

Abstract
Pairs of hydroxysteroid dehydrogenases (HSDs) govern ligand access to steroid receptors in target tissues and act as molecular switches. By acting as reductases or oxidases, HSDs convert potent ligands into their cognate inactive metabolites or vice-versa. This pre-receptor regulation of steroid hormone action may have profound effects on hormonal response. We have identified the HSDs responsible for regulating ligand access to the androgen receptor (AR) in human prostate. Type 3 3α-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) acts solely as a reductase to convert 5α-dihydrotestosterone (DHT), a potent ligand for the AR (Kd = 10−11 M for the AR), to the inactive androgen 3α-androstanediol (Kd = 10−6 M for the AR); while RoDH like 3α-HSD (a short chain dehydrogenase/reductase (SDR)) acts solely as an oxidase to convert 3α-androstanediol back to 5α-DHT. Our studies suggest that AKRs and SDRs function as reductases and oxidases, respectively to control ligand access to nuclear receptors.
Keywords: aldo-keto reductase, short-chain dehydrogenase/reductase, 3α-androstanediol, prostate disease
1. Introduction
Hydroxysteroid dehydrogenases (HSDs) regulate ligand access to nuclear receptors in steroid target tissues (Funder, et al., 1988; Penning, 2003). This is often achieved by HSDs working in pairs to convert potent steroid hormones to their inactive metabolites and vice versa where they act preferentially as ketosteroid reductases or hydroxysteroid oxidases. The prototypic example is the opposing action of 11β-HSD Type 1 and Type 2 (Funder, et al.,1988; Stewart and Whorwood et al., 1994; Seckl and Walker, 2001). The Type 1 enzyme catalyzes the NADPH reduction of cortisone (inactive hormone) to yield cortisol (active hormone) and acts to amplify local glucocorticoid action by regulating ligand access to the glucocorticoid receptor. The Type 2 enzyme catalyzes the NAD+ dependent oxidation of cortisol to cortisone and protects the mineralocorticoid receptor from apparent mineralocorticoid excess in the kidney. Positional specific HSD pairs have also been proposed to regulate the action of the androgen receptor (AR), the estrogen receptor and the progesterone receptor (Penning, et al., 2003).
This concept has been challenged by the observation that many HSDs are bidirectional in transfection paradigms if steroid flux is measured when the enzymes attain equilibrium (Khana, et al., 2004; Agarwal and Auchus, 2005). However, these experiments fail to take into account two important concepts. First, in steroid target tissues equilibrium may not be achieved since the HSDs are part of a metabolic sequence, with other enzymes working either side of the HSD reaction. Second, the HSDs that have preference to act as reductases and oxidases may co-exist in the same cell type and hormone activation/inactivation will be governed by the expression level of each enzyme and their respective kcat/Km values for the opposing reactions.
Regulation of ligand access to the AR by HSDs in human prostate has been our focus. 5α-DHT is the most potent natural male hormone and is produced by the reduction of testosterone catalyzed by 5α-reductase type 2 (Russell and Wilson, 1994). 5α-DHT has a Kd of 10−11 M for the AR and will trans-activate the AR leading to gene transcription and normal and abnormal prostate growth. Our data show that this androgen signal is eliminated by human type 3α-HSD (aldo-keto reductase (AKR) 1C2) which reduces 5α-DHT to 3α-androstanediol (3α-diol). 3α-Diol is a weak androgen Kd of 10−6 M for the AR that is incapable of trans-activation. Studies in rats (Orlowski, et al., 1983), dogs (DeKlerk, et al, 1979; Jacobi, ,et al 1978), marsupials (Shaw, et al., 2000) and humans (Horst, et al., 1975) have demonstrated that 3α-diol can be converted back to 5α-DHT to stimulate growth of the prostate, but the identity of the oxidative 3α-HSD responsible for this back reaction has remained elusive. Our data shows that “RoDH like 3α-HSD” (a short-chain dehydrogenase /reductase (SDR)) (Biswas and Russell, 1997) is responsible for this reaction in human prostate. Thus the HSD pair that regulates ligand access to the AR has been identified, Figure 1.
Fig. 1.
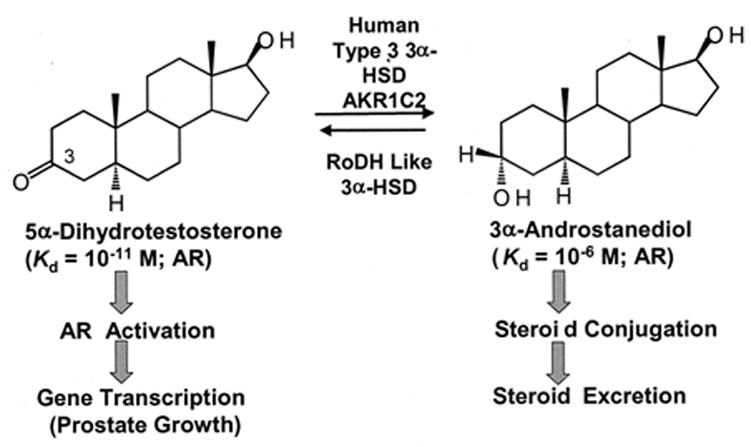
Regulation of ligand occupancy of the AR by HSDs in human prostate.
2. AKR1C2 and elimination of the androgen signal
2. 1. 3a-Androstanediol formation and AKR tissue distribution
In human tissues four AKR1C isoforms exist that are capable of reducing 5α-DHT to 3α-diol. These enzymes vary in their 3-, 17- and 20-ketosteroid reductase activity (Penning, et al., 2000) such that: AKR1C1 is predominately a 20-ketosteroid reductase that inactivates progesterone by forming 20α-hydroxyprogesterone; AKR1C2 is predominately a 3-ketosteroid reductase that will convert 5α-DHT to 3α-diol; AKR1C3 will act as a 17-ketosteroid reductase and will convert Δ4-androstene-3,17-dione to testosterone and estrone to 17β-estradiol; and AKR1C4 is a liver specific 3-ketosteroid reductase (Penning, et al., 2000). Using more discriminating TLC systems the products of the 3-ketosteroid reductase activities of the four AKR1C isoforms were re-examined. Each human enzyme produced two isomeric products, 3α-diol and 3β-diol, in different ratios indicating that they were not stereospecific (Steckelbroeck, et al., 2004). By contrast rat 3α-HSD (AKR1C9) showed strict stereochemical preference and reduced 5α-DHT only to 3α-diol. Ratios of the specific activities for 3α-diol : 3β-diol formation at saturating 5α-DHT concentrations showed that AKR1C2 favored 3α-diol formation Table 1. Isoform specific RT-PCR was performed to determine the level of AKR1C expression across nine human tissues. AKR1C1-AKR1C3 were all expressed in adult human prostate. Since AKR1C2 was present in prostate it became the candidate isoform for the elimination of 5α-DHT (Penning, et al., 2000).
Table 1.
Specific activities for the reduction of 5α-DHT to 3α-diol and 3β-diol catalyzed by human AKR1C isoforms
| Specific Activity [nmoles min−1mg−1]1 | ||||
|---|---|---|---|---|
| Product | AKR1C1 | AKR1C2 | AKR1C3 | AKR1C4 |
| 3α-Diol | 2.8 | 75.6 | 2.7 | 102.0 |
| 3β-Diol | 12.4 | 4.4 | 2.0 | 29.8 |
Assayed with 40 μM 5α-DHT plus 180 μM NADPH at 37°C. From Steckelbroeck et al. 2004.
2. 2. Kinetic considerations for AKR1C2
AKR1C2 like all AKRs catalyzes a sequential ordered bi bi mechanism in which NADPH binds first, followed by 5α-DHT, the ternary complex forms, chemistry occurs, and the 3α-diol and NADP+ products are released in that order (Askonas, et al., 1991). The steady state kinetic parameters for 5α-DHT reduction were kcat = 0.033 s−1, Km = 2.9 μM, and kcat/Km = 1.1 x 104 M−1 s−1; and for 3α-diol oxidation the steady state parameters were kcat = 0.008 s−1, Km = 3.1 μM and kcat/Km = 3.2 x 103 M−1 s−1, Table 2. The kinetic Haldane (Keq = kcat reduction x KiNADP+ x Km3α-diol/kcat oxidation x Ki NADPH x KmDHT) for the reaction gave a Keq of 8.0 indicating that the reduction of 5α-DHT is easily preferred (Jin and Penning, 2006).
Table 2.
Steady state kinetic parameters for reductive and oxidative hydroxysteroid dehydrogenases
| Enzyme | Reaction | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| AKR1C2 | 5α-DHT + NADPHa | 0.033 | 2.9 | 1.1 x 104 |
| AKR1C2 | 3α-Diol + NADP+ | 0.008 | 3.1 | 3.2 x 103 |
| Enzyme | Reaction | Vmax (nmoles min−1 mg−1) | Km | Vmax/Km (nmoles min−1 mg−1 /μM) |
|
| ||||
| RL-HSD | 3α-Diol + NAD+ | 5.9 ± 0.26 | 0.4 ± 0.04 | 14.8 |
| RODH5 | 3α-Diol + NAD+ | 18.5 ± 4.1 | 39.6 ± 3.3 | 2.1 |
| RODH4 | 3α-Diol + NAD+ | 8.9 ± 0.19 | 0.27 ± 0.03 | 32.4 |
These steady state kinetic constants are consistent with the re-estimation of these values
described by Steckelbroeck et al. 2004.
The AKR1C2 reaction was also dissected by transient kinetics using stopped-flow spectroscopy. The binding of NADP(H) measured by quenching the intrinsic protein tryptophan fluorescence showed that a tight E•NADP(H) complex was formed. This results in a 80-fold increase in affinity for NADPH where the Kd decreases from 9.6 μM to 120 nM and a 219-fold increase in affinity for NADP+ where the Kd decreases from 46 μM to 210 nM. These high affinities for NADP(H) have metabolic consequences for the enzyme (see later).
In the reduction direction the smallest microscopic rate constant that governs product (NADP+) release was 0.66 s−1 which was 20-times greater than kcat suggesting that other steps must contribute to rate determination. Multiple turnover experiments showed burst-phase kinetics, which indicated that slow product release steps occur. Global fitting (DynaFit) of the transient kinetic data to the minimal equation for an ordered bi bi reaction predicted that three slow- steps of similar magnitude accounted for the low kcat = 0.033 s−1. They are the chemical event (0.12 s−1), the release of 3α-diol (0.081 s−1), and the release of NADP+ (0.21 s−1) (Jin and Penning, 2006).
2. 3. Directionality of AKR1C2 in a cellular context
Human AKR1C2 and its rat homolog AKR1C9 were transiently transfected into COS-1 cells (monkey kidney fibroblasts) using pcDNA3 constructs. Lysates from transfected cells were able to catalyze the NADPH-dependent reduction of 5α-DHT to yield 3α-diol and the NAD+ dependent oxidation of 3α-diol to yield 5α-DHT whereas mock-transfected cells showed no catalysis (Rizner, et al., 2003). Metabolism studies were then performed in intact cells where either AKR1C2 or AKR1C9 would utilize the prevailing pyridine nucleotide concentrations, Figure 2. While intact cells were able to reduce 5α-DHT to 3α-diol, no oxidative reaction could be detected with either enzyme. Based on the kinetic characterization of AKR1C2 we tested whether its preferred direction was governed by its high affinity for NADPH. The NADPH-dependent reduction of 5α-DHT catalyzed by AKR1C2 was not inhibited by 1mM NAD+ (a physiological concentration). By contrast the NAD+-dependent oxidation of 3α-diol catalyzed by AKR1C2 was potently inhibited by 10 μM NADPH. Thus AKR1C2 acts predominately as a 3-ketosteroid reductase in cells due to a favorable Keq = 8.0 and the potent inhibition of the oxidative reaction by NADPH.
Fig. 2.

AKR1C2 and AKR1C9 preferentially catalyze the reduction of 5α-DHT to 3α-diol in COS-1 cells. Percent conversion of 5α-DHT to 3α-diol in COS-1 cells stably transfected with pcDNA3 (mock), pcDNA3-AKR1C9 (rat 3α-HSD; positive control) or pcDNA3-AKR1C2 (human type 3 3α-HSD). Cells were incubated with 5 μM [14C]-5α-DHT. Adapted from Rizner at al. 2003.
3. RoDH Like 3α-HSD (RL-HSD) and the Formation of 5α-DHT
3. 1. Comparison with other oxidative 3α-HSDs
One or more SDRs could catalyze the back reaction (oxidation of 3α-diol to 5α-DHT) in human prostate: L-3-hydroxyacyl coenzyme A dehydrogenase (endoplasmic reticulum amyloid β-peptide binding protein (ERAB)) (He, et al., 2000); RL-HSD (Biswas and Russell, 1997); 11-cis-retinol dehydrogenase (RoDH 5) (Huang, et al., 2001); novel type of human microsomal 3α-HSD (NT-3α-HSD) (Chetyrkin, et al, 2001); and retinol dehydrogenase 4 (RoDH 4) (Gough, et al., 1998). To compare these enzymes, their respective cDNAs were cloned into bis-cistronic constructs to yield (pcDNA3-3α-HSD-Lac-Z) in which the CMV promoter drives the expression of the 3α-HSD of interest plus β-galactosidase as a single transcript (Bauman, et al., 2006a). The presence of an IRES (internal ribosomal entry sequence) permits the single transcript to be processed as two proteins. Thus the expression of 3α-HSD can be normalized to β-galactosidase (internal standard) in the absence of antibodies for each enzyme. Transient transfection into COS-1 cells followed by measurement of the conversion of 0.1 μM 3α-diol to 5α-DHT showed that three enzymes (RoDH4, RoDH5, and RL-HSD) converted 100% of the steroid substrate within 30 min, whereas NT-3α-HSD and ERAB converted less than 5–10% of this substrate over the same time Figure 3. Transfection studies showed that these enzymes were unable to reduce 5α-DHT to 3α-diol. Steady state kinetic parameters for the NAD+ dependent oxidation of 3α-diol catalyzed by RoDH4, RoDH5 and RL-HSD in the COS-1 cell lysates showed that only RoDH4 and RL-HSD were high affinity low capacity enzymes for the oxidation reaction, Table 2.
Fig. 3.

RL-HSD preferentially catalyzes the oxidation of 3α-diol to 5α-DHT. Percent conversion of 3α-diol to DHT in COS-1 cells stably transfected with bis-cistronic constructs (pcDNA3-3α-HSDisofrom-LacZ) where the 3α-HSD isoform corresponds to RoDH4 (■); RL-HSD (○); RoDH5 (□); NT-3α-HSD (♦); ERAB (▽) and pcDNA3 (▲). Cells were incubated with 0.1 μM [3H]-3α-diol. Percent conversion was normalized to β-galacosidase activity, and conversion to 5α-DHT and 5α-androstane-3,17-dione were combined due to the endogenous 17β-HSD present. Adapted from Bauman, et al. 2006a.
3. 2. Oxidative 3α-HSDs and Trans-activation of the AR
To determine whether the oxidative 3α-HSDs were able to convert sufficient 3α-diol to 5α-DHT to trans-activate the AR, reporter gene assays were performed. COS-1 cells were co-transfected with AR, a p(androgen response element)2-tk-CAT reporter gene construct in the absence or presence of the oxidative 3α-HSD of interest and exposed to 10−12 to 10−6 M 3α-diol. Oxidative 3α-HSDs that regulate ligand occupancy of the AR should convert 3α-diol to 5α-DHT and increase reporter gene activity, and shift the 3α-diol dose response curve to the left (i.e. an apparent increase in affinity). Only RoDH4, RoDH5 and RL-HSD were able to increase the affinity of the AR for 3α-diol by a 100-fold in this assay, Figure 4, (Bauman et al., 2006a).
Fig. 4.

Trans-activation of the AR by 3α-diol in the presence of oxidative 3α-HSDs. (A), Activation of the (ARE)2-tk-CAT reporter gene by the AR in the presence of co-transfected HSDs versus the concentration of 3α-diol (10−12 to 10−6 M); (B) the calculated EC50 values to reach a 100% trans-activation; where 100% trans-activation is the maximal response seen with 5α-DHT. The fold increase in chloramphenicol acetyl transferase (CAT) activity seen at maximal response was 30-fold; and (C) The cellular basis of the assay. Abbreviations, T = testosterone. Adapted from Bauman et al. 2006a.
3. 3. Expression of AKR1C isoforms and oxidative 3α-HSDs in human prostate by real-time RT-PCR
Our studies showed that three oxidative 3α-HSDs may be responsible for the back-reaction in human prostate: RoDH4, RoDH5, and RL-HSD. To determine their expression levels in human prostate we conducted real time RT-PCR assays using RNA pooled from 32 Caucasian males (BD Biosciences, Palo Alto, CA). Only ERAB and RL-HSD were abundantly expressed, of these only RL-HSD is able to oxidize 3α-diol to 5α-DHT efficiently, and in sufficient quantities to trans-activate AR. Thus RL-HSD is the isoform identified as the 3α-HSD responsible for the back reaction in human prostate.
To determine whether the reductive 3α-HSD (AKR1C2) and the oxidative 3α-HSD (RL-HSD) co-exist in the same cell type real time RT-PCR was conducted in primary cultures of prostate epithelial and stromal cells. AKR1C2 was predominately expressed in epithelial cells and no significant differences in levels of expression were found in cells from normal patients and patients with cancer of the prostate (CaP) or benign prostatic hyperplasia (BPH). By contrast RL-HSD was predominately expressed in stromal cells and a significant increase in expression level was noted in BPH. These data imply that the influence of these HSDs on AR ligand occupancy may be affected by stromal-epithelial cell interactions (Bauman et al., 2006b). It is proposed that 5α-DHT is inactivated in the epithelial cells by AKR1C2 to form 3α-diol. In stromal cells 3α-diol is reactivated to 5α-DHT by RL-HSD where 5α-DHT can bind and trans-activate the stromal AR. This could then result in androgen-dependent paracrine effects on prostate epithelial cells.
Fig. 5.
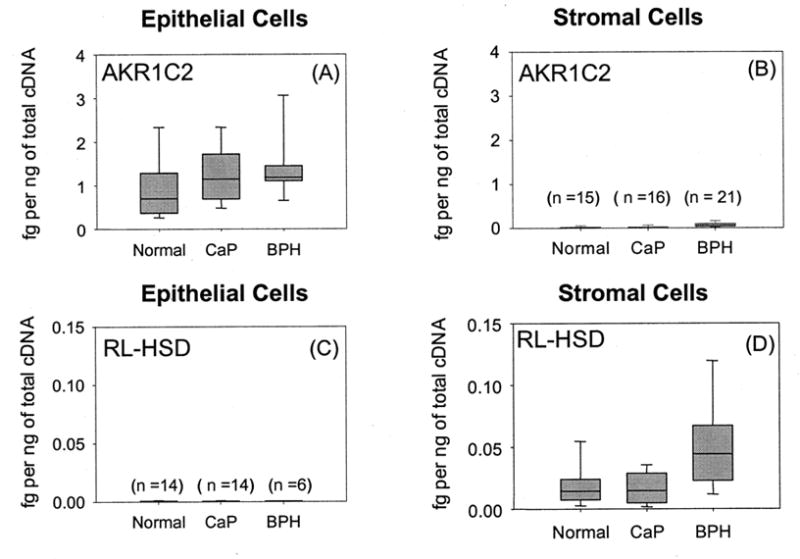
Expression of AKR1C2 and RL-HSD in primary cultures of epithelial and stromal cells taken from normal patients, cancer patients (CaP), and patients with benign prostatic hyperplasia (BPH). Cells were obtained from biopsy samples under an IRB approved protocol obtained by Dr. Donna M. Peehl at the University of Stanford. Cells were cultured as described (Bauman et al, 2006b). AKR1C2 and RL-HSD were quantified by real-time RT-PCR using validated primers (Bauman et al., 2006b) and amounts were normalized to two housekeeping genes (GAPDH-high abundance and porphobilinogen deaminase low abundance) and expressed as fg transcript per ng of total cDNA. The box plots show the median values and their associated standard areas. Adapted from Bauman et al. 2006b.
Acknowledgments
Supported by R01 DK47015, R01 CA 90742 and P30ES013508 awarded to T.M.P. We thank Dr. Donna Peehl, Stanford University for primary cultures of prostate epithelial and stromal cells.
Abbreviations
- AKR
aldo-keto reductase
- AR
androgen receptor
- BPH
benign prostatic hyperplasia
- 5α-DHT
5α-dihydrotestosterone
- 3α-diol or 3α-androstanediol
5α-androstane-3α,17β-diol
- ERAB
endoplasmic reticulum β-peptide binding protein
- HSD
hydroxysteroid dehydrogenase
- NT-3α-HSD
novel type of human microsomal 3α-HSD
- RL-HSD
“RoDH like 3α-HSD”
- RoDH
retinol dehydrogenase
- SDR
short-chain dehydrogenase/reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal AK, Auchus RJ. Minireview: Cellular redox state regulates hydroxysteroid dehydrogenase activity and intracellular hormone potency. Endocrinology. 2005;146:2531–2538. doi: 10.1210/en.2005-0061. [DOI] [PubMed] [Google Scholar]
- Askonas LJ, Ricigliano JW, Penning TM. The kinetic mechanism catalyzed by homogeneous rat liver 3α-hydroxysteroid dehydrogenase. Evidence for binary and ternary dead-end complexes containing non-steroidal anti-inflammatory drugs. Biochem J. 1991;278:835–841. doi: 10.1042/bj2780835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3α-hydroxysteroid dehydrogenase in human prostate that converts 5α-androstane-3α,17β-diol to 5α-dihydrotestosterone: A potential therapeutic target for androgen dependent disease. Mol Endocrinol. 2006a;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- Buaman DP, Steckelbroeck S, Peehl DM, Penning TM. Transcript profiling of the androgen signal in normal prostate, benign prostatic hyperplasia, and prostate cancer. Endocrinology. 2006b doi: 10.1210/en.2006-0627. Epub Ahead of Print September 7, 2006. [DOI] [PubMed] [Google Scholar]
- Biswas MG, Russell DW. Expression cloning and characterization of oxidative 17β– and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J Biol Chem. 1997;272:15959–15966. doi: 10.1074/jbc.272.25.15959. [DOI] [PubMed] [Google Scholar]
- Chetyrkin SV, Belyaeva OV, Gough WH, Kedishvili NY. Characterization of a novel type of human microsomal 3α-hydroxysteroid dehydrogenase: unique tissue distribution and catalytic properties. J Biol Chem. 2001;276:22278–22286. doi: 10.1074/jbc.M102076200. [DOI] [PubMed] [Google Scholar]
- DeKlerk DP, Coffey DS, Ewing LL, McDermott IR, Reiner WG, Robinson CH, Scott WW, Strandberg JD, Talalay P. Comparison of spontaneous and experimentally induced canine prostatic hyperplasia. J Clin Invest. 1979;64:842–849. doi: 10.1172/JCI109532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1998;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- Gough WH, Van Ooteghem S, Sint T, Kedishvili NY. cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all trans-retinol and 3α-hydroxysteroids. J Biol Chem. 1998;273:19778–19785. doi: 10.1074/jbc.273.31.19778. [DOI] [PubMed] [Google Scholar]
- He XY, Merz G, Yang YZ, Pullakart R, Mehta P, Schulz H, Yang SY. Function of human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase in androgen metabolism. Biochim Biophys Acta. 2000;1484:267–277. doi: 10.1016/s1388-1981(00)00014-7. [DOI] [PubMed] [Google Scholar]
- Horst HJ, Denis M, Kaufmann J, Voigt KD. In vivo uptake and metabolism of [3H]-5α-androstane-3α,17β-diol and of [3H]-5α-androstane-3β,17β-diol by human prostatic hypertrophy. Acta Endocrinol. 1975;79:394–402. [PubMed] [Google Scholar]
- Huang XF, Luu-The V. Characterization of the oxidative 3α-hydroxysteroid dehydrogenase activity of human recombinant 11-cis retinal dehydrogenase. Biochim Biophys Acta. 2001;1547:351–358. doi: 10.1016/s0167-4838(01)00200-x. [DOI] [PubMed] [Google Scholar]
- Jacobi GH, Moore RJ, Wilson JD. Studies on the mechanism of 3α-androstanediol-induced growth of the dog prostate. Endocrinology. 1978;102:1748–1758. doi: 10.1210/endo-102-6-1748. [DOI] [PubMed] [Google Scholar]
- Jin Y, Penning TM. Multiple steps determine the overall rate of the reduction of 5α-dihydrotestosterone catalyzed by human type 3 3α-hydroxysteroid dehydrogenase: implications for the elimination of androgens. Biochemistry. 2006 doi: 10.1021/bi060591r. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khana N, Sharma KK, Andersson S, Auchus R. Human 17β-hydroxysteroid dehydrogenase types 1, 2 and 3 catalyze bi-directional equilibrium reactions rather than unidirectional metabolism in HEK-293 cells. Arch Biochem& Biophys. 2004;429:50 –59. doi: 10.1016/j.abb.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Orlowski J, Bird CE, Clark AF. Androgen 5α-reductase and 3α-hydroxysteroid dehydrogenase activities in ventral prostate epithelial and stromal cells from immature and mature rats. J Endocrinol. 1983;99:131–139. doi: 10.1677/joe.0.0990131. [DOI] [PubMed] [Google Scholar]
- Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM. Hydroxysteroid dehydrogenases and pre-receptor regulation of steroid hormone action. Human Reproduction Update. 2003;9:193–205. doi: 10.1093/humupd/dmg022. [DOI] [PubMed] [Google Scholar]
- Rizner T, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3α-hydroxysteroid dehydrogenase (AKR1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- Russell DW, Wilson JD. Steroid 5α-reductase two genes/two enzymes. Ann Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Walker BR. Minireview: 11β-Hydroxysteroid dehydrogenase type 1 a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- Shaw G, Renfree MB, Leihy MW, Shackleton CH, Roitman E, Wilson JD. Prostate formation in a marsupial is mediated by the testicular androgen 5α– androstane-3α,17β-diol. Proc Natl Acad Sci USA. 2000;97:12256–12259. doi: 10.1073/pnas.220412297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: Implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- Stewart PM, Whorwood CB. 11β-Hydroxysteroid dehydrogenase activity and corticosteroid hormone action. Steroids. 1994;59:90–95. doi: 10.1016/0039-128x(94)90082-5. [DOI] [PubMed] [Google Scholar]