

Abstract
Pentalenolactone (1) is an antibiotic that has been isolated from many species of Streptomyces. The putative dehydrogenase encoded by the ptlF gene (SAV2993) found within the Streptomyces avermitilis pentalenolactone gene cluster was cloned and overexpressed in Escherichia coli. PtlF, which belongs to the short-chain dehydrogenase/oxidoreductase superfamily, was shown to catalyze the oxidation of 1-deoxy-11β-hydroxypentalenic acid (9) to 1-deoxy-11-oxopentalenic acid (10), a new intermediate of the pentalenolactone biosynthetic pathway. The methyl ester of 10 was characterized by NMR, GC-MS and high resolution mass spectrometry. PtlF exhibited a 150-fold preference for β-NAD+ over β-NADP+. PtlF had a pH optimum of 8.0 in the physiological pH range, while a significant activity enhancement was observed from pH 9.0 to 11.3. At pH 8.0, PtlF had a kcat of 0.65±0.03 s−1, with a Km for 9 of 6.5±1.5 μM and Km for NAD+ of 25 ± 3 μM.
Keywords: farnesyl diphosphate, pentalenolactone, Streptomyces, biosynthesis, GC-MS, proton NMR, COSY, dehydrogenase
Pentalenolactone (1) is a sesquiterpenoid antibiotic that has been isolated from over 30 Streptomyces species. The antibiotic, which is active against a variety of microorganisms including Gram-positive and Gram-negative bacteria, fungi and protozoa, irreversibly inactivates the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase [1,2]. Pentalenolactone also inhibits the replication of DNA viruses such as HSV-1 and HSV-2 [3] as well as smooth muscle cell proliferation [4]. Isotopic incorporation experiments using intact cultures of Streptomyces UC5319 have firmly established that pentalenene (3), which is formed by the cyclization of farnesyl diphosphate (2), is the parent hydrocarbon of the pentalenolactone family of metabolites [5]. A variety of oxidized metabolites that are plausible intermediates in the conversion of pentalenene to pentalenolactone have been isolated, including 1-deoxypentalenic acid (4, isolated as the glucuronyl ester) [6], pentalenolactone D (5) [7], pentalenolactone E (6) [8], and pentalenolactone F (7) [7], as well as pentalenic acid, a shunt metabolite of the main pentalenolactone biosynthetic pathway [5]. A plausible biosynthetic pathway has been proposed based on the structures of these known pentalenolactone metabolites (Scheme 1). Until recently, however, there had been no direct experimental evidence for the detailed biosynthetic sequence leading from pentalenene (3) to pentalenolactone (1).
Scheme 1.
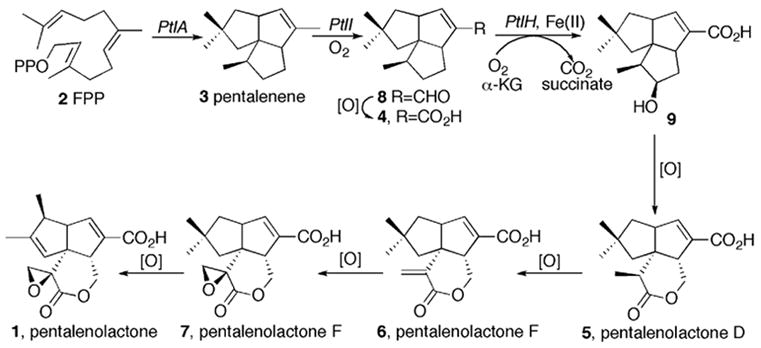
Proposed pentalenolactone biosynthetic pathway.
Streptomyces avermitilis is a Gram-positive soil bacterium responsible for the production of the widely used anthelminthic macrolide avermectins. (S. avermectinius is a junior homotypic synonym of S. avermitilis.) Pentalenolactone F (7) has recently been isolated from S. avermitilis, confirming that the pentalenolactone pathway is functional in this organism [9]. Sequencing of the complete 9.03 Mb genome of S. avermitilis revealed the presence of 30 presumed biosynthetic gene clusters related to secondary metabolism [10], including a 13.4 kb gene cluster containing 13 unidirectionally transcribed open reading frames (ORFs1) corresponding to the apparent operon for pentalenolactone biosynthesis (Figure 1). Deletion of this entire operon from S. avermitilis abolished pentalenolactone formation, while transfer of the intact 13.4 kb cluster to S. lividans 1326 resulted in formation of pentalenic acid, a known pentalenolactone shunt product [9]. Heterologous expression of the ptlA gene (SAV2998) in Escherichia coli and analysis of the resultant protein established that PtlA is a pentalenene synthase that catalyzes the cyclization of farnesyl diphosphate (2) to the sesquiterpene hydrocarbon pentalenene (3) [9], as predicted by the 76% identity of the PtlA gene product to the well characterized pentalenene synthase of Streptomyces UC5319 [10]. The gap1 gene (SAV2990) at the 5′-end of the cluster was shown to encode a pentalenolactone-insensitive glyceraldehyde-3-phosphate dehydrogenase corresponding to the pentalenolactone resistance gene [9]. Seven ORFs correspond to monooxygenases or dioxygenases, including ptlI (SAV2999), a cytochrome p450 that catalyzes the oxidation of pentalenene (3) to 1-deoxypentalenal (8) [11] (and possibly 1-deoxypentalenic acid (4)) and ptlH (SAV2991), a non-heme iron/α-ketoglutarate-dependent hydroxylase, that we have shown catalyzes the oxidative conversion of 1-deoxypentalenic acid (4) to 1-deoxy-11β-hydroxypentalenic acid (9) [12]. Of the remaining ORFs, ptlB corresponds to a typical farnesyl diphosphate synthase, while ptlR is a putative transcriptional regulator and PtlG represents a likely transmembrane efflux protein [10]. Finally, ptlF (SAV2993) appears to encode a typical short-chain dehydrogenase/reductase [10]. As illustrated in Scheme 2, we hypothesized that PtlF would catalyze the nicotinamide-dependent dehydrogenation of 1-deoxy-11β-hydroxypentalenic acid (9) to 1-deoxy-11-oxopentalenic acid (10), which would be oxidized to pentalenolactone D (5) by PtlE (SAV2994), a putative flavin-dependent monooxygenase with similarity to enzymes that catalyze Bayer-Villiger-type oxidations [10].
Figure 1.
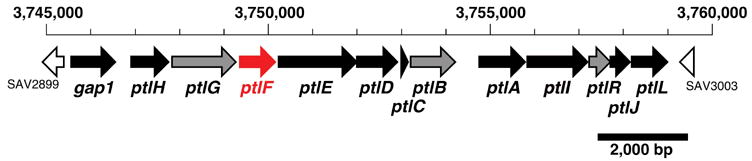
Pentalenolactone biosynthetic gene cluster from Streptomyces avermilitis. See the website of the S. avermitilis Genome Project (http://avermitilis.ls.kitasato-u.ac.jp/) for annotations and detailed sequence alignments.
Scheme 2.
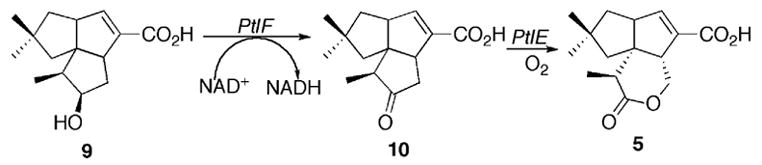
PtlF-catalyzed conversion of 1-deoxy-11β-hydroxypentalenic acid (9) to 1-deoxy-11-oxopentalenic acid (10) and proposed oxidative conversion to pentalenolactone D (5).
PtlF belongs to the short-chain dehydrogenase/reductase (SDR) superfamily [13,14]. Enzymes within the SDR family are known to have low sequence identities (about 15–30%) but to share highly distinct motifs, including the characteristic coenzyme NAD(P)+ binding motif GXXXGXG normally found near the N-terminus and an active site triad of catalytically important Ser, Tyr, Lys residues, of which Tyr is the most conserved residue in the entire family. We now report the molecular cloning, expression and characterization of PtlF, which is shown to catalyze the oxidative conversion of 1-deoxy-11β-hydroxypentalenic acid (9) to 1-deoxy-11-oxopentalenic acid (10).
Materials and Methods
Materials
Streptomyces avermitilis cosmid CL_216_D07 and expression vector pET28e were prepared as described previously [9]. Electrocompetent cells for electroporation were prepared from E. coli BL21(DE3) by standard methods. Restriction enzymes and ligases were purchased from Promega. The DNA purification kit and nickel-nitrilotriacetic acid (Ni-NTA) resin were purchased from Qiagen. PCR primers were ordered from Integrated DNA Technologies (IDT). (±)-1-Deoxypentalenic acid was prepared as previously described [12,15,16]. Reagent grade chemicals were purchased from Sigma-Aldrich.
Methods
Protein concentration was determined by the Bradford assay using bovine serum albumin as standard [17]. DNA sequencing of plasmid constructs was performed by dideoxy sequencing (DNA Sequencing Facility, UC Davis, CA). UV spectra were recorded on an HP 8452A photodiode array spectrophotometer. GC-MS analyses were carried out on an Agilent 6890 Series GC system and a JEOL MS Route JMS 600H mass spectrometer. MALDI-TOF analysis was performed on an Applied Biosystems Voyager DE PRO MALDI-TOF bench top mass spectrometer. NMR experiments were performed on 300 MHz and 400 MHz Bruker high field spectrometers.
PtlF expression vector
The ptlF gene was amplified by PCR using DNA from S. avermitilis cosmid DNA CL_216_D07 as template and the following primers: 5'-CCGCGCGCCATATGCACCTTCAACCGAGTACGG-3' (Forward, Tm 70.1 °C) and 5'-GGCCGGAAGCTTACTAGTCAATTGTCATGGGC-GAGAACTGTTAACGCTCGG-3' (Reverse, Tm 74.3 °C) to introduce NdeI and HindIII restriction sites (underlined) flanking the normal stop and start codons (in bold), respectively. The PCR mixture contained 1 μM of each primer, 2 mM dNTP, 1 μL of DNA template, 5 μL of 10x PCR reaction buffer, 5 U Pfu Ultra DNA polymerase, 4 μL DMSO, and deionized water in a total volume of 50 μL. The first ten cycles employed a denaturing temperature of 95 °C, an annealing temperature of 56 °C and extension temperature of 72 °C, and the next 30 cycles used the same denaturing and extension temperature with an annealing temperature of 66 °C. The amplified PCR product and vector pET28e were each digested with NdeI and HindIII, purified separately with a Qiagen PCR purification kit, and then ligated by DNA T4 ligase at 4 °C overnight. The ligation mixture was used to transform electrocompetent cells of E. coli XL-1 Blue. The sequence of the PCR-derived insert in the resulting plasmid, pET28e-ptlF, was confirmed by sequencing.
Expression and purification of recombinant PtlF
Plasmid pET28e-ptlF was transformed into E. coli BL21(DE3). Expression of PtlF was carried out in LB medium supplemented with kanamycin (50 mg/L). The cells were induced at OD600 = 0.6 with isopropyl-β-D-thiogalactopyranoside (IPTG, 0.4 mM) at 18 °C and harvested after an additional 20 h by centrifugation (6000g, 10 min). The cell pellet was resuspended in 25 mL of lysis buffer (50 mM Tris, 300 mM NaCl, 10% glycerol, 10 mM imidazole, 2.7 mM β−mercaptoethanol, 10 mg/L pepstatin, 10 mg/L PMSF, pH 7.5) and disrupted by French Press (10,000 psi). Cellular debris was removed after centrifugation (20,000g, 50 min) and the supernatant was mixed with 2 mL Ni-NTA resin. The resin slurry was incubated on ice for 1 h before loading into a column. After the flow-through was collected, elution employed a gradient of 10 mM, 20 mM, 30 mM, 40 mM, 60 mM, 100 mM and 200 mM imidazole in buffers containing 50 mM Tris, 300 mM NaCl, 10% glycerol and 2.7 mM β-mercaptoethanol at pH 7.5. The fractions of pure PtlF protein were pooled and concentrated in a Centri-Prep (YM-10) column. Concentrated protein was exchanged into buffer containing 20 mM Tris, 20% glycerol, and 2.7 mM β-mercaptoethanol at pH 7.5, using a PD-10 gel-filtration column. The purified PtlF protein (14 mg from 1 L of LB culture) was flash-frozen in small aliquots in liquid N2 and stored at −80 °C. The homogeneity and molecular weight of purified protein was analyzed by 10% SDS-PAGE and by MALDI-TOF mass spectrometry.
1-Deoxy-11β-hydroxypentalenic acid (9)
1-Deoxy-11β-hydroxypentalenic acid (9) was prepared by hydrolyzing methyl 1-deoxy-11β-hydroxypentalenate (9-Me), which was synthesized enzymatically from (±)-1-deoxypentalenic acid (4) using the non-heme iron dioxygenase PtlH, as previously described [12]. A solution of 9-Me (9 mg, 0.0341 mmol) in 2 mL of 5% aqueous K2CO3 and 4 mL of methanol was heated at reflux for 24 h, and then cooled to room temperature. After the methanol was removed under vacuum, the pH was adjusted to ~ 2 with 10% HCl. The aqueous solution was extracted three times with chloroform. The organic layers were combined, dried over anhydrous MgSO4 and concentrated to yield crystalline1-deoxy-11β-hydroxypentalenic acid (9) (8.4 mg, 0.0336 mmol). 1H NMR (CDCl3, 300.15 MHz): δ 6.78 (t, J = 2.34 Hz, 1H, H-7), 4.08 (m, 1H, H-11), 3.14 (m, 1H, H-5), 2.93 (m, 1H, H-8), 2.04 (m, 1H, H-12), 1.91 (d, J = 13.8 Hz, 1H, H-3, and m, 1H, H-9), 1.70-1.77 (m, 2H, H-12 and H-1), 1.45 (d, J = 13.8 Hz, 1H, H-3), 1.31 (dd, J1 = 12.87 Hz, J2 = 5.34 Hz, 1H, H-1), 1.01 (s, 3H, H-14 or H-15), 1.00 (s, 3H, H-14 or H-15), 0.98 (d, J = 7.68 Hz, 3H, H-10). 13C NMR (CDCl3, 75.48 MHz): δ 169.51 (C-13), 150.16 (C-7), 136.84 (C-6), 75.61 (C-11), 62.52 (C-4), 59.15 (C-8), 55.30 (C-5), 49.85 (C-3), 48.10 (C-9), 45.22 (C-1), 40.87 (C-2), 37.95 (C-12), 29.75 and 29.08 (C-14 and C-15), 10.21 (C-10).
Methyl 1-deoxy-11-oxopentalenate (10-Me)
Methyl 1-deoxy-11β-hydroxypentalenate (9-Me) (2.6 mg, 0.0098 mmol) was dissolved in 0.9 mL of 3:1 CH2Cl2/Et2O. Celite (27 mg) was added and the mixture was stirred in an ice bath, before addition of CrO3 (7.0 mg, 0.07 mmol) [18]. The reaction was complete after 20 min, as monitored by thin-layer-chromatography, after which additional Celite and ether were added to the mixture which was stirred for another 15 min. After filtration, the solvent was removed via rotovap, and the residue was dissolved in CDCl3. Acetic acid (0.5%, v/v) was added to the solution to inhibit the epimerization of the labile methyl group adjacent to the ketone. The 1H and 13 C chemical shift assignments were confirmed by 1H-1H COSY, HMQC, and HMBC spectra. 10-Me: HRMS (EI) 262.1575 (calculated for C16H22O3 262.1569). 1H NMR (CDCl3, 0.5% acetic acid, 300.15MHz): δ 6.79 (t, J = 2.33 Hz, 1H, H-7,), 3.74 (s, 3H, -OCH3), 3.33 (m,1H, H-5), 3.06 (tt, J1 = 2.74 Hz, J2 = 8.46 Hz, 1H, H-8), 2.62 (dd, J1 = 10.55 Hz, J2 =19.39 Hz, 1H, H-12), 2.38 (m, 2H, H-12, H-9), 1.79 (m, 1H, H-1), 1.77 (d, J = 13.8 Hz, 1H, H-3 ), 1.43 (d, J = 13.8 Hz, 1H, H-3), 1.39 (m, 1H, H-1), 1.08 (s, 3H, H-14 or H-15), 1.01 (s, 3H, H-14 or H-15), 1.075 (d, J = 7.13 Hz, 3H, H-10). 13C NMR (CDCl3, 0.5% acetic acid, 75.475 Hz): δ 219.47 (C-11), 165.42 (C-13), 147.66 (C-7), 136.46 (C-6), 61.21 (C-4), 58.08 (C-8), 51.72 (-OCH3), 51.57 (C-5), 51.72 (C-9), 46.62 (C-3), 45.71 (C-1), 41.56 (C-2), 39.51 (C-12), 29.96 (C-14 or C-15), 29.27 (C-14, or C-15), 11.39 (C-10).
PtlF assay
A coupled assay was used to test for the proposed PtlF activity, in which the predicted substrate 1-deoxy-11β-hydroxypentalenic acid (9) was enzymatically generated in situ from (±)-1-deoxypentalenic acid (4). Thus (±)1-Deoxypentalenic acid (4) (0.2 mM) was incubated with the dioxygenase PtlH (1.1 μM) for 50 min in PtlH assay buffer (498 μL, pH 7.5) containing 100 mM Tris, 5 mM α-ketoglutarate, 5 mM L-ascorbate, 2.5 mM FeSO4, 1 mg/mL bovine catalase and 1.2 mM DTT [12]. PtlF and NAD+ were then added to final concentrations of 0.93 μM and 1.6 mM respectively. The increase in NADH concentration was monitored by UV at 340 nm for 10 min before quenching of the reaction with 20 μL of 10% HCl. The reaction mixture was extracted with CH2Cl2 and the organic extract was dried over anhydrous MgSO4, concentrated in a rotovap, and treated with TMS-diazomethane to yield a crude mixture of methyl esters that was analyzed by GC-MS (30 m x 0.25 mm HP5ms capillary column in EI mode, temperature program 60-280 °C). The assay was also repeated using NADP+ as the cofactor.
pH profile of PtlF
The pH optimum for PtlF was determined over a pH range from 5.5 to 11.3 at 0.5 pH intervals using the following buffers: 100 mM sodium acetate, pH 5.5; 100 mM sodium monophosphate, pH 6.0; 100 mM PIPES, pH 6.5 and pH 7.0; 100 mM Tris, pH 7.5, 8.0, 8.5, 9.0; 100 mM sodium bicarbonate, pH 9.5, 10.0, 10.5; 100 mM sodium phosphate, pH 11.3. For each incubation, 485 μL of buffer above was mixed with 5 μL of 200 mM DTT, 5 μL of 100 mM NAD+, and 1 μL of 134 μM PtlF in a 500-μL quartz UV cuvette. Reactions were initiated by addition of 4 μL of 1-deoxy-11β-hydroxypentalenic acid in DMSO to a concentration of 0.24 mM and all the reactions were followed by monitoring the increase of NADH at 340 nm. Initial rates (μM/s) from 10 s to 70 s were calculated with the UV-visible ChemStation software (HP) based on a molar absorption coefficient 6.22 mM−1·cm−1 for NADH. Each reaction was performed in duplicate and the average values of the two measurements were used for the kinetic plot.
Steady-state kinetics
Kinetic assays were performed at 22 °C in 100 mM Tris buffer, pH 8.0, containing 2 mM DTT, 1 mM EDTA, 1 mM β-NAD+, 1% DMSO and 0.268 μM PtlF. The concentration of 1-deoxy-11β-hydroxypentalenic acid (9) was varied from 2.68 to 136 μM. All assays were monitored by UV at 340 nm and carried out in duplicate or triplicate. The initial rates were calculated as described above. The results obtained were the mean of 2 or 3 separate experiments. The kcat and Km for 1-deoxy-11β-hydroxypentalenic acid (9) were determined by fitting the initial rates to the Michaelis-Menten equation using KaleidaGraph software. For determining the Km of NAD+, experiments were performed in a similar manner, but varying NAD+ concentrations from 10 to 600 μM while maintaining the substrate concentration at 26.8 μM.
NADP+ was also evaluated as a PtlF cofactor at a single substrate concentration. To a reaction buffer containing 100 mM Tris (pH 8.0), 0.264 μM PtlF, 1 mM EDTA, 2 mM DTT, and 0.4 mM NADP+, was added 1-deoxy-11β-hydroxypentalenic acid to a concentration of 26.8 μM. The reaction was followed by UV at 340 nm. A parallel procedure was also performed using 0.4 mM NAD+. Each reaction was carried out in duplicate. The initial rates of the reactions were calculated as described above.
Results and Discussion
Expression and purification of recombinant PtlF
Using DNA from S. avermitilis cosmid CL_216_D07 as template [9], the 813-bp ptlF gene (SAV2993) was amplified by PCR while introducing NdeI and HindIII restriction sites at the 5′- and 3′-termini of the ORF, respectively. The amplified DNA was ligated into the expression vector pET28e [9]. The resultant plasmid, pET28e-ptlF, was used to transform the T7 RNA polymerase-based expression host E. coli BL21(DE3). The resulting soluble recombinant protein, carrying an N-terminal His6-TAG, was subjected to Ni2+-affinity purification. The purified PtlF protein was homogeneous by SDS-PAGE (Figure 2). The MALDI-TOF mass spectrum (Figure 3) showed m/z 30183.02 Da, (calculated for PtlF without N-terminal methionine 30190.98 Da). An additional peak at m/z 30364.78 Da [M + 181] could be assigned to the N-terminal gluconyl-modified His6-TAG protein [19]. No species corresponding to protein with bound nicotinamide cofactor was detected in the MALDI-TOF spectrum.
Figure 2.
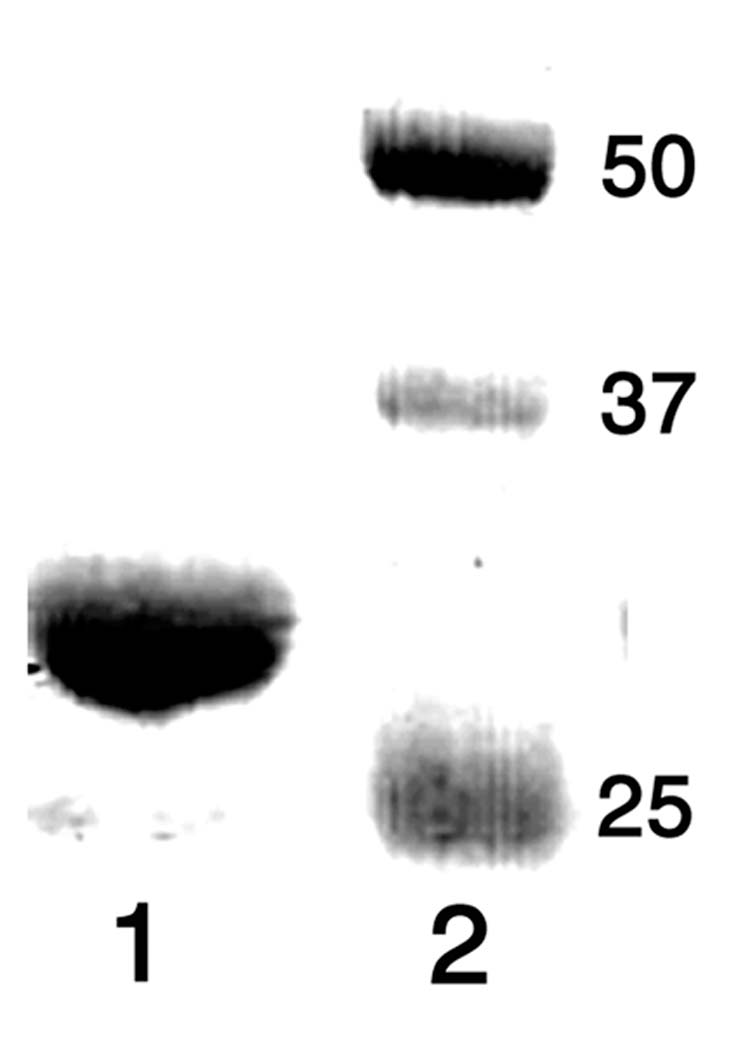
SDS-PAGE analysis of purified PtlF. Lane 1, PtlF; Lane 2, MW markers (kDa).
Figure 3.
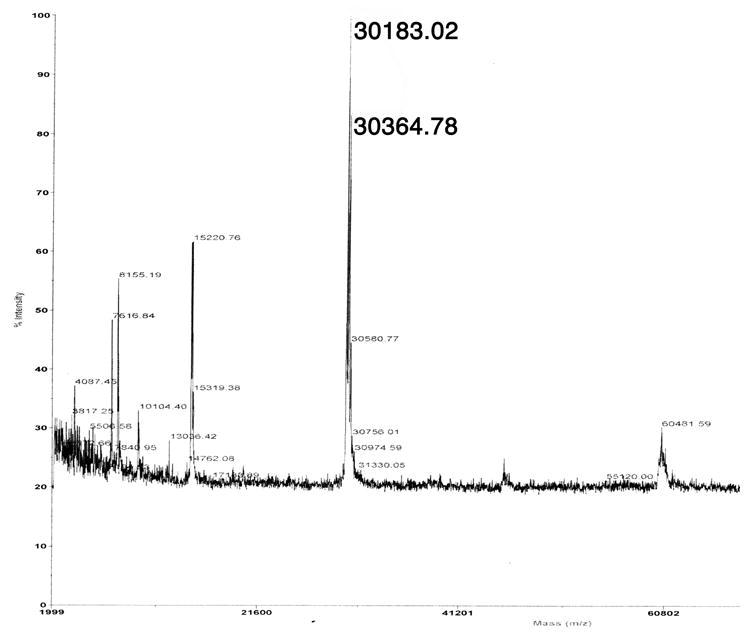
MALDI-TOF mass spectrum of PtlF.
PtlF activity assay
An initial test of the activity of recombinant PtlF was carried out using a coupled assay in which the presumed substrate, 1-deoxy-11β-hydroxypentalenic acid (9), was generated in situ from (±)-1-deoxypentalenic acid (4) using the recombinant dioxygenase PtlH. (±)-1-Deoxypentalenic acid (4) was incubated with purified PtlH for 50 min before the addition of PtlF and NAD+. The absorption at 340 nm increased over time, indicating the generation of NADH. No such increase was observed in control experiments carried out in the absence of PtlF, NAD+, or (±)-1-deoxypentalenic acid (4). These results confirmed that the oxidation product generated by PtlH is the required substrate for PtlF. The crude extract from the reaction mixture was treated with TMS-diazomethane and the resulting methyl esters were analyzed by GC-MS. A new peak at retention time 8.36 min was observed (m/z 262), corresponding to the methyl ester of the expected dehydrogenation product 10-Me, representing a loss of 2 hydrogens from the transiently generated substrate 1-deoxy-11β-hydroxypentalenic acid (m/z 264 for 8-Me) (Figure 4A). The retention time and mass fragmentation pattern of the new peak were identical to those of the authentic 10-Me prepared by oxidation of 9-Me with CrO3, as described below. When NADP+ was used as cofactor in place of NAD+, the same m/z 262 peak for 10-Me (ret. time 8.36 min) was observed, but with much lower intensity (data not shown). Parallel activity assays using synthetic 9 as substrate showed that NAD+ reacted 150 times faster than NADP+ under identical conditions.
Figure 4.
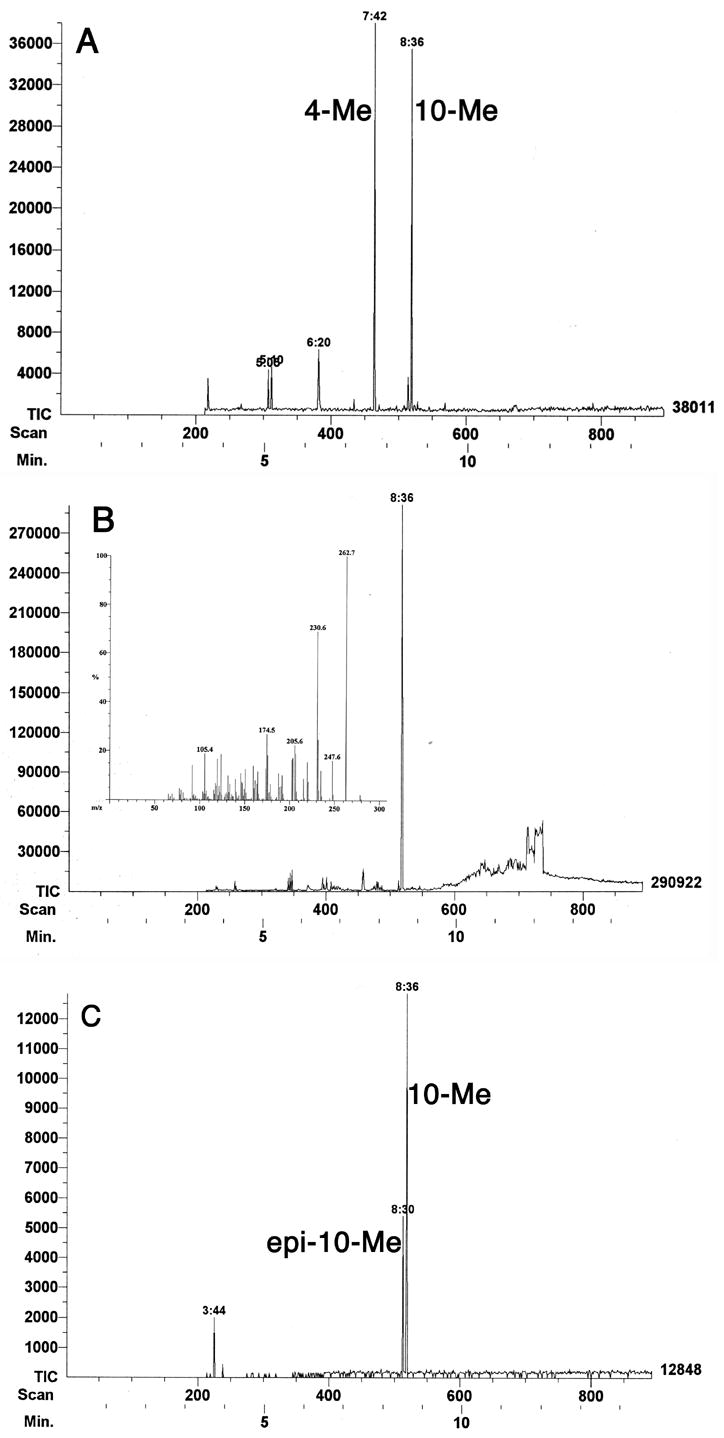
A. GC-MS profile of the methylated organic extract from incubation of 1-deoxypentalenic acid (4) with PtlH and PtlF in the presence of NAD+. B. GC-MS analysis of the CrO3 oxidation of 9-Me to 10-Me. Insert: MS of 10-Me. C. GC-MS after epimerization of 10-Me to epi-10-Me.
Preparation of 1-deoxy-11β-hydroxypentalenic acid (9)
Methyl 1-deoxy-11β-hydroxypentalenate (9-Me) was prepared by incubation of the PtlH and (±)-1-deoxypentalenic acid (4), followed by methylation with TMS-diazomethane and chromatographic purification, as previously described [12]. The requisite synthetic sample of 1-deoxy-11β-hydroxypentalenic acid (9) was obtained by hydrolyzing 9-Me using aqueous methanolic K2CO3 (Scheme 3). The 1H and 13C NMR spectra of the resulting acid 9 were similar to those of the methyl ester 9-Me, with only slight changes, including the absence of the methoxyl singlet at 3.74 ppm in the 1H NMR spectrum and the corresponding signal at 51.8 ppm in the 13C NMR spectrum.
Scheme 3.
Hydrolysis of methyl 1-deoxy-11β-hydroxypentalenate (9-Me).
Methyl 1-deoxy-11-oxopentalenate (10-Me)
Authentic methyl 1-deoxy-11-oxopentalenate (10-Me) was prepared by oxidation of 9-Me with CrO3 (Scheme 4 and Figure 4B). The resulting methylcyclopentanone derivative 10-Me (ret. time 8.36 min) underwent facile epimerization to the epi-10-Me isomer upon exposure to even mild base or common chromatographic media, including silica gel, florisil, and neutral aluminum oxide (Scheme 4), leading to appearance of an additional GC peak (ret. time 8.30 min) with a mass fragmentation pattern essentially identical to that of 10-Me (Figure 4C). Molecular mechanics calculations using MM+ (HyperChem 7.5) indicated that epi-10-Me is 0.25 kCal/mol more stable than 10-Me. This undesired epimerization could be avoided by storing the labile oxoester 10-Me under acidic conditions. An epimerically pure reference sample of 10-Me could be readily prepared, however, by oxidation of synthetic 9-Me using CrO3 in Et2O/CH2Cl2, 12 thereby avoiding contact with base as well as any need for subsequent chromatographic purification. Addition of acetic acid (0.5%, v/v) to the NMR sample prevented the unwanted epimerization. The 1H and 13C chemical shifts of synthetic 10-Me were assigned by a combination of 1D and 2D NMR, including COSY, HMQC and HMBC spectra. In the 1H NMR spectrum of 10-Me, the absence of a multiplet at δ 4.10, corresponding to the H-11 carbinyl proton of 9-Me, confirmed the expected oxidation of the C-11 hydroxyl group. Similarly, the 13C spectrum of 10-Me displayed a new cyclopentanone carbonyl peak at 219.47 ppm, concomitant with the disappearance of the signal at 76.0 ppm for the hydroxyl-bearing C-11 of 9-Me. The 13C signal at 51.72 ppm for C-9 of 10-Me is identical in chemical shift to that of the methoxyl carbon of the methyl ester, as confirmed by 2D NMR. Thus in the HMQC spectrum the 13C signal at 51.72 ppm displays cross peaks with both H-9 (δ 2.38) and -OCH3 (δ 3.74). Similarly, in the HMBC spectrum there is the predicted cross peak between C-9 (51.72 ppm) and H-10 (δ 1.075). Synthetic 10-Me was identical in GC-MS retention time (8.36 min) and MS fragmentation pattern (Figure 4B) with enzymatically-generated 10-Me obtained from the coupled enzymatic incubation of (±)-1-deoxypentalenic acid (4) with PtlH and PtlF.
Scheme 4.
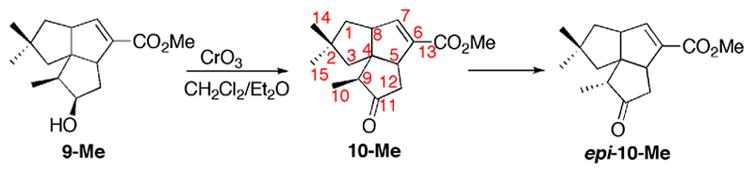
CrO3 oxidation of 9-Me to 1-deoxy-11-oxopentalenic acid (10-Me) and epimerization to epi-10-Me.
pH profile of PtlF
To determine the pH profile for PtlF, incubations were carried out using purified 1-deoxy-11β-hydroxypentalenic acid (9) as substrate and a series of buffers over a pH range of 5.5 to 11.3 at intervals of 0.5 pH units. PtlF showed a local pH optimum of 8.0. Interestingly, the activity also increased markedly beyond pH 9.0 and was >12-fold higher at pH 11.3 than at pH 8.0 (Figure 5). Although unusual, such high pH optima are in fact commonly observed for other enzymes in the SDR superfamily. For example, (−)-isopiperitenol/(−)-carveol dehydrogenase has a pH optimum of 10 [20], 3α-hydroxysteroid dehydrogenase/carbonyl reductase shows a pH optimum of 10.5 [21], and alcohol dehydrogenase from Macaca mulatta has an optimal pH of 11.2 [22]. The unusually high pH optimum is consistent with the proposed catalytic mechanism in which the conserved Tyr residue (pKa 7.2) acts as the general base to extract a proton from hydroxyl group of the substrate, thereby facilitating transfer of the adjacent carbinyl hydride to NAD+ [13].
Figure 5.
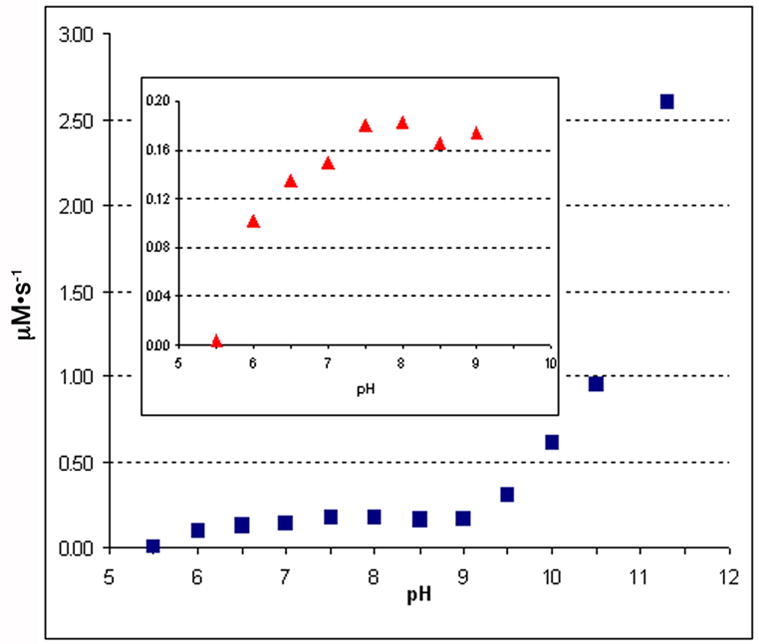
PtlF pH rate profile.
Steady-state kinetics
The steady state kinetic parameters for PtlF were determined at pH 8.0, corresponding to the local pH optimum closest to physiological conditions. The initial velocities measured at varied concentrations of 1-deoxy-11β-hydroxypentalenic acid (9) were fit to the Michaelis-Menten equation, giving a kcat of 0.65 ± 0.03 s−1 and Km of 6.5 ± 1.5 μM. The Km for NAD+ was 25 ± 3 μM. An initial rate of 8.2 ± 3.4 x10−4 μM·s−1 was observed when NADP+ (0.4 mM) was used as the cofactor, which was 150-fold slower compared to that of 1.23 ± 0.05 x 10−1 μM·s−1 observed for NAD+ (0.4 mM) under the same conditions.
Summary and Conclusion
The short-chain dehydrogenase encoded by the ptlF gene (SAV2993) from the S. avermitilis pentalenolactone biosynthetic gene cluster has been cloned, overexpressed, and purified to homogeneity, and its biochemical function has been determined. The experiments described here establish firmly that PtlF catalyzes the NAD+-dependent oxidation of 1-deoxy-11β-hydroxypentalenic acid (9) to 1-deoxy-11-oxopentalenic acid (10), a previously unknown intermediate of pentalenolactone biosynthesis. The dehydrogenase can also utilize NADP+ as a surrogatge cofactor, but shows a 150-fold preference for NAD+. We expect that ketone 10 will be converted to pentalenolactone D (5) by a Bayer-Villiger-like oxidation that is likely to be catalyzed by the presumed monoxygenase PtlE, which has 49% identity and 62% similarity over 591 aa to the cyclodecanone-lauryl lactone monooxygenase of Rhodococcus ruber (CddA gene product, Genbank AY052630.1). Further characterization of the remaining ORFs of the S. avermitilis pentalenolactone biosynthetic gene cluster is in progress.
Acknowledgments
We thank Dr. Tun-Li Shen for assistance with GC-MS analyses. This research was supported by a grant from the U. S. National Institutes of Health GM30301 to D.E.C., by Grant of the 21st Century COE Program, Ministry of Education, Culture, Sports, Science and Technology, Japan to H.I and S.O, and by Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science No. 17510168 to H.I.
Abbreviations used
- IPTG
isopropyl-β-D-thiogalactopyranoside
- LB
Luria-Bertrani
- OD
optical density
- ORF
open reading frame
- PMSF
phenylmethylsulfonyl fluoride
- SDR
short-chain dehydrogenase/reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hartmann S, Neeff J, Heer UM, Mecke D. FEBS Lett. 1978;93:339–342. doi: 10.1016/0014-5793(78)81135-1. [DOI] [PubMed] [Google Scholar]
- 2.Cane DE, Sohng JK. Arch Biochem Biophys. 1989;270:50–61. doi: 10.1016/0003-9861(89)90006-4. [DOI] [PubMed] [Google Scholar]
- 3.Nakagawa A, Tomoda H, Hao MV, Okano K, Iwai Y, Omura S. J Antibiot. 1985;38:1114–1115. doi: 10.7164/antibiotics.38.1114. [DOI] [PubMed] [Google Scholar]
- 4.Ikeda M, Fukuda A, Takagi M, Morita M, Shimada Y. Eur J Pharmacol. 2001;411:45–53. doi: 10.1016/s0014-2999(00)00894-3. [DOI] [PubMed] [Google Scholar]
- 5.Cane DE, Oliver JS, Harrison PH, Abell C, Hubbard BR, Kane CT, Lattman R. J Am Chem Soc. 1990;112:4513–4524. [Google Scholar]
- 6.Takahashi S, Takeuchi M, Arai M, Seto H, Otake N. J Antibiot. 1983;36:226–228. doi: 10.7164/antibiotics.36.226. [DOI] [PubMed] [Google Scholar]
- 7.Cane DE, Sohng JK, Williard PG. J Org Chem. 1992;57:844–852. [Google Scholar]
- 8.Cane DE, Rossi T. Tetrahedron Lett. 1979:2973–2974. [Google Scholar]
- 9.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. Biochemistry. 2006;45:6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M. Proc Natl Acad Sci USA. 2001;98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quaderer R, Omura S, Ikeda H, Cane DE. J Am Chem Soc. 2006;128:13036–13037. doi: 10.1021/ja0639214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.You Z, Omura S, Ikeda H, Cane DE. J Am Chem Soc. 2006;128:6566–6567. doi: 10.1021/ja061469i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jörnvall H, Persson B, Krook M, Atrian S, Gonzàlez-Duarte R, Jeffery J, Ghosh D. Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 14.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jörnvall H. Chem-Biol Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 15.Ohfune Y, Shirahama H, Matsumoto T. Tetrahedron Lett. 1976;17:2869–2872. [Google Scholar]
- 16.Ohtsuka T, Shirahama H, Matsumoto T. Tetrahedron Lett. 1983;24:3851–3854. [Google Scholar]
- 17.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 18.Flatt SJ, Fleet GWJ, Taylor BJ. Synthesis. 1979:815–817. [Google Scholar]
- 19.Yan Z, Galdwell GW, McDonell PA. Biochem Biophys Res Com. 1999;262:793–800. doi: 10.1006/bbrc.1999.1304. [DOI] [PubMed] [Google Scholar]
- 20.Ringer KL, Davis EM, Croteau R. Plant Physiol. 2005;137:863–875. doi: 10.1104/pp.104.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang CC, Chang YH, Hsu CN, Hsu HH, Li CW, Pon HI. J Biol Chem. 2005;280:3522–3528. doi: 10.1074/jbc.M411751200. [DOI] [PubMed] [Google Scholar]
- 22.Dafeldecker WP, Meadow PE, Pares X, Vallee BL. Biochemistry. 1981;20:6729–6734. doi: 10.1021/bi00526a031. [DOI] [PubMed] [Google Scholar]