition has been observed in ALLN treated HEK-293 cells (Iwata et al., 2005).
Figure 2. Changes in the levels, lipidation and localization of LC3 in PMP22 aggresome-containing cells.
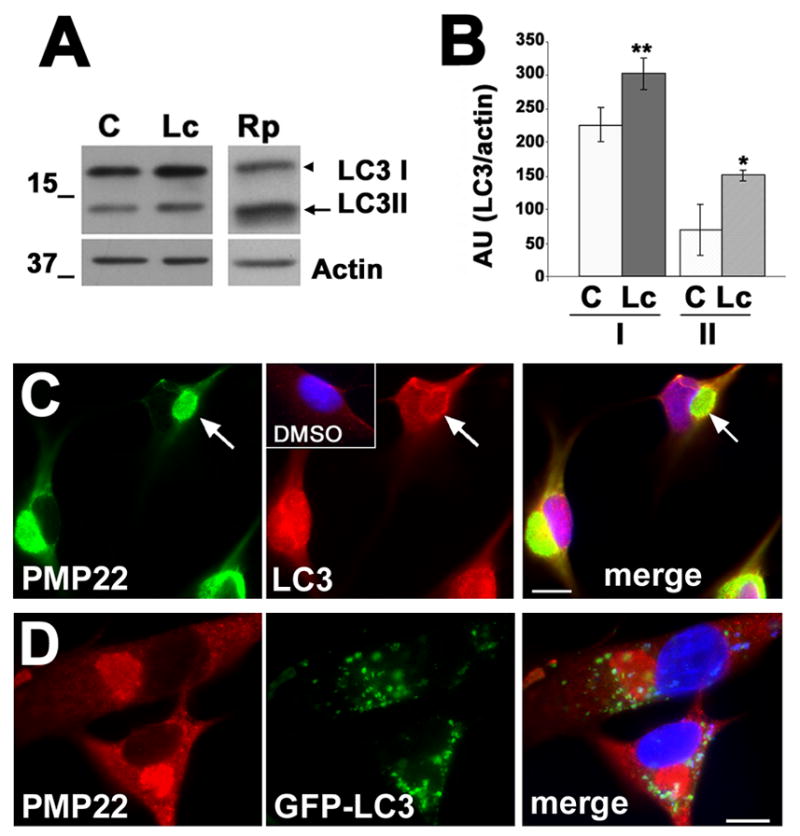
(A) The levels of LC3 and actin in DMSO control (C), Lc or rapamycin (Rp) treated (16 h) cell lysates (20 μg/lane) were determined by western blot. The arrowhead and arrow point at LC3 I and LC3 II, respectively. Molecular mass are indicated at the left, in kDa. (B) The levels of LC3, after correction for actin, were quantified in five independent experiments. AU: absorbance units; p<0.05*; p<0.005**. (C) PMP22 aggresomes (green) recruit endogenous LC3 (red) in Lc-treated (16 h) cells. The localization of LC3 in control SCs is shown (inset, red). The arrows denote LC3 in the periphery of a PMP22 aggresome. (D) Lc-induced PMP22 aggregates (red) are surrounded by GFP-LC3 containing autophagosomes. Nuclei are visualized by Hoechst dye (blue) (merge, C, D). Scale bars, 10 μm (C, D).
Figure 3. An increase in autophagosome number and size correlates with aggresome formation.
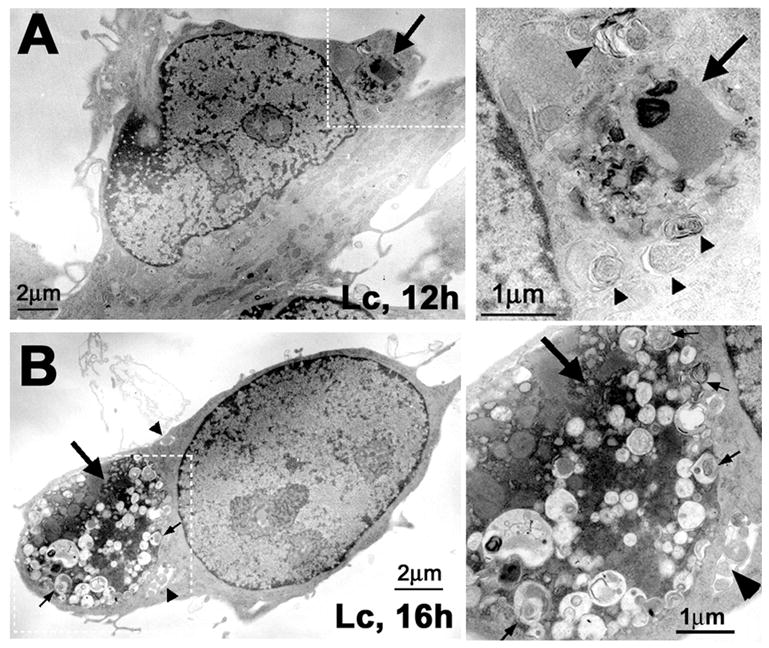
(A) Normal rat SCs, after 12 h or (B) 16 h treatment with the proteasome inhibitor Lc (10 μM) were processed for ultrastuctural analysis. The large arrows represent electron dense inclusions formed at the different time points. Autophagic vacuoles are visible within (thin arrows) and around the inclusions (arrowheads). Enlargements of the boxed areas in A and B are shown at the right. Scale bars, as indicated.
To visualize the recruitment of autophagosomes during the formation of aggresomes, SCs incubated with lactacystin for 12 or 16 h were examined by transmission electron microscopy (Fig. 3). After the 12 h treatment, as determined by anti-ubiquitin and anti-PMP22 immunostainings, about 57% of the cells contain aggregates (see quantification below in Fig. 5B). The aggregates are electron dense amorphous inclusions that form in the vicinity of the nucleus, and many of them are surrounded by autophagosomes (Fig. 3A). The boxed area in Fig. 3A is enlarged on the right to show autophagosomes (arrowheads) at the periphery of the aggregate (arrow). After extended proteasome inhibition (16 h), over 90% of SCs cells form large protein aggregates (Notterpek et al., 1999, and also see Fig. 5 below) many of which contain autophagosomes within (narrow arrows) and around (arrowheads) the inclusion (Fig. 3B). The size and the morphology of the autophagosomes appear heterogeneous, suggestive of the different stages in their formation and maturation (Fig. 3B, right). To quantify the activation of autophagy, the volume of autophagosomes relative to the cytoplasm volume was determined in eight independent samples. After 12 h of proteasome inhibition, the volume of autophagosomes is increased by ~4%, but not significantly (p=0.257), as compared to untreated control cells. However, a 7-fold increase (2.43 ± 1.23 vs. 16.92 ± 8.84) was observed after 16 h (p=0.002), when nearly all the cells contain aggresomes. This data suggest that autophagy is enhanced coincident with the formation of PMP22 aggregates.
Figure 5. The formation of PMP22 aggregates is modulated by autophagy.
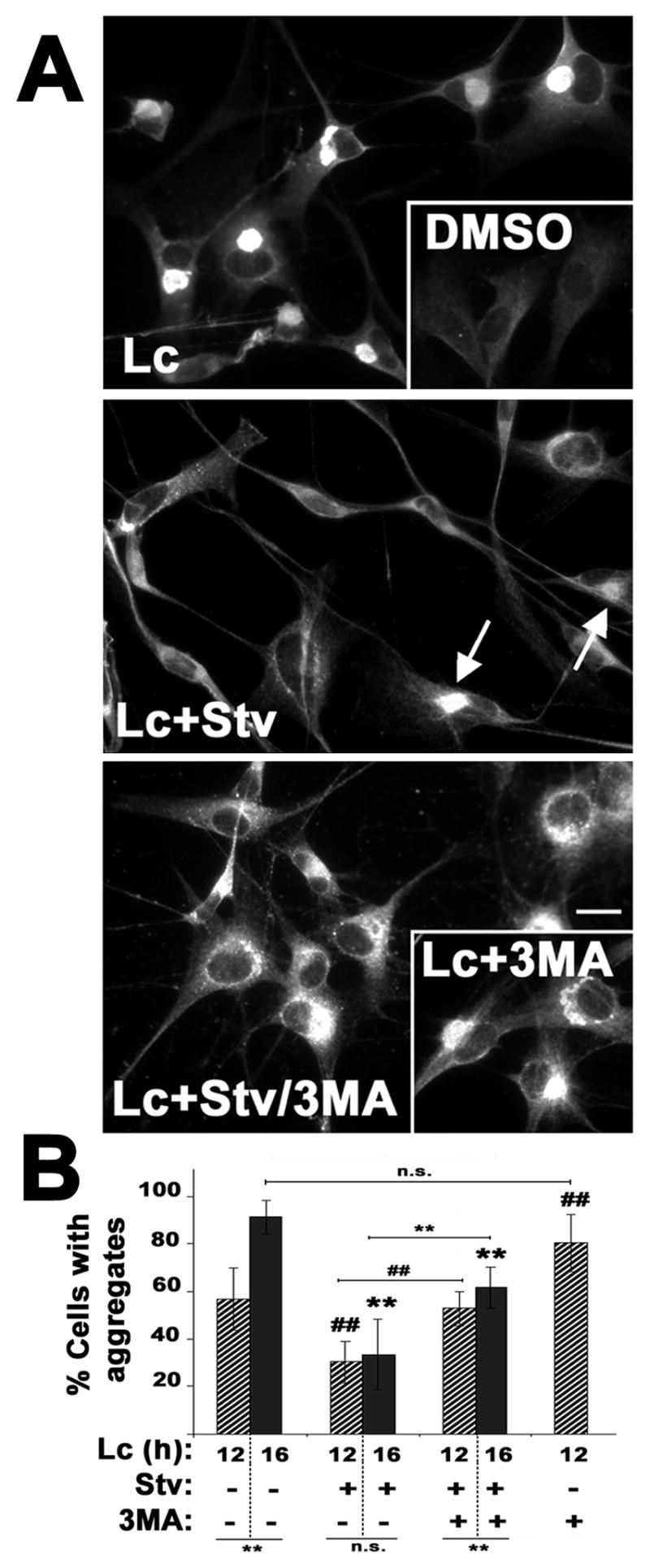
(A) The localization of PMP22 was determined in primary rat SCs treated with Lc (16 h) or DMSO, as control (inset). To stimulate autophagy, Lc treatment was performed under amino acid and serum-depleted starvation conditions (Lc+Stv). Arrows indicate aggregates after simultaneous inhibition of the proteasome and induction of autophagy. To block autophagy, 3-MA was included in proteasome-inhibited cells, during fed (Lc+3-MA, right inset) or starved (Lc+Stv/3-MA) conditions. Scale bar, 10 μm. (B) The percentage of cells with PMP22 aggregates was quantified after 12 h (diagonal bars), or 16 h (solid bars) treatment paradigms. The statistical significance of differences between the various treatment paradigms at 16 h (**, p<0.005), or at 12 h (##, p<0.005) are denoted. Connecting horizontal lines represent the significance between corresponding treatments (**, p<0.005; n.s., no significance).
Autophagosomes after fusing with lysosomes become autolysosomes and acquire acid phosphatase (AcP) and cathepsins (Klionsky and Emr, 2000). Therefore, the presence of the lysosomal enzyme, AcP, in aggresome-containing cells was examined by monitoring enzymatic activity (Lenk et al., 1992) (Fig. 4A). Lysosomal structures, containing AcP (dark reaction product) are seen in control samples (Fig. 4A, control). After treatment with Lc for 16 h, however, more lysosomes (smaller, round, uniform) and autolysosomes (larger, less uniform) are visible, mainly near the nucleus (Fig. 4A, Lc). Furthermore, the Golgi apparatus appears reactive for AcP in Lc-treated, but not in control cells (Fig. 4A, arrows), suggesting the de novo synthesis and transport of AcP through the secretory pathway. The association of another lysosomal enzyme cathepsin D with Lc-induced PMP22 aggregates was examined by double immunolabeling and confocal microscopy (Fig. 4B, C). On representative single plane images, cathepsin D-positive vesicles accumulate in the region of PMP22 aggregates (Fig. 4B). The relationship of lysosomes (red) with PMP22 aggregates (green) is resolved in more detail on single plane rotated sections through a specific aggregate (arrow) (Fig. 3C). The red and yellow color within the green aggregate on the x and y sections is in agreement with the morphological detection of autolysosomes within the inclusions (Fig. 3). Higher numbers and volume of autophagosomes and autolysosomes, as well as increased levels of LC3 II (Figs. 1–3) indicate the induction of autophagy by the accumulation of misfolded PMP22.
Figure 4. Lysosomal enzymes are present at sites of protein aggregation.
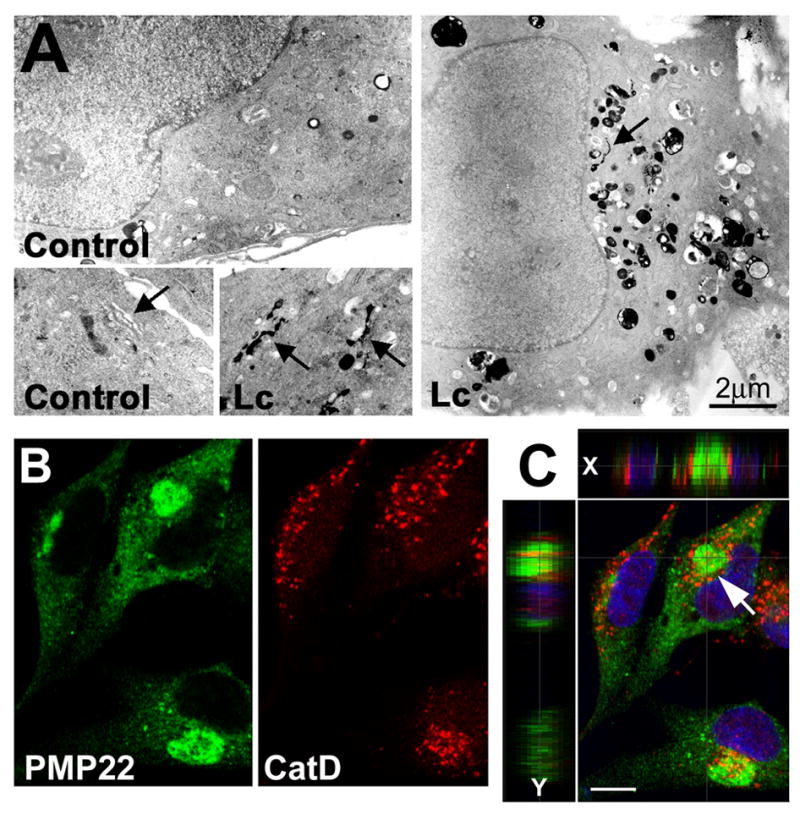
(A) Control (DMSO) or Lc-treated (16 h) primary rat SCs were processed for acid phosphatase (AcP) cytochemistry. Arrows point at Golgi stacks, which are AcP-positive in Lc-treated, but not in control cells. (B) Single plane confocal images of Lc-treated SCs reveal the close association of PMP22 aggresomes (green) and cathepsin D-positive lysosomes (red). (C) A merged image of the same field is shown, with single x and y plane sections across the marked aggresome (arrow). These single plane sections reveal the accumulation of lysosomes (red) around the aggregate (green) (x axis) and their entrapment within the aggregate (y axis slice). Nuclei are visualized by Hoechst stain (blue). Scale bars, as indicated in A, and 10 μm for B and C.
The formation of aggresomes is hindered by autophagy
Previously we have shown that experimental activation of autophagy by nutrient deprivation aids the removal of pre-existing PMP22 aggregates (Fortun et al., 2003). The studies described above indicate (Figs. 1–3) that the formation of protein aggregates in itself, either in response to impaired proteasomal degradation or spontaneously due to mutations, endogenously induces autophagy. Therefore, we wanted to test if further enhancement of autophagy by starvation conditions could potentially prevent or reduce the accumulation of misfolded PMP22 (Fig. 5). Normal rat SCs treated with lactacystin for 16 h form aggresomes in nearly all of the cells (Fig. 5A) (Notterpek et al., 1999), without significant cell death (Fortun et al., 2003). In DMSO carrier-treated cells, PMP22 is detected at low levels in the ER and Golgi apparatus, and at the plasma membrane (Fig. 5A, DMSO inset, also see Pareek et al., 1997). When simultaneous to proteasome inhibition, autophagy is enhanced (starvation, Stv), fewer cells form aggregates (Fig. 5A, Lc+Stv). To confirm the specificity of this effect, the same experiment was carried out in the presence of 3-MA, a known blocker of autophagy (Dorn et al., 2001). When 3-MA is included (Fig. 5A, Lc+Stv/3-MA), the beneficial effect of starvation is partially lost, corroborating that the reduction in the number of aggresomes was autophagy-dependent. Aggregates formed under these conditions appear less compact compared to Lc treatment alone, suggesting that the inhibition of autophagy by 3-MA is incomplete. The levels or the localization of PMP22 are not affected by incubation of the cells in starvation medium, or 3-MA alone (not shown, see Fortun et al., 2003).
The effect of enhanced autophagy on reducing the formation of aggregates was quantified in three independent experiments after 12 h (diagonal bars) or 16 h (solid bars) treatment paradigms (Fig. 5B). The criteria for aggresome counting included immunoreactivity for ubiquitin and PMP22, assembly at the perinuclear region, exclusion from ER and Golgi, and a diameter larger than 1 μm (Notterpek et al., 1999). Note, the lack of statistically significant differences between the 12 h and 16 h Lc+Stv treatments, suggesting that the enhancement of autophagy halts the formation of aggresomes. Even after 24 h of proteasome inhibition and starvation, the percentage of cells with aggregates remains similar to 12 h or 16 h (34%±12; p=0.97). Analogous results were obtained when autophagy was stimulated by rapamycin (Lc+Rp: 27%±4; p=0.32). We also examined if inhibition of autophagy simultaneous to proteasome inhibition favors aggresome formation. For this, the number of aggregate-containing cells was compared between fed cells treated with Lc alone, or simultaneously with 3-MA (first and last bars in Fig. 5B). We choose 12 h treatment because about half of the cells at this time point contain PMP22 aggresomes. After concurrent inhibition of the proteasome and autophagy for 12 h, the number of cells with aggresomes is higher than Lc alone (p=0.004), and is similar to Lc at 16 h (Fig. 5B) (p=0.107). These results indicate that under conditions of reduced proteasome activity, blocking autophagy accelerates the accumulation of aggregated proteins.
The formation of protein aggregates is associated with an overall accumulation of unrelated proteasomal substrates (Johnston et al., 1998; Bence et al., 2001; Fortun et al., 2005). Therefore, a reduction in the number of aggregates upon starvation-induced autophagy should correlate with a decrease in the levels of accumulated UPS substrates. Since polyubiquitination is the primary signal for proteasomal degradation (Goldberg, 2003), the levels of slow-migrating ubiquitinated proteins were evaluated after the indicated treatment paradigms (Fig. 6A). Compared to control cells (C), Lc treatment alone leads to the accumulation of slow-migrating ubiquitinated proteins, indicating overall proteasome inhibition. Coincident with the accumulation of polyubiquitinated chains, the level of the monomeric ubiquitin (Fig. 6A, arrow) is reduced, as compared to control. When autophagy is enhanced by starvation (Stv), concomitant with Lc treatment, the levels of ubiquitinated substrates are slightly reduced. Starvation alone does not alter the levels of ubiquitinated proteins. Therefore, enhancement of autophagy in proteasome-inhibited cells reduces the accumulation of ubiquitinated substrates.
Figure 6. Reduced aggresome formation correlates with lower levels of an unrelated proteasome substrate.
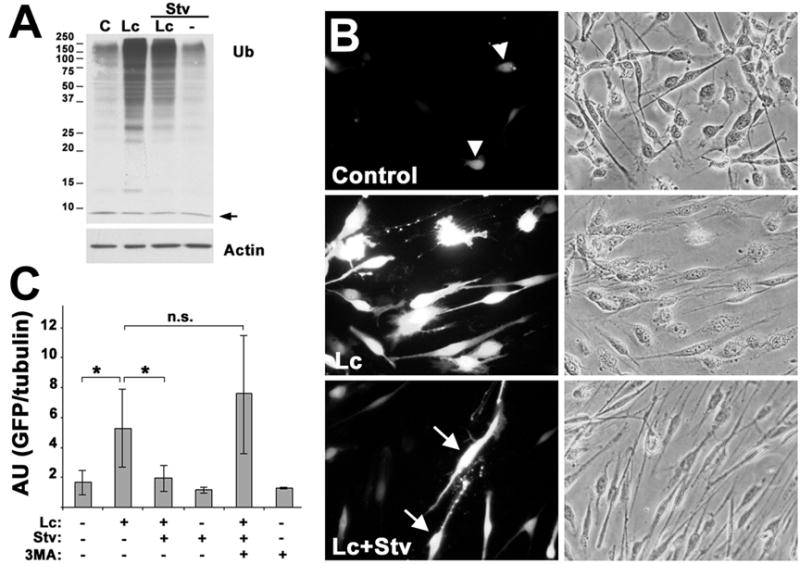
(A) The levels of ubiquitin (Ub) and actin were determined by western blot of total lysates (15 μg/lane) from fed and starved cells (Stv), treated with DMSO control (C) or Lc (16 h). The arrow denotes the monomeric ubiquitin (Ub). Molecular mass are depicted at the left, in kDa. (B) Rat SCs transfected with UbG76V-GFP and treated with the indicated agents (16 h), were imaged for GFP fluorescence (B, left column), or phase contrast (B, right column). Arrowheads indicate the low levels of GFP-like fluorescence in control cells. Arrows point at a small percentage of cells with bright GFP fluorescence, upon enhancement of autophagy during Lc treatment (B, Lc+Stv). (C) The stabilization of GFP signal was quantified by western blots, after correction for tubulin (duplicates of n=3). AU: absorbance units; p<0.05*; n.s., no significance.
To further examine the consequences of autophagy-mediated reduction in protein aggregate formation, we determined the steady-state levels of a short-lived UbG76V-GFP reporter (Fig. 6B). The attachment of the uncleavable UbG76V to GFP targets this otherwise stable protein for polyubiquitination and rapid proteasomal degradation and thus, GFP levels are hardly detected unless the UPS is inhibited (Dantuma et al., 2000). SCs were transiently transfected with UbG76V-GFP, and after a 24 h period, the proteasomal and autophagic pathways were modulated (Fig. 6B and C). After each treatment, the cells were imaged for GFP fluorescence (Fig. 6B, left column) and phase contrast (Fig. 6B, right column) or processed for Western blot with anti-GFP antibodies (Fig. 6C). Because of the rapid turnover rate of UbG76V-GFP, its levels are very low (arrowheads) or undetectable in most control cells (Fig. 6B, control). When the proteasome is inhibited, the UbG76V-GFP protein accumulates (Fig. 6B, 6C). Inhibition or enhancement of autophagy alone by 3-MA or starvation conditions, respectively, has no effect on UbG76V-GFP levels, confirming that this substrate is tagged for proteasomal and not autophagic degradation (Fig. 6C). However, when autophagy is enhanced simultaneous to Lc treatment, the levels of UbG76V-GFP are considerably reduced, as compared to Lc alone (Fig. 6B; 6C). In a small percentage of these cells however, intense UbG76V-GFP signal is still detected (Fig. 5B, Lc+Stv, arrows). As a measure of specificity, the effects of starvation are blocked by the inclusion of 3-MA (Fig. 6C). This result indicates that by experimental enhancement of autophagy, the accumulation of UbG76V-GFP upon proteasome inhibition can be minimized.
The effects of HSPs on aggregate formation
Heat shock proteins (HSPs) are known to influence the aggregation of proteins in cultured cells (Muchowski and Wacker, 2005), and are found within and around PMP22 inclusions in TrJ neuropathic nerves (Fortun et al., 2003). To assess the relationship of HSPs to proteasome-inhibition induced aggregates of endogenous PMP22, normal rat SCs treated with Lc, or DMSO, were immunostained with the indicated antibodies (Fig. 7). In control cells, Hsp70-like immunoreactivity is low (DMSO inset, green), but becomes distinct following proteasome inhibition (Lc) (Fig. 7A). In cells with small ubiquitin-reactive aggregates (red, arrowheads) the levels of Hsp70 are not as dramatically increased. Ubiquitin-like staining however almost exclusively overlaps with PMP22 aggresomes (right inset; also see Notterpek et al., 1999). The small cytosolic chaperone, αB-crystallin is also recruited within and around the aggregates resulting in a yellow color on the merge (Fig. 7B). Hsp40 is found at the centrosome region in control cells (inset, arrow), and accumulates with PMP22 aggregates (Fig. 7B). The association of HSPs with aggresomes likely represents an attempt of the chaperones to prevent the formation and/or favor the dissolution of the inclusions.
Figure 7. Heat shock proteins associate with PMP22 aggresomes.
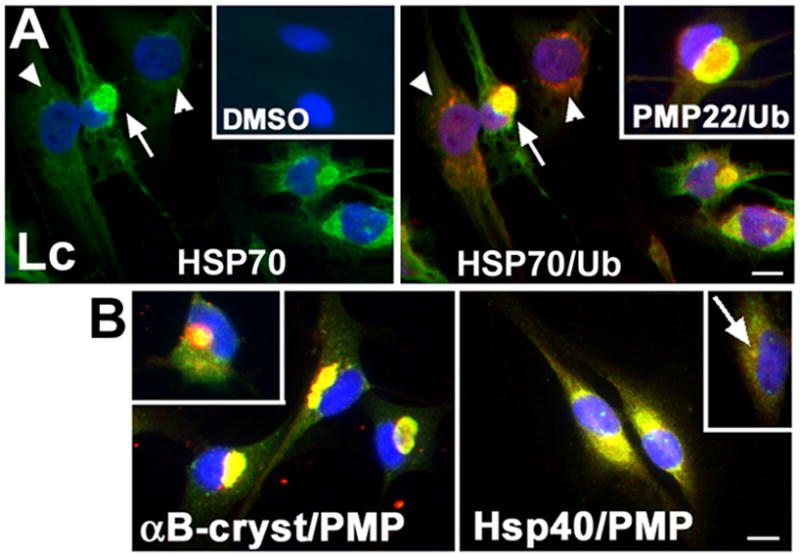
(A) The association of ubiquitin (Ub, red) with Hsp70 (green), or with PMP22 (green) (inset, top row) was determined by double labeling SCs after treatment with Lc or DMSO (16 h). Arrows denote the co-localization of Hsp70 and ubiquitin in aggresomes. Arrowheads point at cells that contain small ubiquitin-positive aggregates, but HSP70 levels remained low, similar to DMSO control (inset). (B) The small chaperones, αB-crystallin (αB-cryst, red) and Hsp40 (red) are recruited to PMP22 aggresomes (green). The αB-cryst inset shows the chaperone mainly around a PMP22 aggresome. The arrow points at Hsp40-like staining at the centrosome (B, inset in right panel). Nuclei are visualized by Hoechst stain (blue). Scale bars, 10 μm.
To investigate whether increasing the levels of HSPs could hamper the formation of PMP22 aggregates, rat SCs were simultaneously treated with geldanamycin (GA) and Lc, or sequentially with a brief preconditioning heat shock (HS) and then Lc (Fig. 8). GA binds to an ATP site on HSP90 and blocks its interaction with HS factor 1, promoting its activation and the synthesis of HSPs (Muchowski and Wacker, 2005). Both approaches lead to a robust increase in the steady-state levels of Hsp70, while the levels of calnexin are unaltered (Fig. 8A). A sixteen hour treatment with GA (250 nM) elevates the levels of Hsp70 by ~7-fold (Fig. 8A, left panel). Similarly, after a brief HS treatment (Allen et al., 2004), Hsp70 expression is transiently increased, peaking at 8 h (Fig. 8A, right panel). Sixteen hours after HS, Hsp70 levels remain slightly elevated, as compared to control.
Figure 8. Enhancement of HSP expression hinders aggresome formation.
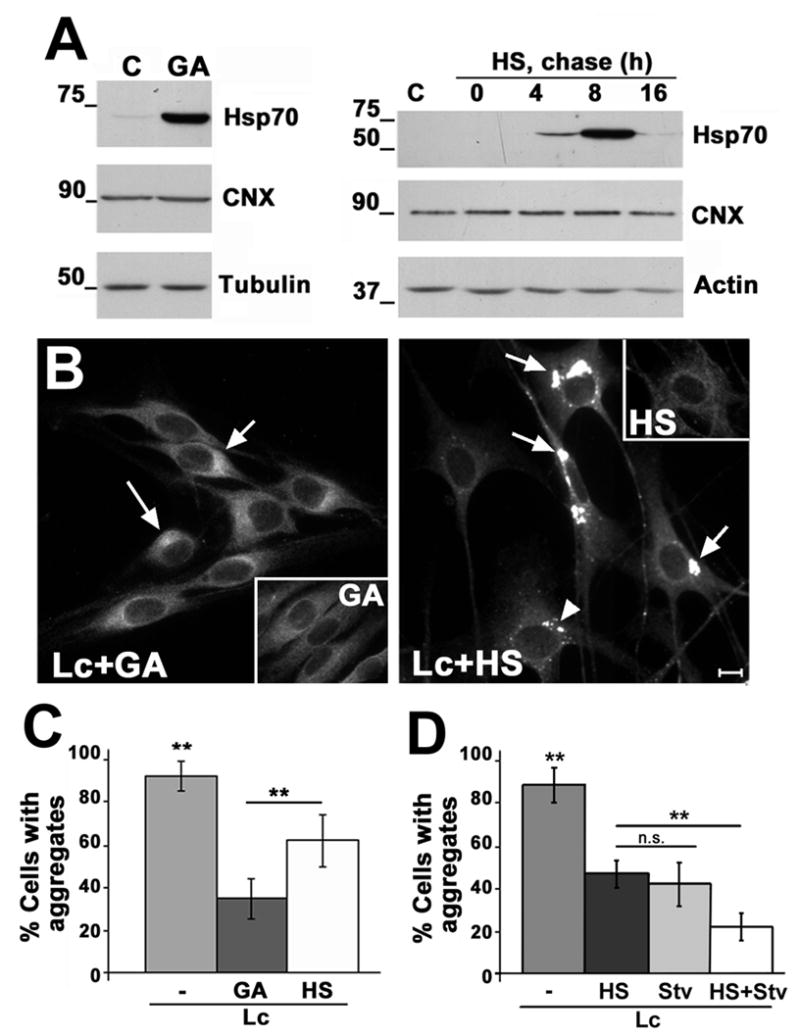
(A) The levels of Hsp70, calnexin (CNX) and tubulin or actin were determined by western blots in total lysates (20 μg/lane) of untreated (C), GA-treated (16 h), or HS preconditioned (HS) cells, followed by the indicated chases at 37°C (0–16 h). (B) The localization of PMP22 after each specified treatment (Lc+GA; Lc+HS) was monitored by immunostaining. Arrows point at remaining PMP22 aggregates with larger than 1 μm diameter and are included in the quantification as cells with aggregates. The arrowhead (Lc+HS) indicates a cluster of small aggregates (less than 1 μm) that are excluded in the quantification. Scale bar, 10 μm. (C) The number of cells with aggregates was quantified in four independent experiments and expressed as percentage of total cells (p<0.005**). (D) The additive effect of HS and Stv was investigated in three independent experiments. The number of cells with aggregates was quantified and expressed as a percentage of total cells (p<0.005**). ns; not significant.
To test the effects of elevated HSPs on PMP22 aggresome formation, SCs were simultaneously treated with GA (250 nM) and Lc for 16 h (Fig. 8B). In contrast to proteasome inhibition alone (Fig. 5A), when GA is included (Lc+GA), most SCs do not form aggregates. In a fraction of the cells, a condensation of PMP22 around the nucleus is seen (arrows), which were considered as aggregates based on co-staining with ubiquitin (not shown) and diameter larger than 1 μm. These aggregates however are less defined than those formed after proteasome inhibition alone (Fig. 5A). Similar results are observed when the cells are preconditioned with HS (Fig. 8B, Lc+HS). Furthermore, the average size of the aggregates formed (arrows) is reduced ~3-fold (p<0.0001). The arrowhead denotes an example of a cluster of small PMP22 aggregates (<1 μm) that are ubiquitin-negative (not shown). Both aggregates and cell death were absent in cells subjected to HS or GA alone and allowed to recover for 16 h without proteasome inhibition (Fig. 8B, insets). Quantification of three independent experiments reveals that GA-treatment or HS preconditioning significantly reduces the formation of protein aggregates in proteasome-inhibited cells (Fig. 8C). While GA is more effective in preventing the formation of PMP22 aggregates, as compared to HS (p<0.005), extended exposure of cells to this compound is known to be toxic (Miyata 2005). Therefore, to explore the effects elevated of HS proteins in combination with enhanced autophagy, cells were first exposed to HS preconditioning followed by proteasome inhibition under starvation conditions (Fig. 8D). Comparing the influence of enhanced autophagy or chaperones on preventing protein aggregate formation, we found that these two treatments are equally effective (p=0.18). However, when the two treatments are combined, the accumulation of misfolded PMP22 is further reduced (p<0.005). While HS preconditioning stimulates autophagy, as measured by an increase in LC3 levels and lipidation (data not shown), when 3-MA was included during the post-HS treatments, the number of cells with aggregates remained the same (p=0.81). Therefore, the effect of HS in hindering aggresome formation is likely independent of macroautophagy, but could possibly involve CMA.
Based on the results described here, a protective nature of PMP22 aggregate formation can be proposed (Fig. 9). Aggresomes may represent a link between proteasome overwhelming/saturation (a) and the activation of autophagy (e), as a secondary degradation mechanism to prevent the accumulation of misfolded PMP22. Increasing the levels of HSPs (f) adds to this protective response and can further reduce aggregate formation by aiding protein re-folding and trafficking, and/or alternate degradative mechanisms.
Figure 9. Model for the protective role of aggresomes.
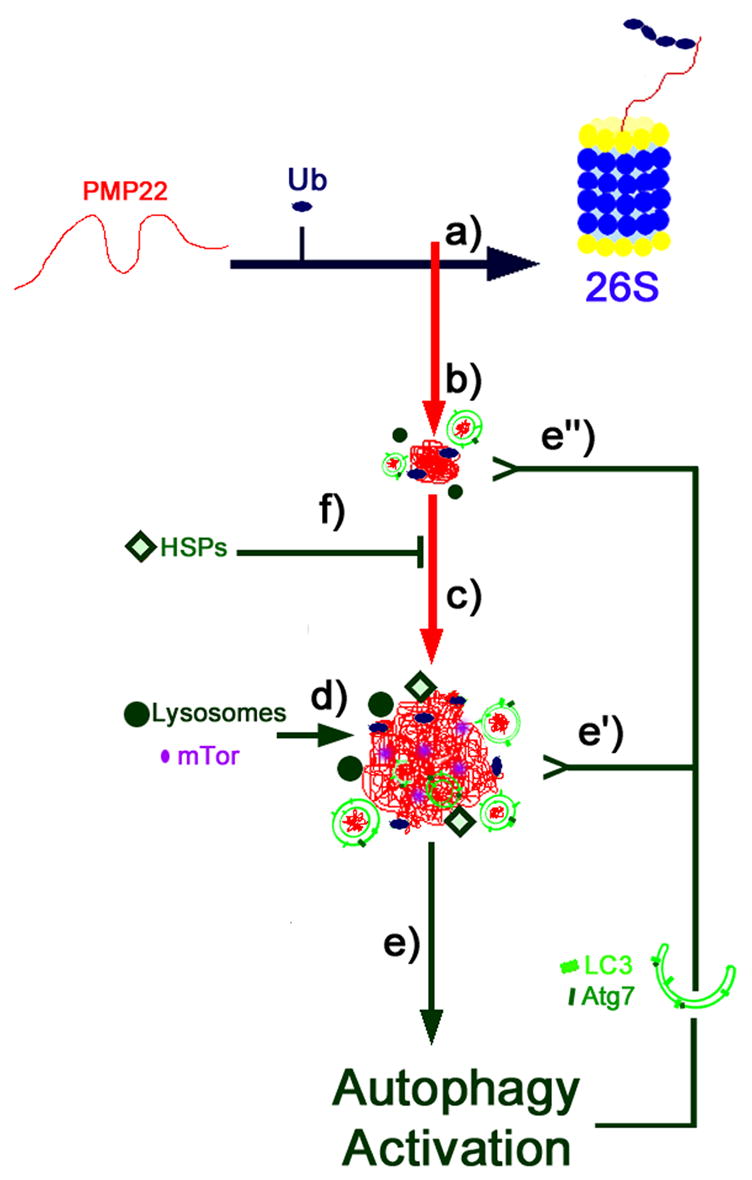
PMP22 is a hydrophobic tetraspan proteasome substrate. Proteasome inhibition (a) in SCs results in the formation of small aggregates containing PMP22 and ubiquitin (Ub) (b). Such aggregates are transported along the microtubules to the centrosome, forming a central inclusion termed aggresome (c). Aggresomes sequester mTOR, and associate with autophagosomes and autolysosomes (d). The activation of the autophagic pathway (e) upon proteasome inhibition and aggresome formation is a protective response of the cell to prevent the accumulation of proteasome substrates. Two mechanisms account for this effect: the clearance of the aggresome (e’) and the removal of small aggregates (e’’), thus hindering the formation of the final inclusion. Moreover, aggregates recruit heat shock proteins (HSPs) that likely add to this protective response (f).
Discussion
Cytosolic protein aggregates form when the quality control mechanisms of the cell fail to ensure the folding and/or degradation of proteins (Goldberg, 2003). Such aggregates are commonly found in neurodegenerative disorders and often associate with malfunction of the proteasome (Keller et al., 2000; Waelter et al., 2001; McNaught et al., 2003; Kabashi et al., 2004; Fortun et al., 2005; 2006). Here we show that the accumulation of endogenous PMP22 in aggresomes in neuropathic samples, or in response to proteasome inhibition, results in the concomitant induction of autophagy. Further enhancement of autophagy, or the levels of HSPs by experimental manipulations hampers the formation of PMP22 aggresomes and prevents the accumulation of unrelated UPS substrates. These results suggest that when proteasome activity is compromised, cellular homeostasis is maintained by re-routing proteasome substrates to an alternative pathway, namely autophagic-mediated lysosomal degradation. The developments of pharmacologic agents to stimulate these pathways therefore could provide a therapeutic approach for protein misfolding disorders.
Autophagic vacuoles have been observed within or adjacent to the surface of cytoplasmic protein aggregates formed upon proteasome inhibition (Wojcik et al., 1996; Harada et al., 2003; Rideout et al., 2004; Iwata et al., 2005). Additionally, autophagosome formation and LC3 lipidation was reported in cells containing ubiquitinated inclusions as a result of oxidative stress (Martinet et al., 2004). In the TrJ neuropathic model, we found the recruitment of Atg7 to PMP22 aggregates and autophagosomes filled with electron dense material (Fortun et al., 2003). Similarly, autophagosomes were detected in SCs of neuropathic mice based on PMP22 overexpression (Fortun et al., 2006). The studies detailed above suggest a cellular response by which, autophagy is activated upon degradative failure of the proteasome to maintain cellular homeostasis and prevent the accumulation of potentially harmful protein aggregates. Indeed, a relationship between degradation pathways has been reported previously, as proteasome inhibition resulted in enhanced macroautophagy (Ding et al., 2003; Iwata et al., 2006). The signal behind this compensatory mechanism is yet uncertain; however, the formation of protein aggregates could represent the missing link. Recent studies have suggested a mechanism for the activation of autophagy by protein aggregates, based on a loss of function of mTOR (mammalian target of rapamycin) (Ravikumar et al., 2004). Although this event may result in the activation of autophagy, it is not yet known how the inclusions are recognized by the autophagic-lysosomal system. Aggresomes are often surrounded by a vimentin cage (Johnston et al., 1998; Notterpek et al., 1999), a factor that is poorly studied, but might have important consequences aside from being a structural constraint. In this regard, previous studies with internalized viral proteins, or microinjected purified proteins, suggested the existence of a sequestration site for protein aggregates in tight association with intermediate filaments at perinuclear regions, as a necessary step preceding autophagic-lysosomal degradation (Earl et al., 1987; Doherty et al., 1987).
The studies mentioned above and described here (Figs. 1–4) show that the aggregation of undegraded proteasome substrates is associated with the induction of an endogenous autophagic response by the cells. In light of these results, why do aggresomes form despite the activation of autophagy? One possibility is that the removal of misfolded proteins by autophagy is insufficient to prevent the formation of aggresomes. Alternatively, since lactacystin has been shown to inhibit the activity of human platelet lysosomal cathepsin A-like enzyme at pH 5.5 (Ostrowska et al., 1997), lysosomal degradation might be incomplete. Nonetheless, we obtained similar results in SCs from neuropathic mice bearing spontaneous aggregates in the absence of proteasome inhibition, or when epoxomicin instead of lactacystin was used, casting doubts on this possibility. Additionally, when autophagy is experimentally enhanced by amino acid and serum deprivation, the formation of de novo aggresomes and the accumulation of proteasomal substrates are hindered. Pulse labeling experiments indicate that this effect is not due to a generalized depression in new protein synthesis, as the incorporation of 35S-methionine/cystine, was similar among cells incubated in control medium or under Lc+Stv conditions for 16 h (p=0.44).
How does experimental activation of autophagy prevent the accumulation of misfolded PMP22? Aggresome assembly is a multi-step process, by which small aggregates throughout the cell are transported along microtubules towards the centrosome, where they form the final inclusion (Johnston et al., 1998; Notterpek et al., 1999). Thus, it is likely that upon activation of autophagy, small aggregates are engulfed within autophagosomes, reducing the load of proteins being transported towards the centrosome. In agreement, the enhancement of autophagy by rapamycin reduced the accumulation of protein aggregates and cellular toxicity in Huntington's disease models (Ravikumar et al., 2002; 2004). The role of autophagy in the degradation of ubiquitinated substrates is also supported by the accumulation of ubiquitinated aggregates in the liver of conditional Atg7-deficient mice, which have defective autophagy, but intact UPS activity (Komatsu et al., 2005). Two recently established transgenic mouse lines further support the essential role of autophagy in preventing the accumulation of abnormal proteins (Komatsu et al., 2006; Hara et al., 2006). In agreement, the simultaneous inhibition of the UPS and autophagy in our SC model is associated with more pronounced accumulation of PMP22, as compared to Lc treatment alone (Fig. 5).
The consequences of inclusion body formation within the cytoplasm are still debated. It has been proposed that the inclusion bodies in neurodegenerative conditions are protective, whereas the intermediate aggregates trigger neuronal toxicity by an unknown cascade of events (Muchowski and Wacker, 2005). As cell death is not a prominent feature of PMP22-associated neuropathies, and is absent in our cell culture model (Fortun et al., 2003), our results support a protective role for protein aggregates. The lack of cellular toxicity in our studies could be related to the endogenous protein background rather than recombinant protein overexpression, and/or the cell type used, as well as the shorter periods of proteasome inhibition and autophagy stimulation. It is also feasible to speculate that the collapsed vimentin cage around PMP22 aggresomes (Notterpek et al., 1999) prevents the mobility of proteins, whereas small aggregates are free to associate with additional molecules. Indeed, small PMP22 aggregates could be detrimental for SCs by providing a core for trapping other ubiquitinated substrates, based on specific, or hydrophobic protein-protein interactions (Fortun et al., 2005). Although we did not detect SC death in the time frame used for proteasome inhibition (16 h) or in the neuropathic models (Fortun et al., 2003; 2006), it is possible that PMP22 aggregates could lead to toxicity if not eliminated by autophagy.
In addition to autophagy, other events might be activated to prevent the accumulation of aggregates and the disruption of cellular homeostasis. A common response to proteasome inhibition and formation of aggregates is the increase in the expression of molecular chaperones (Muchowski and Wacker, 2005). Proteasome impairment in SCs overexpressing wt and mutant PMP22s is associated with the elevation of HSPs and their recruitment to aggresomes (Ryan et al., 2002). HSPs are also recruited to aggregates of the endogenous PMP22 (Fig. 7). The reason for this event is uncertain, but it might involve the refolding and/or the targeting of the misfolded proteins for degradation (Muchowski and Wacker, 2005). Indeed, the kinetics of association and dissociation of Hsp70 with polyglutamine protein aggregates is fast and similar to the rate for the folding of thermally denatured substrates, suggestive of an active role in the refolding of the aggregate (Kim et al., 2002). On the other hand, the protection observed in Drosophila models of Parkinson’s and polyglutamine expansion diseases due to the elevation of HSPs is not accompanied by a reduction in the number of inclusions (Kazemi-Esfarjani and Benzer, 2000; Auluck et al., 2002)
A protective role for chaperones in preventing the accumulation of misfolded proteins is supported by the present study. The formation of PMP22 aggresomes is hindered by a preconditioning HS or GA treatment, likely due to the enhancement of cytoplasmic molecular chaperones. Preventing the formation and/or aiding the refolding of small aggregates before the assembly of a large inclusion can explain this result. In culture models of Hsp40 and/or Hsp70 overexpression, the intracellular aggregation of several unrelated proteins was also suppressed (Cummings et al., 1998; Stenoien et al., 1999; Dul et al., 2001; Abu-Baker et al., 2003). In our model, GA-treatment, as compared to HS was more effective in preventing Lc-induced aggresome formation, likely because of the sustained expression of HSPs over time. The effects of GA on hindering aggresome formation does not correlate with a pronounced reduction in the steady-state levels of ubiquitinated substrates however (not shown), suggesting that the action of GA may be based on the physical disassembly of the aggregates by chaperones and/or promoting the trafficking of PMP22 to the plasma membrane. Alternatively, given the various Hsp90 substrates and the different processes in which they are involved with, including signal transduction and cell cycle regulation (Neckers, 2002), GA-treatment may influence unknown factors of protein aggregation. Nonetheless, as judged from anti-LC3 Western blots (data not shown) GA treatment does not appear to stimulate macroautophagy. Additionally, as PMP22 lacks a KFERQ-like targeting motif for CMA (Majesky and Dice, 2004), the observed benefits of GA treatment are likely independent from effects on CMA.
Based on the results described here, we propose a model where the formation of aggresomes is a protective response of the cell that concentrates misfolded proteins in a central location to activate an autophagic response (Fig. 9). This model entails that the monomeric PMP22 is turned-over by the proteasome (Ryan et al., 2002), whereas the aggregated forms resulting from proteasome inhibition or saturation, are degraded by the autophagic-lysosomal pathway. Concomitant with the reduction in aggregates within the cells, the levels of slow migrating ubiquitinated substrates and of an unrelated proteasome reporter are diminish. This suggests that substrates originally tagged for proteasomal degradation, upon inhibition of this system, are re-routed for breakdown within lysosomes through activation of autophagy. An interaction between the two degradative pathways is not specific for PMP22, but rather appears to represent a general response of cells, as suggested by studies with other aggregation-prone proteins, such as wt α-synuclein and its A53T or A30P mutants (Tofaris et al., 2001; Webb et al., 2003; Rideout et al., 2004; Iwata et al., 2005). Based on our results, we also propose that the chaperone system is activated to aid in the refolding of PMP22 and dissolution of small aggregates.
Acknowledgments
The authors wish to thank Debbie Akin for technical assistance with the ultrastructural studies and N. Mizushima (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) and T. Yoshimori (National Institute of Genetics, Mishima, Japan) for providing the GFP-LC3 plasmid. These studies were supported by the National Muscular Dystrophy Association and the NIH-NINDS (LN) and the NIH-NCI (WAD).
Abbreviations
- PMP22
peripheral myelin protein 22
- SC
Schwann cell
- wt
wild type
- HSP
heat shock protein
- Lc
lactacystin
- DMSO
dimethyl sulfoxide
- Stv
starvation
- 3MA
3-methyladenine
- GA
geldanamycin
- HS
heat shock
- GFP
green fluorescent protein
- CMT1A
Charcot-Marie-Tooth disease type 1A
- P
postnatal
- UPS
ubiquitin-proteasome system
- CMA
chaperone-mediated autophagy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abu-Baker A, Messaed C, Laganiere J, Gaspar C, Brais B, Rouleau GA. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum Mol Genet. 2003;12(20):2609–2623. doi: 10.1093/hmg/ddg293. [DOI] [PubMed] [Google Scholar]
- 2.Allen TA, Von Kaenel S, Goodrich JA, Kugel JF. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol. 2004;11(9):816–821. doi: 10.1038/nsmb813. [DOI] [PubMed] [Google Scholar]
- 3.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295(5556):865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 4.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 5.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40(2):427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 6.Colby J, Nicholson R, Dickson KM, Orfali W, Naef R, Suter U, Snipes GJ. PMP22 carrying the trembler or trembler-J mutation is intracellularly retained in myelinating Schwann cells. Neurobiol Dis. 2000;7:561–573. doi: 10.1006/nbdi.2000.0323. [DOI] [PubMed] [Google Scholar]
- 7.Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY. Chaperone suppression of aggregation altered subcellular proteasome localization implying protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 8.Dantuma NP, Heessen S, Lindsten K, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 9.Ding Q, Dimaguya E, Martin S, Bruce-Keller AJ, Nukula V, Cuervo AM, Keller N. Characterization of chronic low level proteasome inhibition on neuronal homeostasis. J Neurochem. 2003;86:489–497. doi: 10.1046/j.1471-4159.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 10.Doherty FJ, Wassell JA, Mayer RJ. A putative protein-sequestration site involving intermediate filaments for protein degradation by autophagy. Studies with microinjected purified glycolytic enzymes in 3T3-L1 cells. Biochem J. 1987;241(3):793–800. doi: 10.1042/bj2410793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69(9):5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dul JL, Davis DP, Williamson EK, Stevens FJ, Argon Y. Hsp70 and antifibrillogenic peptides promote the degradation and inhibit intracellular aggregation of amyloidogenic light chains. J Cell Biol. 2001;152 (4):705–715. doi: 10.1083/jcb.152.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Urso D, Prior R, Greiner-Petter R, Gabreels-Festen AA, Muller HW. Overloaded endoplasmic reticulum-Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J Neurosci. 1998;18(2):731–740. doi: 10.1523/JNEUROSCI.18-02-00731.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Earl RT, Mangiapane EH, Billett EE, Mayer RJ. A putative protein-sequestration site involving intermediate filaments for protein degradation by autophagy. Studies with transplanted Sendai-viral envelope proteins in HTC cells. Biochem J. 1987;241(3):809–815. doi: 10.1042/bj2410809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fortun J, Go JC, Li J, Amici SA, Dunn WA, Jr, Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis. 2006;22(1):153–64. doi: 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem. 2005;92:1531–1541. doi: 10.1111/j.1471-4159.2004.02987.x. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 19.Groll M, Clausen T. Molecular shredders: how proteasomes fulfill their role. Curr Opin Struct Biol. 2003;13(6):665–673. doi: 10.1016/j.sbi.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 21.Harada M, Kumemura H, Omary MB, Kawaguchi T, Maeyama N, Hanada S, Taniguchi E, Koga H, Suganuma T, Ueno T, Sata M. Proteasome inhibition induces inclusion bodies associated with intermediate filaments and fragmentation of the Golgi apparatus. Exp Cell Res. 2003;288(1):60–69. doi: 10.1016/s0014-4827(03)00162-9. [DOI] [PubMed] [Google Scholar]
- 22.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280(48):40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 23.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;89:1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x. [DOI] [PubMed] [Google Scholar]
- 25.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;122(17):4577. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazemi-Esfarjani P, Benzer S. Suppression of polyglutamine toxicity by a Drosophila homolog of myeloid leukemia factor 1. Hum Mol Genet. 2002;11(21):2657–2672. doi: 10.1093/hmg/11.21.2657. [DOI] [PubMed] [Google Scholar]
- 27.Keller JN, Hanni KB, Markerbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, Nollen EAA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 32.Lenk SE, Dunn WA, Jr, Trausch JS, Ciechanover A, Schwartz AL. Ubiquitin-activating enzyme, E1, is associated with maturation of autophagic vacuoles. J Cell Biol. 1992;118(2):301–308. doi: 10.1083/jcb.118.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majesky AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36(12):2435–44. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Martinet W, De Bie M, Schrijvers DM, De Meyer GR, Herman AG, Kockx MM. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24(12):2296–2301. doi: 10.1161/01.ATV.0000146266.65820.a1. [DOI] [PubMed] [Google Scholar]
- 35.Mclean PJ, Klucken J, Shin Y, Hyman BT. Geldanamycin induces Hsp70 and prevents α-synuclein aggregation and toxicity in vitro. Biochemical and Biophysical Res Com. 2004;321:665–669. doi: 10.1016/j.bbrc.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 36.McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasome function in sporadic Parkinson’s disease. Exp Neurol. 2003;179:38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- 37.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6(1):11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 38.Munafo BD, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 39.Naef R, Suter U. Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol Dis. 1999;6(1):1–14. doi: 10.1006/nbdi.1998.0227. [DOI] [PubMed] [Google Scholar]
- 40.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8(4 Suppl):S55–61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 41.Notterpek L, Snipes G, Shooter EM. Upregulation of the endosomal-lysosomal pathway in the Trembler-J neuropathy. J Neurosci. 1997;17(11):4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Notterpek L, Ryan MC, Tobler AR, Shooter EM. PMP22 accumulation in aggresomes: implications for CMT1A pathology. Neurobiol Dis. 1999;6:450–460. doi: 10.1006/nbdi.1999.0274. [DOI] [PubMed] [Google Scholar]
- 43.Ostrowska H, Wojcik C, Omura S, Worowski K. Lactacystin, a specific inhibitor of the proteasome, inhibits human platelet lysosomal cathepsin A-like enzyme. Biochem Biophys Res Commun. 1997;234(3):729–732. doi: 10.1006/bbrc.1997.6434. [DOI] [PubMed] [Google Scholar]
- 44.Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, Iacampo S, Suter U, Shooter EM, Murphy RA. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17:7754–7762. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 46.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 47.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36(12):551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Ryan MC, Shooter EM, Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis. 2002;10:109–118. doi: 10.1006/nbdi.2002.0500. [DOI] [PubMed] [Google Scholar]
- 49.Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13) J Cell Biol. 1992;117:225–238. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8(5):731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 51.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tobler AR, Notterpek L, Naef R, Taylor V, Suter U, Shooter EM. Transport of Trembler-J mutant peripheral myelin protein 22 is blocked in the intermediate compartment and affects the transport of the wild-type protein by direct interaction. J Neurosci. 1999;19:2027–2036. doi: 10.1523/JNEUROSCI.19-06-02027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tobler AR, Liu N, Mueller L, Shooter EM. Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc Natl Acad Sci USA. 2002;99:483–488. doi: 10.1073/pnas.012593399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tofaris GK, Layfield R, Spillantini MG. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509(1):22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 55.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278(27):25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 57.Wojcik C, Scroeter D, Wilk S, Lamprecht J, Paweletz N. Ubiquitin-mediated proteolysis centers in Hela cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur J Cell Biol. 1996;71:311–318. [PubMed] [Google Scholar]
- 58.Young P, Suter U. Disease mechanisms and potential therapeutic strategies in Charcot-Marie-Tooth disease. Brain Res Brain Res Rev. 2001;36:213–221. doi: 10.1016/s0165-0173(01)00097-2. [DOI] [PubMed] [Google Scholar]