

Abstract
Regulated proteolysis of the polyprotein precursor of West Nile virus (WNV) by the essential NS2B-NS3(pro)tease, a promising drug target for WNV inhibitors, is required for the propagation of infectious virions. Structural and drug design studies, however, require pilot-scale quantities of a pure and catalytically active WNV protease that is resistant to self-proteolysis. Autolytic cleavage at the NS2B-NS3 boundary leads to individual, non-covalently associated, NS2B and NS3 domains, together with residual amounts of the intact NS2B-NS3, in the NS2B-NS3pro samples. We modified the cleavage site sequence of the NS2B-NS3 junction region and then developed expression and purification procedures to prepare a covalently linked, single-chain, NS2B-NS3pro K48A mutant construct. This construct exhibits high stability and functional activity and is thus well suited for the follow-up purification and structural and drug design studies.
Introduction
West Nile virus (WNV), a member of the Flaviviridae family and an emerging pathogen in the US, is transmitted to animals, including humans, by mosquito bites [1, 2]. There are no specific countermeasures against WNV infection in humans. The WNV capsid encloses a single-stranded RNA encoding a 3,400 amino acid residue polypeptide precursor. The precursor is comprised of three structural proteins [C, capsid, M, membrane, and E, envelope] and seven non-structural (NS) proteins arranged in the order C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5 [3, 4]. Proteases from the host (furin and secretase) and from the virus [NS3 serine proteinase (NS3pro)] are required to process the polyprotein precursor into individual functional proteins [5, 6]. NS3pro is responsible for the cleavage of the capsid protein C and at the NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/NS4B and NS4B/NS5 boundaries [7–14].
The full-length NS3 peptide sequence is a multifunctional protein. The N-terminal 184 amino acid residue fragment represents the serine proteinase NS3pro with a conventional His-Asp-Ser catalytic triad. The C-terminal portion of the NS3 protein encodes an RNA helicase. During virus propagation, protease and helicase normally work together in a coordinated fashion [15]. As is the case with a number of flaviviruses, the NS2B protein that is located in the polypeptide precursor upstream of the NS3pro domain functions as a cofactor and promotes the folding and the functional activity of the NS3pro [16–18]. The individual NS3pro domain, lacking the NS2B part, is catalytically inert. The cofactor activity of the 35–48-residue central portion is approximately equivalent to that of the entire NS2B sequence [19, 20]. Inactivating mutations of the NS3pro cleavage sites in the polyprotein precursor abolish viral infectivity [3]. These data suggest that the two-component NS2B-NS3pro is a promising drug target for WNV inhibitors. The sequence of WNV NS2B-NS3pro is 56% identical to that of Dengue virus (DV), thus suggesting a high degree of structural similarity that exists between these two proteinases. A crystal structure of the individual DV NS3pro complexed with mung-bean Bowman-Birk type trypsin inhibitor has been available for several years [21, 22]. This structure, however, represents a virtually inactive protease. When our work was in progress, the high resolution structures of the two-component WNV and DV NS2B-NS3pro in complex with the substrate-based inhibitor benzoyl-norleucine (P4)-lysine (P3)-arginine (P2)-arginine (P1)-aldehyde (Bz-Nle-Lys-Arg-Arg-H) became available [17].
Structural and drug design studies require the availability of substantial amounts of recombinant NS2B-NS3pro with high proteolytic activity and resistance to self-proteolysis. Normally, because of self-proteolysis of the NS2B-NS3 junction region, individual, non-covalently associated NS2B and NS3 domains, together with residual amounts of intact NS2B-NS3, are present in the NS2B-NS3pro samples [18]. Their presence complicates the isolation and analysis of NS2B-NS3pro. To overcome this difficulty, we modified the cleavage site sequence of the NS2B-NS3 junction region in order to obtain covalently linked, single-chain NS2B-NS3pro that exhibits high stability and functional activity, well suited for follow-up purification and structural and drug design studies.
Materials and Methods
Reagents
Reagents were purchased from Sigma-Aldrich (Milwaukee, WI) unless indicated otherwise. Pyroglutamic acid-RTKR-7-amino-4-methylcoumarin (Pyr-RTKR-AMC) was purchased from American Peptide (Sunnyvale, CA).
Enzyme cloning, expression and purification
Cloning of the DNA sequence encoding the wild-type two-component NS2B-NS3pro from WNV was described earlier [18]. The 48 amino acid residue central portion of NS2B (residues 1393–1440 of the WNV polyprotein precursor) and the NS3 (residues 1476–1687 of the WNV polyprotein precursor) sequences were linked by a flexible GGGGSGGGG linker. The WNV autolytic site-deficient NS2B-NS3pro(K48A) construct was prepared with 5’-CCAGGAGCACCTTGGGCGGGCGGGGGAGGT-3’ and 5’-ACCTCCCCCGCCCGCCCAAGGTGCTCCTGG-3’ forward and reverse primers, respectively (mutant nucleotides are underlined) using a QuickChange mutagenesis kit (Stratagene, San Diego, CA). After confirming their authenticity by sequencing, the constructs were re-cloned into the pET101 expression vector. Competent E.coli BL21 (DE3) Codon Plus cells (Stratagene, San Diego, CA) were transformed with the recombinant pET101 vectors. Transformed cells were grown at 30°C in a Luria–Bertani broth containing ampicillin (0.1 mg/ml). Cultures were induced with 0.6 mM isopropyl β-D-thiogalactoside for 16 h at 18°C. E.coli cells (6 g/L of E.coli culture) were then collected by centrifugation (5,000xg; 15 min), resuspended in 20 ml PBS containing 1 M NaCl, 1 mM phenylmethylsulfonyl fluoride and lyzozyme (5 mg/ml), and disrupted by sonication (30 sec pulse, 30 sec interval; 8 pulses) on ice. The debris were then removed by centrifugation at 20,000xg, 30 min.
The NS2B-NS3pro WT and K48A constructs, C-terminally tagged with a 6xHis tag, were each purified from the soluble fraction of E.coli cell extract using affinity chromatography on a 1.6x10 cm Co2+-chelating Sepharose Fast Flow column (Amersham, Piscataway, NJ) equilibrated with PBS - 1 M NaCl - 1 mM phenylmethylsulfonyl fluoride. NS2B-NS3pro was eluted with 100 ml of 10–500 mM gradient of imidazole in PBS - 1 M NaCl - 1 mM phenylmethylsulfonyl fluoride buffer (elution rate 0.5 ml/min). The fractions (1.5 ml each; 10 fractions total) containing NS2B-NS3pro were collected and diluted 10-fold with PBS and the protease samples were then re-chromatographed under the same conditions on a Co2+-chelating Sepharose Fast Flow column. The presence of NS2B-NS3pro in the fractions was confirmed by 4–20% gradient gel electrophoresis and also by the activity assay. In the activity assays 1 μl fraction aliquots and a Pyr-RTKR-AMC substrate were used. The NS2B-NS3pro K48A protein fractions were concentrated with a 5 kDa-cutoff concentrator (Millipore, Billerica, MA) and dialyzed against 10 mM Tris-HCl, pH 8.0, containing 0.005% Brij35. Typical yields of the purified NS2B-NS3pro K48A were 10 mg/L of E. coli culture.
Protease assays with fluorogenic peptides
The assay for NS3pro cleavage activity was performed in 10 mM Tris-HCl buffer, pH 8.0, containing 20% glycerol and 0.005% Brij 35. The Pyr-RTKR-AMC substrate and enzyme concentrations, unless indicated otherwise, were 24 μM and 10 nM, respectively. The total assay volume was 0.1 ml. Initial reaction velocities were monitored continuously at λex 360 nm and λem 460 nm on a Spectramax Gemini EM fluorescence spectrophotometer (Molecular Devices, Sunnyvale, CA). All assays were performed in triplicate in a 96 well plate. The values of Km and kcat were derived from a double reciprocal plot of 1/V0 vs. 1/[S], using the Lineweaver-Burk transformation: 1/V0 = Km/Vmax x 1/[S] + 1/Vmax, where V0 is the initial velocity of the substrate hydrolysis and [S] is the substrate concentration, Vmax is the maximum rate of hydrolysis, and Km is the Michaelis-Menten constant. The concentration of the catalytically active proteinase was measured using a fluorescence assay by titration against a standard aprotinin solution of a known concentration.
Circular dichroism spectroscopy of the NS2B-NS3pro constructs
CD spectra (190–250 nm) of the NS2B-NS3pro and NS2B-NS3pro K48A purified samples (both at 1.4 mg/ml in 15 mM potassium phosphate buffer, pH 7.8 ) were collected in a 1 mm pathlength quartz cuvette at ambient temperature using a 62ADC spectrometer (AVIV Instruments, Lakewood, NJ). Prior to the CD spectra tests, the NS2B-NS3pro sample was incubated for 16 h at 24°C to complete its autolytic conversion into the individual NS3pro enzyme and the NS2B domain. The resulting samples were re-checked by SDS gel-electrophoresis to confirm that autolytic conversion had been completed. The results are expressed as the [Θ]MRW (Mean Residue Weight) ellipticity.
Size-exclusion chromatography of the NS2B-NS3pro constructs
Size-exclusion chromatography of the NS2B-NS3pro and NS2B-NS3pro K48A purified samples was performed on a Superdex 200HR 10/30 pre-packed column (Amersham) on an FPLC System. The column was equilibrated with 50 mM Tris-HCl buffer, pH 7.5, containing 2 mM DTT and 0.2 M NaCl at a constant flow rate of 0.5 ml/min.
N-terminal sequencing
To determine the N-terminal sequence of the NS3pro domain and, consequently, the autolytic cleavage site, following SDS gel-electrophoresis the purified NS2B-NS3pro sample (4 μg) was transferred onto an Immobilon membrane support. The 30 kDa NS3pro band was subjected to N-terminal sequencing at ProSeq (Boxford, MA).
Results and Discussion
Previous studies showed that the presence of the 48 residue central NS2B domain linked to the N-terminus of NS3 significantly enhanced the accumulation of soluble recombinant NS3pro in E.coli [18, 20, 23]. We used a similar approach to express the WNV NS2B-NS3pro. The 48 residue central portion of the NS2B sequence was linked with the NS3pro domain via a GGGGSGGGG linker. To facilitate its isolation, the NS2B-NS3pro construct was C-terminally tagged with a (His)x6 tag and then expressed in E.coli. After induction with isopropyl-β-D-thiogalactoside, large amounts of NS2B-NS3pro were produced as a soluble protein. The soluble fraction was loaded onto a Co2+-agarose affinity column. The NS2B-NS3pro protein was then eluted with a 10–500 mM gradient of imidazole concentrations (Fig. 1). The pooled, partially purified, NS2B-NS3pro fractions were then re-chromatographed on a Co2+-agarose column to acquire the homogenous NS2-NS3pro samples. During purification, we observed an incomplete autolytic conversion of the 36 kDa NS2B-NS3pro construct into the individual 30 kDa NS3pro and the NS2B sequence (5–6 kDa).
Fig. 1.
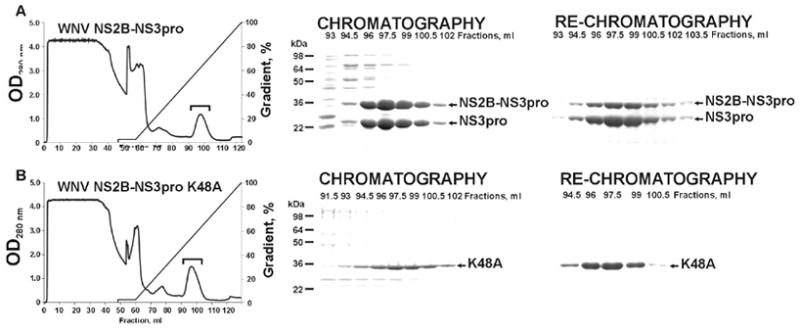
Purification of recombinant, C-terminally tagged, NS2B-NS3pro and NS2B-NS3 K48A constructs on a Co2+-chelating Sepharose Fast Flow column (A and B, respectively). The left panels show the profile of elution of the constructs. To remove impurities, the protease was eluted with 100 ml of 10–500 mM gradient of imidazole (100 ml) at a 0.5 ml/min flow rate. Fractions containing the protease are indicated by a bracket. The middle and the right panels show the gel electrophoresis analysis of the fractions eluted from the column during chromatography and re-chromatography of the protease samples, respectively.
To complete the autolytic conversion, the purified samples (0.02–0.2 mg/ml) were incubated overnight at pH 8 and 24°C yielding the individual NS3pro domain and the NS2B (the latter is not visible in Fig. 2 because of its low molecular mass). The resulting NS3 samples retained the C-terminal His tag since the NS3 moiety was readily recognized by a His-tag antibody. By using N-terminal microsequencing, we determined that in the course of autolysis the NS3pro activity cleaved the linker sequence and that the N-terminal sequence of the individual NS3pro was GGGSGG suggesting cleavage at the K48G↓GGGSGGGG site (the linker sequence is underlined). Consistent with our data, Erbel et al. [17] also determined that the WNV NS2B-NS3pro construct undergoes self-cleavage during expression and purification at the K48G↓G site of the glycine linker. We believe that the cleavage at this unconventional K48G↓G site occurs because of disordered loop structure of the linker region. The structure of the WNV constructs and the relative positions of the mutations are shown in Fig. 2. Inhibitors of the NS3pro activity, including aprotinin (Ki = 25 nM), nona-D-Arg-amide (Ki = 6 nM) and decanoyl-Arg-Val-Lys-Arg-chloromethylketone (Ki = 400 nM) completely inhibited autolysis of the NS2B-NS3pro construct (data not shown). Size-exclusion chromatography confirmed that the NS2B sequence remained non-covalently bound with the NS3pro domain following autocatalytic cleavage of the NS2B-NS3pro construct (Fig. 3).
Fig. 2.
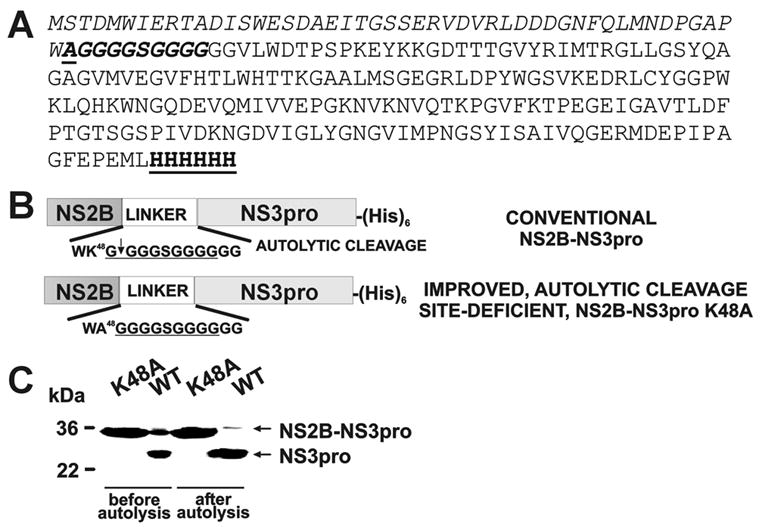
NS2B-NS3pro and NS2B-NS3pro K48A constructs. (A) The peptide sequence of the WNV NS2B-NS3pro K48A construct. The N-terminal NS2B sequence is italisized. The linker sequence with the K48A mutation (underlined) is italisized and bolded. The NS3pro is shown in a regular font. The C-terminal Hisx6 tag is bolded and underlined. (B) The 48 amino acid residue-long, central portion of NS2B (NS2B) was linked with the NS3pro sequence via a GGGGSGGGG linker (in the figure, the linker sequence is underlined). The constructs were C-terminally tagged with a (His)x6 tag. After the isolation, the NS2B-NS3pro construct autolytically cleaved the G49↓G50 bond and generated the individual NS3pro domain commencing at Gly50. To inactivate the autolytic cleavage site Ala was substituted for Lys48 in the C-terminal region of the NS2B moiety. Because of the furin-like cleavage specificity of the WNV NS2B-NS3pro, the presence of either Lys or Arg at the P2 position is essential for substrate cleavage by the protease. (C) Autolytic conversion of the NS2B-NS3pro construct. The purified NS2B-NS3pro (WT; wild type) and NS2B-NS3pro K48A (K48A) samples were each incubated overnight (0.1 mg/ml; pH 8, 24°C) to generate the NS3pro domain. The digest samples were analyzed by SDS gel electrophoresis followed by Coomassie staining of the gel. In contrast to WT, K48A is resistant to self-proteolysis.
Fig. 3.
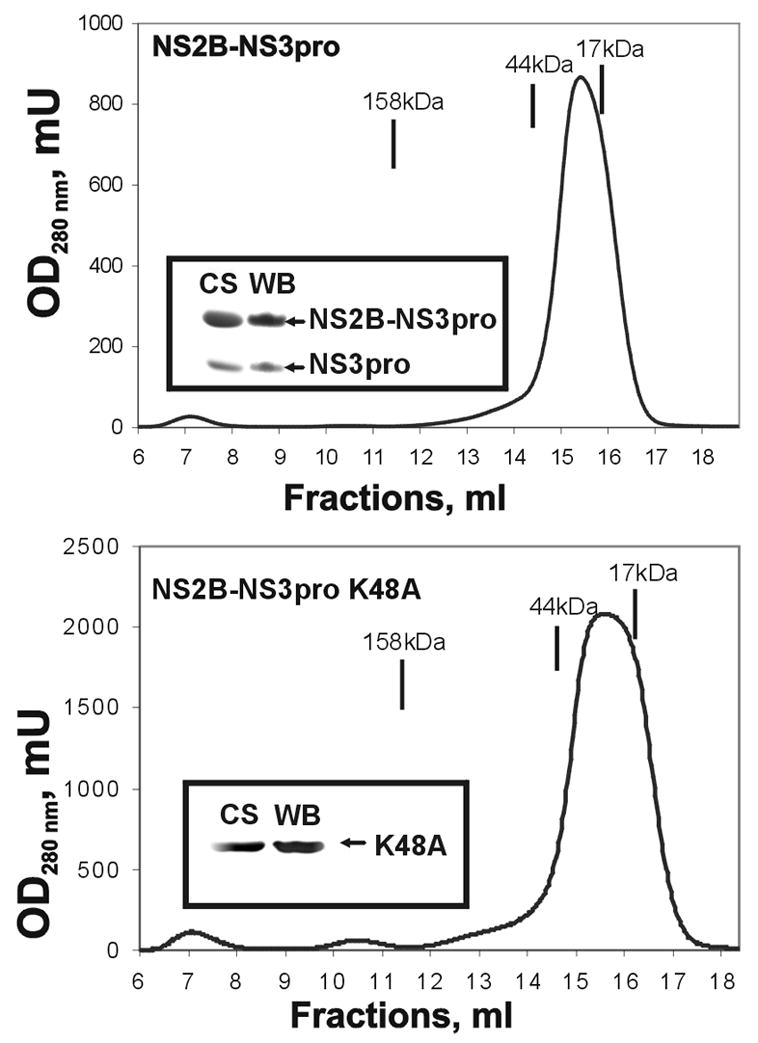
Size-exclusion chromatography of NS2B-NS3pro and NS2B-NS3pro K48A. The protease samples were chromatographed on a Superdex 200HR 10/30 column at a 0.5 ml/min flow rate. The insets show the results of Coomassie staining following SDS gel-electrophoresis of the peak fractions and Western blotting using an antibody to a Hisx6 tag (CS and WB, respectively). The elution positions of the molecular mass markers are shown by the lines above the elution profile.
Based on these sequence data, we constructed the autolysis-resistant K48A mutant. Metal-chelating chromatography followed by re-chromatography of the pooled fractions was sufficient for isolation of the homogenous NS2B-NS3pro K48A construct with a high (45%) yield (Fig. 1 and 2; Table 1). The data from gel-electrophoresis and size-exclusion chromatography confirmed that the K48A mutant represented a single-chain protein and that it was fully resistant to autolysis at the linker region (Fig. 2 and 3). We conclude that the K48A mutation of the C-terminal amino acid residue of the NS2B sequence inactivated the autolytic cleavage site in the NS2B-NS3pro construct. The K48A mutation, however, did not have any effect on the catalytic activity of the NS2B-NS3pro construct, and the K48A construct was highly efficient in cleaving Pyr-RTKR-AMC (Table 2). Circular dichroism studies also confirmed structural integrity of the wild-type and the K48A mutant constructs (Fig. 4).
Table 1.
Purification of recombinant WNV NS2B-NS3pro and NS2B-NS3pro K48A from E. coli cell extract1
| Total protein, mg | Total activity, U2 | Specific activity, U/mg | Purification, -fold | Yield, % | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | K48A | WT | K48A | WT | K48A | WT | K48A | WT | K48A | |
| Crude extract | 248 | 234 | 26.2 | 25,7 | 0.106 | 0.11 | 1 | 1 | 100 | 100 |
| Chromatography | 47.4 | 43.7 | 18.1 | 18.9 | 0.38 | 0.43 | 3.6 | 3.9 | 69.1 | 73.5 |
| Re-chromatography | 11.2 | 13.1 | 10,7 | 12 | 0.9 | 0.9 | 8.5 | 8.2 | 40.8 | 46.7 |
| 5 kDa-cut off concentration | 10.5 | 12 | 10.2 | 11.5 | 1 | 1 | 9.4 | 9.1 | 38.9 | 44.7 |
E.coli cells (5 g wet weight) were used for purification. NS2B-NS3pro, WT; NS2B-NS3pro K48A, K48A.
One unit (U) of enzymatic activity corresponds to the liberation of 1 μmol of 7-amino-4-methylcoumarin (AMC) per min/mg from the Pyr-RTKR-AMC substrate.
Table 2.
NS2B-NS3pro and NS2B-NS3pro K48A cleave the Pyr-RTKR-AMC substrate
| WNV protease | Km (μM) | kcat (s−1) | Kcat/Km (M−1·s−1) |
|---|---|---|---|
| NS2B-NS3pro | 71±15 | 6.25±0.35 | 88000±12,000 |
| NS2B-NS3pro K48A | 58.8±10 | 5.25±0.25 | 89300±11,000 |
Fig. 4.
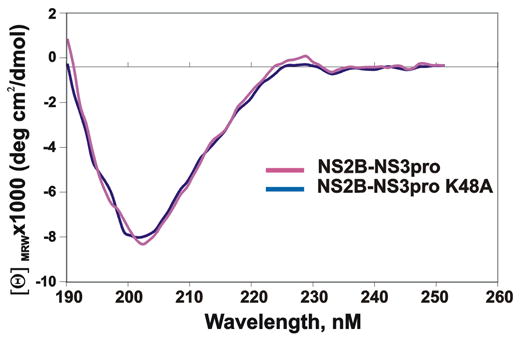
CD spectra of the NS2B-NS3pro and NS2B-NS3pro K48A purified samples. Before the tests, the NS2B-NS3pro sample was incubated for 16 h at 24°C to complete its autolytic conversion into the non-covalently associated NS3pro and the NS2B domains. The similarity of the CD spectra suggests that the structure of the non-covalently associated NS2B-NS3pro enzyme is similar to that of the single chain NS2B-NS3pro K48A with the inactivated autolytic cleavage site.
Overall, we conclude that the NS2B-NS3pro K48A mutant, which exhibits high stability and functional activity and which can be purified in pilot-scale quantities from E.coli cells as a soluble recombinant protein, is well suited for follow-up structural and drug design studies. Consistent with this suggestion, we have already accomplished the crystallization and X-ray structural analysis of the NS2B-NS3pro K48A construct with an 1.8Ǻ resolution, while our previous attempts with the wild-type NS2B-NS3pro were unsuccessful. Because the structural analysis was performed using the K48A construct complexed with aprotinin rather than with the substrate-based inhibitor, a highly valuable information about the P1’–P4’ subsites of NS2B-NS3pro has been acquired in the course of our work (A. Aleshin, S. Shiryaev, A. Strongin, R. Liddington, Structural evidence for multiple levels of regulation of flaviviral protease activity, submitted for publication).
The structural organization of the two-component NS2B-NS3pro is similar in many flavivirus types, including WNV, Dengue types 1–4, Yellow fever, Japanese and Murray valley encephalitis and Kunjin viruses. Accordingly, we predict that similar modifications of the NS2B-NS3pro boundary (which may involve residues distinct from K48 because this lysine residue is conserved only in WNV and Japanese encephalitis viruses) will be sufficient to inactivate the autolytic cleavage site, facilitate isolation and improve the stability and presentation of many flaviviral NS2B-NS3 constructs. These mutant, autolysis-resistant flaviviral protease constructs can then be efficiently used in drug design and structural studies.
Acknowledgments
This work was supported by NIH Grants AI056385, AI61139 and U54 RR020843 to AYS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hayes CG. West Nile virus: Uganda, 1937, to New York City, 1999. Ann NY Acad Sci. 2001;951:25–37. doi: 10.1111/j.1749-6632.2001.tb02682.x. [DOI] [PubMed] [Google Scholar]
- 2.van der Meulen KM, Pensaert MB, Nauwynck HJ. West Nile virus in the vertebrate world. Arch Virol. 2005;150:637–657. doi: 10.1007/s00705-004-0463-z. [DOI] [PubMed] [Google Scholar]
- 3.Beasley DW. Recent advances in the molecular biology of west nile virus. Curr Mol Med. 2005;5:835–850. doi: 10.2174/156652405774962272. [DOI] [PubMed] [Google Scholar]
- 4.Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 5.Samuel CE. Host genetic variability and West Nile virus susceptibility. Proc Natl Acad Sci USA. 2002;99:11555–11557. doi: 10.1073/pnas.202448899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahour A, Falgout B, Lai CJ. Cleavage of the dengue virus polyprotein at the NS3/NS4A and NS4B/NS5 junctions is mediated by viral protease NS2B-NS3, whereas NS4A/NS4B may be processed by a cellular protease. J Virol. 1992;66:1535–1542. doi: 10.1128/jvi.66.3.1535-1542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambers TJ, Nestorowicz A, Amberg SM, Rice CM. Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J Virol. 1993;67:6797–6807. doi: 10.1128/jvi.67.11.6797-6807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elshuber S, Allison SL, Heinz FX, Mandl CW. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J Gen Virol. 2003;84:183–191. doi: 10.1099/vir.0.18723-0. [DOI] [PubMed] [Google Scholar]
- 10.Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jan LR, Yang CS, Trent DW, Falgout B, Lai CJ. Processing of Japanese encephalitis virus non-structural proteins: NS2B-NS3 complex and heterologous proteases. J Gen Virol. 1995;76(Pt 3):573–580. doi: 10.1099/0022-1317-76-3-573. [DOI] [PubMed] [Google Scholar]
- 12.Preugschat F, Yao CW, Strauss JH. In vitro processing of dengue virus type 2 nonstructural proteins NS2A, NS2B, and NS3. J Virol. 1990;64:4364–4374. doi: 10.1128/jvi.64.9.4364-4374.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stocks CE, Lobigs M. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J Virol. 1998;72:2141–2149. doi: 10.1128/jvi.72.3.2141-2149.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamshchikov VF, Trent DW, Compans RW. Upregulation of signalase processing and induction of prM-E secretion by the flavivirus NS2B-NS3 protease: roles of protease components. J Virol. 1997;71:4364–4371. doi: 10.1128/jvi.71.6.4364-4371.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tackett AJ, Chen Y, Cameron CE, Raney KD. Multiple full-length NS3 molecules are required for optimal unwinding of oligonucleotide DNA in vitro. J Biol Chem. 2005;280:10797–10806. doi: 10.1074/jbc.M407971200. [DOI] [PubMed] [Google Scholar]
- 16.Droll DA, Krishna Murthy HM, Chambers TJ. Yellow fever virus NS2B-NS3 protease: charged-to-alanine mutagenesis and deletion analysis define regions important for protease complex formation and function. Virology. 2000;275:335–347. doi: 10.1006/viro.2000.0488. [DOI] [PubMed] [Google Scholar]
- 17.Erbel P, Schiering N, D’Arcy A, Renatus M, Kroemer M, Lim SP, Yin Z, Keller TH, Vasudevan SG, Hommel U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol. 2006;13:372–373. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- 18.Shiryaev SA, Ratnikov BI, Chekanov AV, Sikora S, Rozanov DV, Godzik A, Wang J, Smith JW, Huang Z, Lindberg I, Samuel MA, Diamond MS, Strongin AY. Cleavage targets and the D-arginine-based inhibitors of the West Nile virus NS3 processing proteinase. Biochem J. 2006;393:503–511. doi: 10.1042/BJ20051374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- 20.Nall TA, Chappell KJ, Stoermer MJ, Fang NX, Tyndall JD, Young PR, Fairlie DP. Enzymatic characterization and homology model of a catalytically active recombinant West Nile virus NS3 protease. J Biol Chem. 2004;279:48535–48542. doi: 10.1074/jbc.M406810200. [DOI] [PubMed] [Google Scholar]
- 21.Murthy HM, Clum S, Padmanabhan R. Dengue virus NS3 serine protease. Crystal structure and insights into interaction of the active site with substrates by molecular modeling and structural analysis of mutational effects. J Biol Chem. 1999;274:5573–5580. doi: 10.1074/jbc.274.9.5573. [DOI] [PubMed] [Google Scholar]
- 22.Murthy HM, Judge K, DeLucas L, Padmanabhan R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: implications for flaviviral polyprotein processing and drug design. J Mol Biol. 2000;301:759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- 23.Chappell KJ, Nall TA, Stoermer MJ, Fang NX, Tyndall JD, Fairlie DP, Young PR. Site-directed mutagenesis and kinetic studies of the West Nile Virus NS3 protease identify key enzyme-substrate interactions. J Biol Chem. 2005;280:2896–2903. doi: 10.1074/jbc.M409931200. [DOI] [PubMed] [Google Scholar]