

Abstract
Frequency-dependent acceleration of relaxation (FDAR) is an important intrinsic mechanism that allows for diastolic filling of the ventricle at higher heart rates, yet its molecular mechanism is still not understood. Previous studies showed that FDAR is dependent on functional sarcoplasmic reticulum (SR) and can be abolished by phosphatase or by Ca/CaM kinase (CaMKII) inhibition. Additionally, CaMKII activity/autophosphorylation has been shown to be frequency-dependent. Thus, we tested the hypothesis that CaMKII phosphorylation of SR Ca2+-handling proteins (Phospholamban (PLB), Ca2+ release channel (RyR)) mediates FDAR. Here we show that FDAR occurs abruptly in fluo-4 loaded isolated rat ventricular myocytes when frequency is raised from 0.1 to 2 Hz. The effect is essentially complete within four beats (2 s) with the τ of [Ca2+]i decline decreasing by 42±3%. While there is a detectable increase in PLB Thr-17 and RyR Ser-2814 phosphorylation, the increase is quantitatively small (PLB<5%, RyR~8%) and the time-course is clearly delayed with regard to FDAR. The low substrate phosphorylation indicates that pacing of myocytes only mildly activates CaMKII and consistent with this CaMKIIδ autophosphorylation did not increase with pacing alone. However, in the presence of phosphatase 1 inhibition pacing triggered a net-increase in autophosphorylated CaMKII and also greatly enhanced PLB and RyR phosphorylation. We conclude that FDAR does not rely on phosphorylation of PLB or RyR. Even though CaMKII does become activated when myocytes are paced, phosphatases immediately antagonize CaMKII action, limit substrate phosphorylation and also prevent sustained CaMKII autophosphorylation (thereby suppressing global CaMKII effects).
Keywords: frequency, relaxation, phosphorylation, calmodulin-dependent kinase, phospholamban, ryanodine receptor, autophosphorylation, protein phosphatase 1
Introduction
The heart rate-dependent shortening of the twitch duration and Ca2+ transient (or frequency-dependent acceleration of relaxation (FDAR)) is a fundamental mechanism found in all mammalian hearts. It is important in limiting a decline in the diastolic interval at higher heart rates thereby allowing the ventricle sufficient filling time and coronary blood flow. However, the molecular basis for FDAR is not yet resolved.
An attractive hypothesis is that FDAR is mediated by Ca2+/Calmodulin (Ca/CaM)-dependent kinase II (CaMKII). De Koninck and Schulman [1] showed that CaMKII autonomous activity increases upon repeated activation by Ca/CaM, dependent on number and frequency of stimuli. Thus, CaMKII is a prime candidate in sensing and mediating frequency-dependent effects.
In the heart, CaMKII regulates several of the Ca2+ handling proteins involved in excitation-contraction coupling and there is evidence that CaMKII is involved in the mediation of FDAR [2–5]. It has been shown that FDAR can be suppressed by CaMKII inhibition and can be abolished by the phosphatase inhibitor okadaic acid [2]. However, the involvement of CaMKII in FDAR is still controversial as several other studies did not observe an effect of CaMKII inhibition on rate-dependent changes in relaxation or Ca2+ transient decline [6–10].
The next question is which of the CaMKII substrates can mediate the effect on relaxation and Ca2+ uptake. A plausible and very likely target is phospholamban (PLB), an inhibitor of the SR Ca2+ pump (for overview see [11]). When PLB becomes phosphorylated at Ser-16 by PKA or Thr-17 by CaMKII the inhibition is relieved and Ca2+ uptake is accelerated.
Hagemann et al. [10] showed that CaMKII phosphorylation of phospholamban at Thr-17 increased in a frequency dependent manner when myocytes are electrically stimulated. On the other hand we found when using PLB (−/−) mice, that FDAR was present to some degree in the absence of PLB, while FDAR could still be abolished by CaMKII inhibition [5]. Zhao et al. [13] reported recently that mutating the CaMKII phosphorylation site on PLB to Alanine (T17A) significantly blunted FDAR, again indicating PLB involvement in FDAR. Thus, the role of CaMKII and in particular PLB phosphorylation in FDAR is incompletely resolved.
On the other hand it is conceivable that other CaMKII targets contribute to FDAR. It is possible that the hastening of Ca2+ transient decline is mediated in part by more complete or rapid shut-off of SR Ca2+ release during excitation-contraction coupling. The cardiac RyR has been clearly identified as an CaMKII substrate, but the number and exact locations of the target sites is only incompletely known and is in part controversial [9, 14].
To further elucidate the link between CaMKII and FDAR we compared the time-course of FDAR development and CaMKII phosphorylation of PLB and RyR. Here we show that the time-courses of PLB as well as RyR phosphorylation and FDAR are clearly dissociated and that the net-increase in substrate phosphorylation is rather low. We also provide evidence that long lasting activation of CaMKII (due to autophosphorylation) and significant substrate phosphorylation in response to pacing is actively limited by phosphatases, most importantly phosphatase 1 (PP1).
Materials and Methods
Myocyte isolation
Isolation of rat ventricular myocytes was carried out essentially as previously described [15] and approved by the Loyola University Chicago animal welfare committee. Briefly, rats were injected with Heparin (about 2 Units/g bodyweight) 30 min prior to anesthesia by i.p. injection of Nembutal ( 0.1 mg/g). Hearts were excised quickly and perfused according to Langendorff for about 5 min with nominally Ca-free MEM (M-0518, SIGMA, St. Louis, MO) supplemented with (in mM): HEPES 10, Na-HEPES 10, Sodium Bicarbonate 4.8, and Pyruvic Acid 2. Additionally 20 U Insulin and 50.000 U Penicillin/Streptomycin were added and the pH adjusted to 7.35 with NaOH. The enzyme used for tissue dissociation was Liberase blendzyme 3 or 4 (13–20 Wuensch Units/heart) sometimes supplemented with 5–10 Units of dispase II (both Roche Diagnostics, Indianapolis, IN). When the heart became flaccid, the ventricle was cut into small pieces for further incubation (5 to 10 min at 37°C) in fresh enzyme solution. The tissue was dispersed, filtered, and suspensions were rinsed several times before use for experiments.
Batch field stimulation experiments were limited to 5 min at 2 Hz and pulse duration was 3 ms to minimize cell damage. The experimental chamber was mounted on an inverted microscope and cell response and survival of each sample monitored. A 70% yield of viable cells that contracted in response to electrical stimulation was typical (isolations containing <50% of rod-shaped cells were not used for phosphorylation experiments).
[Ca2+]i measurements and analysis
Isolated rat ventricular myocytes were then plated onto superfusion chambers, with the glass bottoms treated with natural mouse laminin (Invitrogen, Carlsbad, CA) to increase cell adhesion. The standard tyrode's solution used in all experiments contained (in mM): NaCl 140, KCl 4, MgCl2 1, glucose 10, HEPES 5, and CaCl2 1, pH = 7.4. Myocytes were loaded with 6 μM fluo-4/AM for 25 min and subsequently perfused for at least 30 min to allow for deesterfication of the dye. All experiments were done at room temperature (23–25°C) using field stimulation. Fluo-4 was excited at 480±5 nm and emission measured using a 535±20 bandpass-filter. Ca2+-transients were recorded with Clampex 8.0 and data analyzed with Clampfit.
Western blot analysis
For Western blot analysis, 6 X sample buffer (30 % Glycerol, 10 % SDS, 600 mM Dithiothreitol, 350 mM Tris·Cl pH 6.8, trace of Bromophenol Blue) supplemented with NaF (6 mM) and protease inhibitors (P-8340, SIGMA, St. Louis, MO) was directly added to the cells after treatment and the samples were immediately frozen in liquid N2. Protein was quantitated via a modified Lowry assay (PIERCE, Rockford, IL) and the amount of protein loaded onto the gel was within the linear range for the specific antibody. Primary antibodies used were a custom anti-PLB antibody (a gift from Dr. M. Periasamy, Columbus, OH), anti-phospho-PLB Ser-16 (PS16), anti-phospho-PLB Thr-17 (PT17; both from Badrilla, UK), anti-RyR 5029, anti-phospho-RyR Ser-2808 (PS2808), anti-phospho-RyR-2814 (PS2814; all RyR antibodies a gift from Dr. A. Marks, New York, NY), a custom anti-CaMKIIδ antibody (made against the C-terminal sequence by ZYMED Inc, San Francisco, CA), a monoclonal anti-CaMKII antibody (G1; Santa Cruz Biotechnology, Santa Cruz, CA) and a monoclonal anti-phospho-CaMKII Thr-287 antibody (Affinity BioReagents, Golden, CO). Protein bands were visualized using the SuperSignal West Dura Kit (PIERCE, Rockford, IL). Films were scanned using an UMAX Astra 2400S Scanner and quantified with Image J (NIH).
Immunoprecipitations
After treatment, cells were added directly to lysis buffer (8 % Triton X-100, 9 mM EGTA, 5 mM NaF, protease inhibitors) and immediately frozen in liquid N2. Protein was quantitated using the BCA assay (PIERCE, Rockford, IL). Samples were diluted to 1 μg/μl protein with RIPA-buffer (1 % Nonidet P-40, and in (mM) NaCl 150, Tris 10, EGTA 2, NaF 50, Na2H2P2O7 1, protease inhibitors, pH 7.2) and pre-cleared with Protein A-Sepharose CL-4B (Amersham, Upsala, Sweden). Tubes were rotated overnight at 4°C with 5–8 μg of peptide-affinity purified custom anti-CaMKIIδ antibody. Protein A-Sepharose beads were washed 3X with RIPA-buffer and then incubated in sample buffer for 10 min at 95ºC.
Statistical analysis
Results are expressed as means ± SEM. Significance was estimated by one-way repeated measures ANOVA and/or Student's t-test for paired observations as appropriate. P ≤ 0.05 was considered significant.
Results
FDAR occurs abruptly and is complete within four beats
First we measured the time-course of FDAR development in single fluo-4 loaded rat cardiomyocytes. Myocytes were paced at 0.1 Hz to steady-state before stimulation frequency was increased to 2 Hz (Fig. 1a). The sudden increase in frequency led to a rapid decrease in the τ of intracellular Ca2+ concentration ([Ca2+]i) decline by 42 ± 3 % within four beats or two seconds (Fig. 1b). FDAR was complete at this point, the τ of [Ca2+]i decline did not decrease any further within the next 200 s of continuous stimulation at 2 Hz (176±13 ms (at 2 s) vs 174±13 ms (at 200 s); p>0.05). After 200 s however there was an additional slight but significant decrease in the τ of [Ca2+]i decline (to 166±12 ms (at 300 s); p<0.05).
Figure 1.

Changes in Ca2+ decline and transient amplitude in response to an increase in frequency. (a) Original representative recording showing FDAR development after switching to 2 Hz in a Fluo-4 loaded rat ventricular cardiomyocyte. The τ-values (ms) are indicated for each beat. (b) Decrease in the τ of [Ca2+]i decline in response to an increase in stimulation frequency from steady-state 0.1 Hz (pre) to 2 Hz (n= 8 cells/3 rats). τ decreases rapidly over the first four beats (1–4) and reaches a plateau after 5 s. An additional small decrease in τ occurs after more than 3 min of stimulation at 2 Hz (see inset). Please note that the first Ca2+ transient at 0.5 s interval (2) declines significantly faster than the last at 0.1 Hz (228±19 ms vs 309±28 ms (pre); p < 0.001). (c) Diastolic (dotted line) and peak systolic (solid line) [Ca2+]i as F/F0 after increasing to 2 Hz frequency. After switching to 2 Hz frequency the amplitude (ΔF/F0) is decreased for 5 s and then recovers (inset). The amplitude at 0.1 Hz and after 5 min at 2 Hz is similar (4.45±0.25 (n=9) at 0.1 Hz vs 4.92±0.36 (n=9) at 2 Hz).
The amplitude of the [Ca2+]i transients decreased rapidly in the first two seconds after switching to 2 Hz and then again increased steadily between 2 and 300 s (p<0.001), regaining on average about the same amplitude as at 0.1 Hz steady-state stimulation (0.1 vs 2 Hz (after 5 min); p>0.05; flat [Ca2+]i -frequency response; Fig. 1c). Note that FDAR is essentially stable despite gradual changes in Ca2+ transient amplitude.
PLB-Thr 17 phosphorylation increases in response to pacing, but the increase is quantitatively low
Batches of cells were stimulated for 1 min at 0.1 Hz and then from 0–300 s at 2 Hz and immediately frozen in liquid nitrogen (time between last twitch and frozen condition about 5 s). Then the cell suspension was boiled in sample buffer and phosphorylation of monomeric PLB was analyzed using phosphorylation-site specific antibodies. Western blot analysis revealed that PLB-Thr 17 phosphorylation (barely detectable at rest) increases linearly, proportional to the duration of pacing at 2 Hz (Fig. 2a, b). Phosphorylation did not increase within the first 10 s of fast stimulation and did not reach steady-state within 5 min. These data clearly show that there is a marked temporal dissociation between FDAR and PLB phosphorylation.
Figure 2.
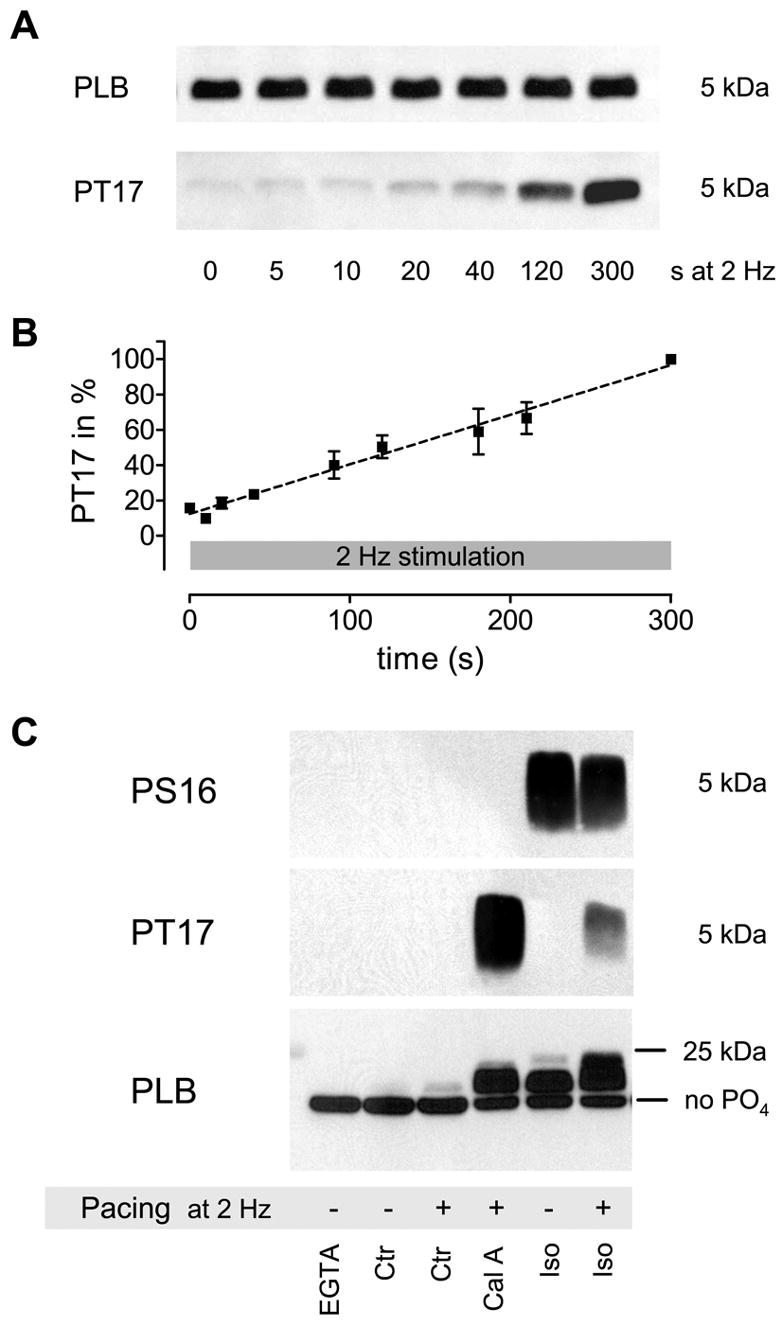
Increase in PLB Threonine 17 phosphorylation (PT17) in response to pacing at 2 Hz (a, b). Phosphorylation increases linear and does not reach steady-state within 300 s (n = 2–10; all signals normalized to 300 s (=100 %)). Total PLB was measured in the same samples on a different blot and used as loading control. PLB Ser-16 phosphorylation did not change within that timeframe (data not shown). For total PLB quantification 4 μg of protein were loaded into each lane, while for PLB phosphorylation (PS16 and PT17) 40 μg were used. Semi-quantitative analysis of total PLB-phosphorylation levels (c). The increase in the apparent molecular weight and subsequent upward shift of the PLB pentamer in SDS-PAGE relative to its phosphorylation level revealed that only a very small amount becomes phosphorylated in response to pacing alone that is likely less than 5% of total PLB (the position of unphosphorylated PLB is indicated (no PO4)). This estimate is derived from the long exposure times needed to detect a signal in the paced control (Ctr+) sample in panel 2 (actually not visible due to a shorter exposure shown here) and the upward shift in panel 3 (some bands are overexposed to visualize the difference between Ctr- and Ctr+). Higher PLB phosphorylation is achieved by phosphatase-inhibition plus pacing (Cal A; mostly Thr-17 phosphorylation), Iso without pacing (Iso-; mostly Ser-16 phosphorylation) and Iso plus pacing (Iso+; some dual phosphorylation). This result is representative of five similar experiments.
Moreover, the gradual increase in PLB phosphorylation during increased frequency was not accompanied by a significant acceleration of [Ca2+]i decline ([Ca2+]i decline is largely stable). One possible explanation is that only a very small amount of PLB becomes phosphorylated, not enough to significantly increase SR Ca2+ uptake and hasten [Ca2+]i decline. To estimate the total amount of pacing-induced phosphorylated PLB we used a semi-quantitative approach (Fig. 2c). This is based on the distinct property of PLB pentamer to disproportionally shift upwards in an SDS-Polyacrylamidgel (displaying higher apparent molecular weight) upon phosphorylation (see [11] for high-resolution example). The PLB pentamer can be phosphorylated five times at Ser-16 and five times at Thr-17 (ten phosphorylation sites total) and each additional phosphorylation causes a further shift upwards. We found that only few PLB shift upwards (Fig. 2c, lower panel (PLB): grey area above black band in lane 3) when cells are paced at 2 Hz for 300 s and the majority of PLB molecules remains unphosphorylated. On the other hand large amounts of phosphorylated PLB are produced by pacing in the presence of a phosphatase inhibitor (Calyculin A (Cal A); 1 μM) or Isoproterenol (Iso; 0.1 μM with or without pacing). The antibody used in these experiments is not affected by the phosphorylation state of PLB [16]. It is unfortunately not possible to exactly quantify the amount of phosphorylated PLB using this approach, but it appears to be very low and we cautiously estimate that it does not exceed 5% of total PLB after 5 min pacing at 2 Hz (for a more detailed explanation see Fig.2(c)). This indeed supports the notion that the lack of a functional effect matching the time-course of PLB phosphorylation is due to low absolute levels of phosphorylated PLB that are generated by pacing at 2 Hz.
We also investigated the reversal of FDAR and dephosphorylation of PLB (Fig. 3). When the frequency of stimulation is lowered to 0.2 Hz, more than 40% of FDAR dissipate between the last fast twich and the first slow twitch (fast phase). Afterwards [Ca2+]i decline slows with a τ of 88 s (n = 7 cells; slow phase). The de-phosphorylation of PLB after stimulation was discontinued roughly concurs with the slow phase of FDAR reversal (τ = 109 s; n = 6), but not with the initial very rapid dissipation of FDAR.
Figure 3.

FDAR reversal and PLB dephosphorylation. (a) Cells were stimulated for 5 min at 2 Hz and then frequency was lowered to 0.2 Hz (n = 7 cells/3 rats). FDAR reversal has an initial very fast and then a long slow component. The slow phase was fitted using a monoexponential equation with τ = 88 s. (b) Representative Western blot showing the increase in PLB Thr-17 phosphorylation upon stimulation at 2 Hz and dephosphorylation upon rest. (c) Summary of six similar experiments. All signals were normalized to phosphorylation after 300 s of stimulation at 2 Hz (=100 %). Total PLB was measured in the same samples on a different blot and used as loading control. The decrease in phosphorylation was fitted using a monoexponential equation with τ = 109 s.
Taken together, the very rapid acceleration of [Ca2+]i decline that is observed in response to pacing at 2 Hz is temporally distinct from the very gradual and modest increase in PLB Thr-17 phosphorylation. Thus, FDAR cannot be mediated by PLB Thr-17 phosphorylation.
RyR Ser-2814 is a CaMKII phosphorylation site and responds to pacing
Another CaMKII substrate also located in the SR membrane is the RyR. CaMKII co-purifies with the RyR [17], appears to directly associate with it [9, 18] and also regulates the receptor through phosphorylation [9, 14, 19]. There has, however, been controversy regarding the exact site or sites phosphorylated by CaMKII. Using a phosphorylation site specific antibody one study found that Ser-2808 (Ser-2809 in rabbit) is phosphorylate-able by both, PKA and CaMKII [14]. Another study describes Ser-2808 as an exclusive PKA site and a different nearby Serine (site 2814; Ser-2815 in rabbit) as the CaMKII site by using mutational analysis and a different set of phosphorylation site specific antibodies [9]. A third study even proposed that Ser-2808 can be phosphorylated by PKA, CaMKII and PKG, with an additional and still to be identified kinase that may be responsible for the baseline phosphorylation level in intact cells [20]. Fig. 4(a,b) shows that when using the same set of antibodies as Wehrens et al. 2004 [9] we found (in agreement with them) that Ser-2808 behaved like a PKA-phosphorylation site (no effect of EGTA incubation (99±5% (n=4), but Iso increased phosphorylation independent of pacing (Iso (−) 192±8% Iso (+) 179±5% (n=4)) and Ser-2814 responded to changes in [Ca2+]i consistent with a CaMKII phosphorylation site (lower phosphorylation after EGTA incubation (53±2% (n=4)), increased phosphorylation in response to pacing in the absence (see below) and presence of Iso (Iso (−) 71±9%; Iso (+) 125±8% (n=4)) and particularly when paced in the presence of the phosphatase inhibitor okadaic acid (275±45% (n=7)). Interestingly, Iso exposure of resting myocytes decreased phosphorylation at Ser-2814 to 71±9% of control indicating an effect of PKA activation on CaMKII. It is possible that Iso inhibits CaMKII by lowering resting [Ca2+]i (by stimulating SR Ca2+ uptake), as has been observed in mouse cardiomyocytes [21]. However, this result could also be explained by sensitivity of the PS2814 antibody to phosphorylation at Ser-2808 (amino acid 2808 is included in the antigenic peptide sequence for PS2814; [9]) and further studies are needed to clarify this issue. It is also important to note that when we used the Ser-2808 antibody described by Rodriguez et al. 2003 [14] to analyze identical samples results were slightly different and maybe consistent with Ser-2808 being susceptible to phosphorylation by both kinases. The reason for this discrepancy is currently unknown.
Figure 4.


Ryanodine Receptor (RyR) phosphorylation at sites Ser-2809 and Ser-2814 measured with phosphorylation-site specific antibodies (PS2809, PS2814; (a)). Where indicated cells were paced (5 min at 2 Hz) and/or exposed to EGTA (4 mM for 6 min), isoproterenol (Iso; 0.1 μM for 6 min) or okadaic acid (OA; 2 μM for 36 min total). Total RyR was measured in the same samples on a different blot and used as loading control. (b) summarizes the results from 4–7 independent experiments. Signals were normalized to Control (sample #2 = 100 %). (c) is representative for 3 independent experiments where cells were paced (5 min at 2 Hz) or treated with EGTA (4 mM for 6 min) or calyculin A (Cal A; 1 μM for 6 min). Assuming that Cal A treatment increases the receptor PS2814 phosphorylation to 90%, than 16±4 % are phosphorylated under resting conditions and this increases to 24±4 % in response to pacing. The timecourse of the effect of pacing alone is shown in (d). Please note that phosphorylation at Ser-2814 due to pacing does not increase within the first 30 s after the start of stimulation.
The time-course of phosphorylation at site Ser-2814 in response to pacing at 2 Hz was different compared to PLB, but similarly did not match the time-course of FDAR (Fig. 4(d)). RyR Ser-2814 phosphorylation was unaltered for 30 s at 2 Hz, but then increased rapidly to 151±19% after 90 s without further increase within 300 s (152±15%; n = 8). We roughly estimate that this increase corresponds to about 8 % of the total RyR pool (Fig. 4c; procedure explained in figure legend). Within the same timeframe there was no change in phosphorylation at Ser-2808.
Taken together we conclude that FDAR does not rely on phosphorylation of PLB or the RyR by CaMKII.
CaMKII- autophosphorylation is unaffected by pacing
Another CaMKII substrate is CaMKII itself (autophosphorylation at Thr-287 (δ-isoform)). Its unique hub-and-spoke architecture permits for intersubunit autophosphorylation upon activation of two neighboring subunits, which results in Ca/CaM independent or autonomous activity. Therefore, we predict that significant physiological activation is accompanied by CaMKII autophosphorylation (an increase in autonomous activity has been shown in isolated perfused rat hearts upon Iso-treatment which was reversible by PP2A; [22]) and accordingly the level of autophosphorylation should reflect the immediately preceding level of activation.
Most often we observed no autophosphorylation in quiescent myocytes, but sometimes a slight signal was detected that decreased with EGTA treatment (data not shown). Most important, using a highly sensitive assay (see Fig. 5; PT287 in immuneprecipitated samples), pacing did not produce any detectable increase in CaMKII–autophosphorylation, even in the presence of high extracellular [Ca2+] or Iso.
Figure 5.
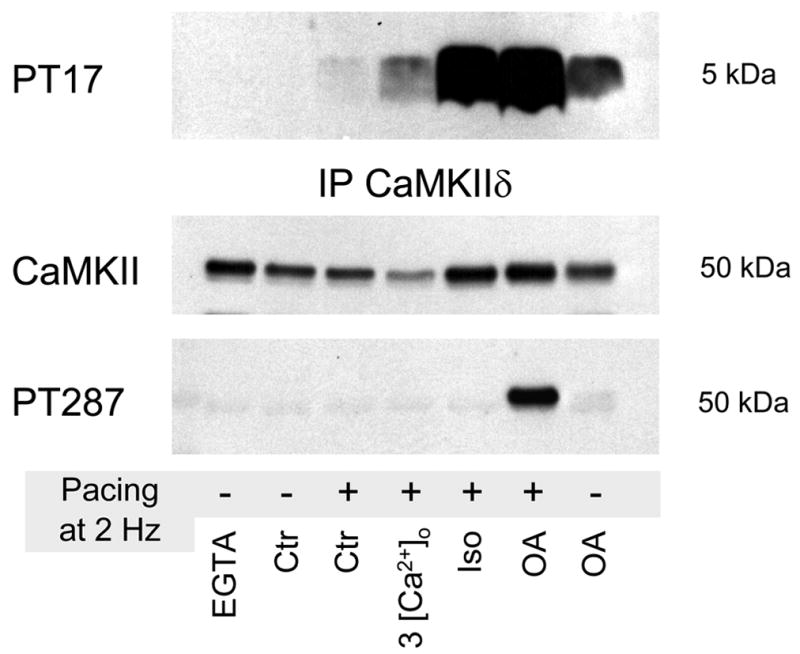
No change in CaMKII autophosphorylation at Threonine-287 in response to pacing alone (5 min at 2 Hz), pacing in the presence of 3 mM [Ca2+]o or of 0.1 μM isoproterenol (Iso). Autophosphorylation was observed after treatment with phosphatase inhibitor okadaic acid (OA; 2 μM for 36 min total). Western blots results from cell lysates (not shown) and immuneprecipitated samples (for increased sensitivity) matched each other. Please note that phosphatase inhibition plus pacing increased autophosphorylation to much higher levels than phosphatase inhibition alone.
This finding could indicate that the global [Ca2+]i fluctuations during each excitation-contraction cycle in cardiomyocytes fail to activate CaMKII to the point where autophosphorylation occurs. This would be consistent with the generally low levels of substrate phosphorylation (PLB/RyR) we observed. A second possibility is that powerful phosphatases rapidly reverse any autophosphorylation (and most of the substrate phosphorylation), thereby preventing autonomous activity of the kinase. To test the role of phosphatases we preincubated myocytes with the broad spectrum phosphatase inhibitor okadaic acid (OA; 2 μM) and analyzed quiescent and paced cells (paced for 5 min at 2 Hz). In the presence of OA a detectable level of autophosphorylated CaMKII is generated in quiescent cells (revealing some basal activity of CaMKII), but autophosphorylation is clearly enhanced when the cells were paced (Fig. 5; penultimate lane). This result is consistent with the idea that in cardiomyocytes, in which [Ca2+]i increases during every contraction, phosphatases prevent sustained CaMKII autophosphorylation and thereby suppress Ca/CaM-independent CaMKII activity.
Pacing increases CaMKII autophosphorylation when PP1 is inhibited
To investigate in detail which phosphatases are involved in antagonizing CaMKII activity we used a pharmacological approach. Phosphatases (PPs) are classified into PP1–PP7, while PP1–PP6 are found in the heart (for overview see [23]). okadaic acid (OA) in nM concentrations inhibits all but PP1, PP2B (Calcineurin) and PP2C and in μM concentrations also inhibits PP1. Calyculin A (Cal A) has been shown to be a potent and equally effective inhibitor of PP1 and PP2A (IC50 1–14 nM; see [23]). Cantharidin at a concentration of 1 μM inhibits PP2a [24]. PP2B can be specifically inhibited by low nM concentrations of cypermethrin [25] and cyclosporin A [26].
To the best of our knowledge, the main phosphatase dephosphorylating endogenous CaMKII in intact cardiac cells or cardiac tissue has not yet been identified. PP1 can dephosphorylate CaMKII in vitro [27], but in mouse hearts overexpressing CaMKIIδB PP2A directly associates with CaMKII [28]. In the rat forebrain PP2a-like enzymes pre-dominantly dephosphorylate soluble CaMKII, while PP1 predominantly dephosphorylates CaMKII associated with postsynaptic densities [29].
When using electrical stimulated rat cardiomyocytes, the inhibition of PP2A (and PP3–PP6) by 200 nM okadaic acid was not enough to generate autophosphorylated CaMKII (Fig. 6). Neither was inhibition of PP2A by 1 μM cantharidin or PP2B by 1 μM cypermethrin and 1 μM cyclosporin A. In our hands strong inhibition of PP1 was required to allow net-autophosphorylation of CaMKII. Unfortunately all cell-permeable PP1 inhibitors available also inhibit PP2A with higher or similar potency and we cannot specifically inhibit PP1 alone. Therefore we cannot rule out some involvement of PP2A. However, our experiments identify PP1 as the major phosphatase dephosphorylating CaMKII in intact cardiomyocytes.
Figure 6.

Phosphatase 1 appears to be the major phosphatase dephosphorylating CaMKII in cardiomyocytes. Pacing triggers autophosphorylation of CaMKII only when phosphatase 1 (and 2a) is inhibited, but not when phosphatase 2b (calcineurin) or 2a alone are inhibited. Phosphatase inhibitors used were cypermethrin (1 μM; 30 min preincubation), okadaic acid (200 nM; 30 min precincubation) and calyculin A (1 μM; 10 min precincubation). Phosphatase 2b was also inhibited by cyclosporin A, phosphatase 2a by cantharidin and phosphatase 1 by higher concentrations of OA yielding the same result (data not shown).
Time-course of CaMKII autophosphorylation in the presence of PP1/2a inhibition
We further analyzed the effect of pacing on CaMKII activation by measuring CaMKII autophosphorylation after 1 min of pre-incubation with 1 μM Cal A (0) and after 0.5, 1.5, 3 and 5 min of pacing (Fig. 7). We chose a very short pre-incubation time to minimize any increase in protein phosphorylation due to baseline kinase activity. Pacing increased PT287 autophosphorylation to 125±24% (0.5 min), 209±37% (1.5 min), 404±106% (3 min) and 442±130% (5 min; with 0 min = 100% and n = 3), which is similar to the RyR PS2814 in response to pacing without phosphatase inhibition. One noteable difference however is that phosphatase inhibition caused a significant increase in RyR phosphorylation within 30 s (to 220±80%; n=3). This is the fastest increase in phosphorylation we observed within this study. However, with or without phosphatase inhibition, the phosphorylation time-courses of PLB, RyR and CaMKII do not match the time-course of FDAR.
Figure 7.

Timecourse of PLB- PT17 (also semiquantitative as pentamer-shift), RyR-PS2814 and CaMKII-PT287 phosphorylation in the presence of Cal A in response to pacing. Autophosphorylation of CaMKII increases between 30 and 180 s and then remains steady. Phosphatase inhibition increases the phosphorylation at all targets dramatically. Please note that PLB PT17 phosphorylation cannot be seen in several samples due to the short exposure time of the blot.
Discussion
FDAR is a phenomenon that has been described for decades but its molecular mechanism is still unresolved. In the present study we tested if pacing (or more specifically the resulting rise in [Ca2+]i and its frequency) activates CaMKII and mediates FDAR by phosphorylation of specific target proteins.
PLB phosphorylation
We first investigated if CaMKII-dependent PLB phosphorylation contributes significantly to the development of the functional effect, an idea that has been tested previously yielding different results [6, 10, 12]. Our group repeatedly showed that CaMKII inhibitors KN-62 and KN-93 can abolish FDAR [2–4] and this is also true in PLB null mice in which FDAR still occurs [5]. Those results indicated that while it is possible that PLB phosphorylation contributes to some degree to FDAR we found it not to be essential for FDAR to develop.
Consistent with these earlier observations we describe here that even though PLB-Thr17 phosphorylation increases when myocytes are paced, the time-courses of FDAR and PLB phosphorylation are dissociated and the absolute amount of PLB that becomes phosphorylated is very small (likely less than 5% after 5 min at 2 Hz). This is similar to recent observations by Valverde and coworkers [10], who also noted a delayed onset of PLB Thr-17 phosphorylation with respect to FDAR. Our finding here that the increase in PLB phosphorylation is detectable using highly sensitive assays, but that the actual amount of phosphorylated PLB is very low, might explain why results have been variable in the past. The amount of PLB phosphorylated appears to be functionally almost insignificant as the only effect that correlates with its time-course is the minor decrease in the τ of Ca2+ decline between 200 and 300 s of pacing (see Fig.1b inset). Thus, FDAR could still be mediated by CaMKII, but PLB phosphorylation does not appear to be involved.
This conclusion was recently further supported by the finding that FDAR is associated with an increase in the maximal velocity (Vmax) of Ca2+ uptake without a change in the apparent Ca2+ affinity of SERCA pump activity [30]. The effect of PLB phosphorylation is primarily an increase in the apparent Ca2+ affinity of SERCA pump (most often no effect on Vmax is observed; [11]), and is therefore not likely to be responsible for the observed effect.
It has been reported that SERCA pump itself can be phosphorylated by CaMKII and that this increases the Vmax of Ca2+ uptake [31]. However, both aspects have been challenged [32, 33] and satisfactory evidence for phosphorylation of the pump in intact tissue or intact cells has not yet been provided. The phosphorylation of SERCA pump was recently investigated and could not be demonstrated at low or higher pacing frequencies [10]. Therefore we are not aware of an alternative CaMKII substrate that might be a good candidate to increase SR Ca2+ uptake upon phosphorylation.
RyR phosphorylation
A CaMKII substrate located in the SR membrane that has been shown to co-immunoprecipitate with CaMKII is the Ryanodine Receptor [17, 18]. Since CaMKII and substrate appear to be co-localized we expected that phosphorylation is triggered more quickly upon CaMKII activation because a diffusion step is not necessary. However, the study of the phosphorylation kinetics was complicated by the fact that the exact CaMKII target site/sites within the RyR is/are still somewhat controversial. RyR PS2808 has been described as a unique CaMKII target site [19], a site phosphorylated by CaMKII as well as PKA [14], a site phosphorylated by at least four different kinases including CaMKII and PKA [20], and as a unique PKA site [9]. Using a phosphorylation site-specific antibody generated by A. Marks’ laboratory we found that the phosphorylation of this site could be influenced by Isoproterenol and phosphatase inhibition, but not by Ca2+-buffering via EGTA or Ca2+-increase due to pacing, indicating that this site is not significantly phosphorylated by CaMKII in intact cells (although we cannot rule out that this happens under certain conditions). On the other hand we found that RyR Ser-2814 phosphorylation decreased when cells were incubated with EGTA and phosphorylation increased when cells were paced, consistent with a CaMKII phosphorylation site as previously described [9]. Stimulation of the cells for 5 min at a frequency of 2 Hz increased phosphorylation by 50% and we estimated that this corresponds to roughly 8% of the total RyR (Fig. 3c,d). In contrast to PLB-Thr17 phosphorylation, the increase in phosphorylation at RyR Ser-2814 in response to pacing reached a plateau rapidly (within 90 s), but similarly to PLB the time-course was completely dissociated from FDAR development. It is very striking that we did not observe a net-increase in Ser-2814 within the first 30 s of pacing.
Furthermore, while the overall functional effects of CaMKII phosphorylation on the RyR are controversial [17, 34, 35], phosphorylation at Ser-2814 has been associated with increased open probability of the channel [9]. Generally it can be expected that this prolongs relaxation time rather than shortens it. Thus, also the modification of channel function appears to be inconsistent with a role of Ser-2814 phosphorylation in the mediation of FDAR.
Does pacing activate CaMKII and increase autophosphorylation?
CaMKII itself is, like the RyR, a substrate that is directly associated with the kinase and thus has the potential to become very quickly phosphorylated upon CaMKII activation. We expect that significant activation of CaMKII should be accompanied by increasing levels of CaMKII autophosphorylation. It has been observed in vitro that the initial rates for CaMKII autophosphorylation are higher than the rates for exogenous substrate phosphorylation [36, 37]. So it is possible that CaMKII autophosphorylation has a timecourse that matches FDAR.
One important finding of this study is that pacing of myocytes does not produce detectable levels of Thr-287 autophosphorylated CaMKII. CaMKII autophosphorylation was only observed when phosphatase 1 was inhibited, but then pacing strongly enhanced autophosphorylation. The increase in CaMKII autophosphorylation is likely due to direct inhibition of phosphatases dephosphorylating CaMKII, but it is also possible that phosphatase inhibition enhances CaMKII activation by increasing [Ca2+]i (via an increase in the phosphorylation level of Ca2+-regulatory proteins; [38–40]). However, as we demonstrate in this study, increasing the intracellular [Ca2+] via increased extracellular [Ca2+] or stimulation with Iso is not sufficient to net-autophosphorylate CaMKII.
We also show however that in the presence of okadaic acid the RyR Ser-2814 was the first site significantly increased in response to pacing, not global CaMKII Thr-287. This does not rule out that CaMKII specifically associated with the RyR does become significantly transiently activated and/or autophosphorylated sooner, especially when considering that the local [Ca2+] around the RyR in the junctional cleft might reach very high concentrations during each excitation-contraction cycle [41–43].
General implications about CaMKII regulation in the heart
The emerging picture from these experiments is that CaMKII does get activated when myocytes are paced, and that this activation does result in autophosphorylation at Thr-287, but any build up of autophosphorylated CaMKII is strongly limited by PP1. It appears that PP1 effectively counteracts the accumulation of autophosphorylated CaMKII and therefore CaMKII remains dependent on Ca/CaM for activation. This would allow autophosphorylation to specifically occur only in locations where phosphatase activity is low and/or Ca/CaM concentration is high. Similar aspects apply to CaMKII target phosphorylation.
Our results regarding the regulatory role of phosphatases are consistent with in vitro results about the effect of phosphatases on CaMKII autophosphorylation [27]. Bradshaw and coworkers [27] describe that in the presence of phosphatases the Ca2+ dependence of CaMKII autophosphorylation is shifted towards higher Ca2+ concentrations and that autophosphorylation occurs in a switch-like fashion over a narrow range of physiological Ca2+-concentration. This ultrasensitive switch behaviour allows CaMKII to transition from non-autophosphorylated to almost completely autophosphorylated once a critical Ca2+ concentration is reached (all-or-none response), providing for multiple pathways to be specifically activated within the physiological range of Ca2+.
On the other hand our results are not consistent with observations from Baltas et al. [22], who described autonomous CaMKII activity upon stimulation with Iso in isolated perfused rat hearts that was reversible by in vitro PP2A treatment. Even though the authors did not measure CaMKII Thr-287 autophosphorylation directly, this could indicate that there are differences in isolated cells compared to whole hearts and maybe more specifically in the localization and activation of kinases and phosphatases. To address this issue further studies are needed. However, despite these potential differences, FDAR is clearly present in cardiomyocytes and therefore we can safely assume that the underlying mechanism is not interrupted.
Could CaMKII still be involved in FDAR?
The experiments presented in this study clearly show that CaMKII phosphorylation of PLB and/or RyR (S2808/S2814) is not responsible for the development of FDAR, but we cannot rule out that CaMKII is involved. The existence of two separate phases (fast and slow phase) during FDAR reversal suggests that possibly more than one mechanism produces or maintains FDAR. Therefore it is conceivable that FDAR relies in part on CaMKII, but additional factors could contribute as well (e.g. intrinsic features of the Ca2+ handling proteins and/or myofilaments may participate).
On the other hand the L-type Ca2+ channel is a known CaMKII substrate. There is cumulative evidence that CaMKII phosphorylation produces Ca2+-dependent ICa facilitation, which is typically observed as an increased ICa amplitude and slower inactivation over 2 to 5 pulses [44–46]. So here is an example for a CaMKII dependent effect with a time-course that closely resembles FDAR (albeit this target is an unlikely participant in FDAR). Thus, it is feasible that FDAR relies on CaMKII phosphorylation of a yet to be identified target with kinetics as rapid as that of the L-type Ca-channel that is protected from rapid phosphatase action.
Acknowledgments
We are grateful to Dr. Andrew Marks for providing us phosphorylation-site specific RyR antibodies. We would also like to thank Dr. Kenneth Ginsburg for numerous helpful discussions and Dr. Eckard Picht for critical reading of the manuscript. The excellent technical assistance of Jayme O’Brien was very much appreciated. This work was supported by grants from the NIH (HL30077 and HL80101).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Koninck P, Schulman H. Sensitivity of CaM Kinase II to the frequency of Ca2+ Oscillations. Science. 1998;279:227–30. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 2.Bassani RA, Mattiazzi A, Bers DM. CaMKII is responsible for activity-dependent acceleration of relaxation in rat ventricular myocytes. Am J Physiol. 1995;268:H703–12. doi: 10.1152/ajpheart.1995.268.2.H703. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca2+-calmodulin-dependent protein kinase on cardiac excitation-contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501:17–32. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L, Chu G, Kranias E, Bers DM. Cardiac myocytes calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am J Physiol. 1998;43:H1335–47. doi: 10.1152/ajpheart.1998.274.4.H1335. [DOI] [PubMed] [Google Scholar]
- 5.DeSantiago, Maier LS, Bers DM. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J Mol Cell Cardiol. 2002;34:975–84. doi: 10.1006/jmcc.2002.2034. [DOI] [PubMed] [Google Scholar]
- 6.Hussain M, Drago GA, Colyer J, Orchard CH. Rate-dependent abbreviation of Ca2+ transient in rat heart is independent of phospholamban phosphorylation. Am J Physiol. 1997;273:H695–706. doi: 10.1152/ajpheart.1997.273.2.H695. [DOI] [PubMed] [Google Scholar]
- 7.Layland J, Kentish JC. Positive force and [Ca2+]i-frequency relationships in rat ventricular trabeculae at physiological frequencies. Am J Physiol. 1999;276:H9–18. doi: 10.1152/ajpheart.1999.276.1.H9. [DOI] [PubMed] [Google Scholar]
- 8.Kassiri Z, Myers R, Kaprielian R, Banijamali HS, Backx PH. Rate-dependent changes of twitch force duration in rat cardiac trabeculae: a property of the contractile system. J Physiol. 2000;524:221–31. doi: 10.1111/j.1469-7793.2000.t01-3-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wehrens XHT, Lehnart SE, Reiken SR, Marks AR. Ca2+/Calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 10.Valverde CA, Mundina-Weilenmann C, Said M, Ferrero P, Vittone L, Salas M, et al. J Physiol. 2005;562:801–13. doi: 10.1113/jphysiol.2004.075432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmerman HKB, Jones LR. Phospholamban: Protein structure, Mechanism of Action and Role in cardiac function. Physiol Rev. 1998;78:921–47. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- 12.Hagemann D, Kuschel M, Juramochi T, Zhu W, Cheng H, Xiao RP. Frequency-encoding Thr17 Phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J Biol Chem. 2000;275:22532–6. doi: 10.1074/jbc.C000253200. [DOI] [PubMed] [Google Scholar]
- 13.Zhao W, Uehara Y, Chu G, Song Q, Qian J, Young K, Kranias EG. Threonine-17 phosphorylation of phospholamban: a key determinant of frequency-dependent increase of cardiac contractility. J Mol Cell Cardiol. 2004;37:607–12. doi: 10.1016/j.yjmcc.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on Serine 2809 by Calmodulin-dependent Kinase II and Protein Kinase A. J Biol Chem. 2003;278:38593–600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 15.Bassani JWM, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–93. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huke S, Periasamy M. Phosphorylation-status of phospholamban and calsequestrin modifies their affinity towards commonly used antibodies. J Mol Cell Cardiol. 2004;37:795–9. doi: 10.1016/j.yjmcc.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, et al. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–19. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 18.Currie S, Loughrey CM, Craig M-A, Smith GL. Calcium/calmodulin-dependent protein kinase IIδ associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem J. 2004;377:357–66. doi: 10.1042/BJ20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–52. [PubMed] [Google Scholar]
- 20.Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, et al. Serine-2030, but not Serine-2808, is the major phosphorylation site in cardiac ryanodine receptor responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 22.Baltas LG, Kraczewski P, Bartel S, Krause EG. The endogenous cardiac sarcoplasmic reticulum Ca2+/calmodulin-dependent kinase is activated in response to β-arenergic stimulation and becomes Ca2+-independent in intact beating hearts. FEBS Let. 1997;409:131–136. doi: 10.1016/s0014-5793(97)00470-5. [DOI] [PubMed] [Google Scholar]
- 23.Neumann J, Herzig S. Effects of Serine/Threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–204. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- 24.Neumann J, Herzig S, Boknik P, Apel M, Kaspareit G, Schmitz W, et al. On the cardiac contractile, biochemical and electrophysiological effects of cantharidin, a phosphatase inhibitor. J pharmacol Exp Ther. 1995;274:530–9. [PubMed] [Google Scholar]
- 25.Enan E, Matsumara F. Specific inhibition of calcineurin by type II synthetic pyrethroid insecticides. Biochem Pharmacol. 1992;43:1777–84. doi: 10.1016/0006-2952(92)90710-z. [DOI] [PubMed] [Google Scholar]
- 26.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA. 1992;89:3686–90. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradshaw JM, Kubota Y, Meyer T, Schulman H. An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc Natl Acad Sci USA. 2003;100:10512–7. doi: 10.1073/pnas.1932759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang T, Johnson EN, Gu Y, Morrisette MR, Sah VP, Gigena MS, et al. The cardiac-specific nuclear δB isoform of Ca2+/Calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–7. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 29.Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/Calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J Neurochem. 1997;68:2119–28. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- 30.Picht E, DeSantiago J, Huke S, Kaetzel MA, Dedman JR, Bers DM. CaMKII inhibition targeted to the sarcoplasmic reticulum inhibits frequency dependent acceleration of relaxation and Ca2+ current facilitation. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2006.09.007. (in revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu A, Hawkins C, Narayanan N. Phosphorylation and activation of the Ca(2+)-pumping ATPase of cardiac sarcoplasmic reticulum by Ca2+/calmodulin-dpendent protein kinase. J Biol Chem. 1993;268:8394–7. [PubMed] [Google Scholar]
- 32.Odermatt A, Kurzydlowski K, MacLennan DH. The Vmax of the Ca2+-ATPase of cardiac sarcoplasmic reticulum (SERCA2a) is not altered by Ca2+/calmodulin dependent phosphorylation of by interaction with phospholamban. J Biol Chem. 1996;271:14206–13. doi: 10.1074/jbc.271.24.14206. [DOI] [PubMed] [Google Scholar]
- 33.Reddy LG, Jones LR, Pace RC, Stokes DL. Purified reconstituted cardiac Ca2+-ATPase is regulated by phospholamban but not by direct phosphorylation with Ca2+/calmodulin dependent protein kinase. J Biol Chem. 1996;271:14964–70. doi: 10.1074/jbc.271.25.14964. [DOI] [PubMed] [Google Scholar]
- 34.Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J Physiol. 1995;487:609–622. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, et al. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:165–168. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 36.Kwiatkowski AP, Shell DJ, King MM. The role of autophosphorylation in activation of the type II Calmodulin-dpendent protein kinase. J Biol Chem. 1987;263:6484–6. [PubMed] [Google Scholar]
- 37.Ishida A, Kitani T, Fujisawa H. Evidence that autophosphorylation at Thr-286/Thr-287 is required for full activation of calmodulin-dependent protein kinase II. Biochim Biophys Acta. 1996;1311:211–7. doi: 10.1016/0167-4889(95)00197-2. [DOI] [PubMed] [Google Scholar]
- 38.Lee JA, Takai A, Allen DG. Okadaic acid, a protein phosphatase inhibitor, increases the calcium transients in isolated ferret ventricular muscle. Exp Physiol. 1991;76:281–4. doi: 10.1113/expphysiol.1991.sp003495. [DOI] [PubMed] [Google Scholar]
- 39.Boknik P, Khorchidi S, Bodor GS, Huke S, Knapp J, Linck B, et al. Role of protein phosphatases in regulation of cardiac inotropy and relaxation. Am J Physiol. 2001;280:H786–94. doi: 10.1152/ajpheart.2001.280.2.H786. [DOI] [PubMed] [Google Scholar]
- 40.duBell WH, Gigena MS, Guatimosim S, Long X, Lederer WJ, Rogers TB. Effects of PP1/PP2A inhibitor calyculin A on the E-C coupling cascade in murine ventricular myocytes. Am J Physiol. 2002;282:H38–48. doi: 10.1152/ajpheart.00536.2001. [DOI] [PubMed] [Google Scholar]
- 41.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soeller C, Cannell MB. Analysing cardiac excitation-contraction coupling with mathematical models of local control. Prob Biophys Mol Biol. 2004;85:141–62. doi: 10.1016/j.pbiomolbio.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J. 2005;89:4096–110. doi: 10.1529/biophysj.105.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacDougall LK, Jones LR, Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem. 1991;196:725–34. doi: 10.1111/j.1432-1033.1991.tb15871.x. [DOI] [PubMed] [Google Scholar]
- 45.Hubbard MJ, Cohen P. On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem Sci. 1993;18:172–7. doi: 10.1016/0968-0004(93)90109-z. [DOI] [PubMed] [Google Scholar]
- 46.Wu Y, Dzhura I, Colbran RJ, Anderson ME. Calmodulin kinase and a calmodulin binding ‘IQ’ domain facilitate L-type Ca2+ current in rabbit ventricular myocytes by a common mechanism. J Physiol. 2001;535:679–87. doi: 10.1111/j.1469-7793.2001.t01-1-00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]