

Abstract
The number of species of Bacteria and Archaea (ca 5000) is surprisingly small considering their early evolution, genetic diversity and residence in all ecosystems. The bacterial species definition accounts in part for the small number of named species. The primary procedures required to identify new species of Bacteria and Archaea are DNA–DNA hybridization and phenotypic characterization. Recently, 16S rRNA gene sequencing and phylogenetic analysis have been applied to bacterial taxonomy. Although 16S phylogeny is arguably excellent for classification of Bacteria and Archaea from the Domain level down to the family or genus, it lacks resolution below that level. Newer approaches, including multilocus sequence analysis, and genome sequence and microarray analyses, promise to provide necessary information to better understand bacterial speciation. Indeed, recent data using these approaches, while meagre, support the view that speciation processes may occur at the subspecies level within ecological niches (ecovars) and owing to biogeography (geovars). A major dilemma for bacterial taxonomists is how to incorporate this new information into the present hierarchical system for classification of Bacteria and Archaea without causing undesirable confusion and contention. This author proposes the genomic–phylogenetic species concept (GPSC) for the taxonomy of prokaryotes. The aim is twofold. First, the GPSC would provide a conceptual and testable framework for bacterial taxonomy. Second, the GPSC would replace the burdensome requirement for DNA hybridization presently needed to describe new species. Furthermore, the GPSC is consistent with the present treatment at higher taxonomic levels.
Keywords: bacterial species, speciation, microbial species, species concepts, co-speciation
1. Introduction
The heyday of bacterial taxonomy was the twentieth century. During this period, many breakthroughs were made that have resulted in our present nomenclature and classification of the Bacteria and Archaea. Early in the century, great emphasis was placed on metabolic and physiological features to characterize bacteria. These features, combined with morphological traits determined by light microscopy, formed the basis for the early classifications such as those found in Bergey's Manual of Determinative Bacteriology culminating in the seventh edition in 1957 (Breed et al. 1957). Since the 1960s, a number of technical innovations have led to discoveries that have transformed bacterial taxonomy into a science. Among these was the widespread use of electron microscopy to examine microbial cell fine structure, introduction of computers to develop phenetic approaches to taxonomy and the analysis of DNA whose sequence represents the ‘chemical formula’ of an organism. For the first time, genotypic characterizations, including mol% G+C composition of DNA and DNA–DNA hybridization, could be used to better define taxonomic entities. Indeed, DNA hybridization, developed in the 1980s (Brenner et al. 1982; Johnson 1984), became the basis for a bacterial species definition that is still used today (Wayne et al. 1987; Stackebrandt & Goebel 1994; Stackebrandt et al. 2002).
Another important finding of the latter half of the twentieth century was the discovery of microbial fossils (Barghorn & Tyler 1965). For the first time, microbiologists were able to study micro-organisms within the context of evolution. Subsequent developments enabled geochemists to date microbial activities and hence early life, deep into the Precambrian era, more than 3 Gyr before the evolution of plants and animals.
One particularly important breakthrough of the late twentieth century was the introduction of phylogenetic analyses to examine evolution through the sequences of macromolecular subunits, especially protein and DNA, a concept introduced by Zuckerkandl & Pauling (1965). The analysis of molecular sequences through phylogenetic approaches, such as the sequencing of the small subunit ribosomal RNA (16S rRNA and 18S rRNA), has led to a revolution in the classification of all living organisms into a Tree of Life (Woese et al. 1990). In support of the fossil record, the Tree of Life has provided additional and independent evidence that microbial life originated first on Earth, well before that of macrobial life.
Accompanying these scientific discoveries was the adoption of some practices, which clarified and furthered the taxonomy of Bacteria and Archaea. One of these was the drafting of the Code of Nomenclature for Bacteria and Archaea. Now in its 2nd edition, the Code contains all the rules necessary for the formulation of proper names at all levels of classification. Another remarkably pragmatic effort led by V. B. D. Skerman was the adoption of an approved list of bacterial names that dated from 1980 (Skerman et al. 1980). The authorities for each taxonomic group were notified and requested to provide recommendations for the treatment of all present species. This resulted in a list of approved names of species that was regarded as acceptable to the knowledgeable experts. This logical artifice resulted in discarding unused, uncertain and disputed names of taxa that cluttered the taxonomy. Since then, taxa from the level of species to order that are properly named, described and published in the International Journal of Systematic and Evolutionary Microbiology (formerly the International Journal of Systematic Bacteriology) are listed as validly published.
2. Where are all the bacterial and archaeal species?
Despite the early evolution of microbial life, which occurred about 3 Gyr before the evolution of plants and animals, about 5000 bacterial species have been named. In contrast, at least a million animal species have been described. How can this great disparity in species numbers be explained considering the vastly longer time that the Bacteria and Archaea have been evolving, their relatively short generation times and their adaptation to fine-scale micro-environments in all ecosystems?
The low numbers of microbial species also seem anomalous considering that most of the major divisions, phyla and kingdoms in the Tree of Life are micro-organisms. This vast genetic diversity of microbial life is incongruous in the light of their low species numbers (Staley 2002). It is difficult to imagine a tree with so many branches but very few twigs.
Consistent with these arguments is one that is based on the organism size. A very large number of microbial species, in particular Bacteria and Archaea, would be predicted to occur owing to the increasing numbers of species that are found among organisms as the size of macro-organisms decreases (May 1988; Hedlund & Staley 2004). Is there a break in the trend because micro-organisms speciate differently from macro-organisms? Or, is this prediction actually true and possibly explained by a number of factors such as:
differences in species concepts between the Bacteria and macro-organisms,
difficulties in growing, isolating in pure culture and characterizing bacteria, a prerequisite to naming new species, and
extensive horizontal gene transfer (HGT) and recombination within the Bacteria and Archaea that blur the distinction of species.
These three issues are considered individually as follows.
(a) Species concept differences
There is no concept that has been advocated for or accepted by bacteriologists for the bacterial species (Rosselló-Mora & Amann 2001). This is probably a result of the inability of bacterial taxonomists, at least until recently, to investigate the evolution of bacterial and archaeal species. There is, however, a bacterial species definition. Bacteria are described on the basis of both genotypic and phenotypic properties. The primary genotypic feature is the degree of DNA–DNA hybridization between the strains within a purported species (Wayne et al. 1987). If an unknown strain shows 70% or greater hybridization to a designated ‘type strain’ that is deposited in a culture collection, then the strain is considered to be a member of the same species; however, if the unknown strain shows a lower level of hybridization, it can be named as a separate species. However, it is incumbent upon the scientist who is naming a bacterial species to also describe as many phenotypic properties as possible to enable the delineation of the new species from closely related species that have already been described. These include morphological and physiological features as appropriate for the taxonomic group. Therefore, each species must have its own distinctive phenotype to permit it to be identified (table 1). The polyphasic species definition for bacteria refers to the combination of features, including molecular and phenotypic properties that are used in naming species (e.g. Gevers et al. 2005).
Table 1.
Techniques used presently for the classification of Bacteria and Archaea. (Their utility for description of a level in the taxonomic hierarchy is noted by a +, − or ±.)
| present taxon level | phenotypic properties | 16S rRNA sequence | DNA–DNA hybridization | genomics | MLSA |
|---|---|---|---|---|---|
| domain | + | + | − | + | + |
| kingdoma | + | + | − | + | + |
| phylum | + | + | − | + | + |
| class | + | + | − | + | + |
| order | + | + | − | + | + |
| family | + | + | − | + | + |
| genus | + | ± | ± | + | + |
| species | + | − | + | + | + |
| subspecies | ± | − | − | + | + |
Note that the present classification does not include the kingdom level (Garrity & Holt 2001).
Clearly, the bacterial species definition has served bacterial taxonomists very well during the past quarter century (Brenner et al. 2005). First, it was based upon and largely consistent with the previous taxonomy that was in place at the time of its introduction. The major accomplishment was that it initiated a standard that was then applied to the naming of all Bacteria and Archaea and therefore led to uniformity within the Bacteria and Archaea. The idea of using an organism's entire genomic DNA in the DNA–DNA hybridization analysis as the basis for the species definition is logical as it permits a comparison of the relatedness of all the genetic features among strains. In addition, having phenotypic properties that are correlated with the genotypic properties enabled labs, such as clinical labs where rapid identification was important, and which were not able to carry out DNA–DNA hybridization analyses, to use more traditional phenotypic approaches. Therefore, the polyphasic definition has not only standardized bacterial taxonomy but stabilized it as well.
Importantly, experiments on the diversity of micro-organisms in soils support the much greater diversity of bacterial species than that represented by culture collections (Torsvik et al. 1990). Indeed, based upon the present 70% DNA–DNA hybridization threshold used to describe bacterial species, over 100 000 species have been reported in a single gram of soil. If this is true, then the species definition cannot be the sole reason for the huge discrepancy in the numbers of bacterial species. These data indicate that bacteriologists need to seek better methods for recovery of the organisms that are difficult to grow (Staley & Konopka 1985).
Some have challenged the bacterial species definition for a variety of reasons (Staley 1997; Ward 1998; Cohan 2002). First, it is based on a threshold that is artificially derived and is dependent largely upon previous bacterial species definitions that were based entirely on phenotypic properties. Second, from a comparative standpoint, it is much broader than that used to describe animal species. For example, virtually all primates from lemurs to Homo sapiens would be considered the same species using the bacterial species definition of the mammalian commensal bacterium, Escherichia coli (Staley 1997). As discussed previously, this has made it impossible to compare fairly the biodiversity of species among disparate biological groups. The result is that erroneous conclusions have been drawn about microbial diversity (Mayr 1998). Third, the present definition does not take evolutionary concepts into consideration in the taxonomy of all organisms because speciation is, largely, an evolutionary process. Fourth, the definition is typological in that it relies on a single type reference strain used for comparative purposes that is placed in a culture collection (Ward 1998); furthermore, this type strain may unknowingly undergo mutations and deletions during transfers and growth subsequent to its description.
The advent of 16S rRNA gene sequencing transformed bacterial taxonomy. It has been widely accepted as a primary technique to identify bacteria. Prior to the introduction of 16S rRNA gene sequencing, the classification of many groups was extremely difficult. For example, aerobic Gram-negative bacteria that use acetate as a sole carbon source were placed in a single group, the pseudomonads, which comprised several genera including Pseudomonas. However, when 16S rRNA gene sequencing was introduced, it was discovered that these organisms arose by three different evolutionary routes that are now recognized as the Alpha-, Beta- and Gammaproteobacteria. As a consequence, many new genera have been identified and named. This is but one example of how 16S rDNA sequencing has not only eased but also greatly enriched the taxonomy of Bacteria and Archaea. Indeed, it is one of the first tests one uses now when a possible novel bacterium has been isolated from nature.
Unlike DNA–DNA hybridization, the 16S rRNA gene sequence as well as other gene and protein sequence information is archival. Once it has been determined, it is a definitive comparative feature of the strain of interest. In contrast, DNA–DNA reassociation experiments must be performed each and every time a purportedly novel strain has been isolated to determine its relatedness to known type species. Owing to the procedural difficulties associated with the DNA hybridization approach, very few laboratories still perform it.
Although 16S rRNA gene sequencing is important in identifying a strain to the family or genus level, it is of very little utility for differentiation of species (table 1). For example, it is well known that organisms can have essentially identical 16S rRNA gene sequences and still belong to separate species based upon DNA–DNA hybridization (Fox et al. 1992).
Nonetheless, one particularly useful property of the 16S rRNA gene sequence analysis is that it has allowed a more rapid determination of whether there is a need for carrying out DNA–DNA hybridization. This is owing to a known demarcation within existing species that have been analysed by both the procedures. If a strain shows less that 97% 16S rRNA gene homology with its highest match-described species, then it can be declared a novel species without carrying out DNA–DNA hybridization (Stackebrandt & Goebel 1994). The establishment of this boundary condition has resulted in the naming of many more species without the need for DNA–DNA hybridization.
(b) Difficulties in cultivation and description
Because animal taxonomists have traditionally described species based upon morphology, the criteria for identification can be simply achieved by macroscopic observation alone. The morphospecies concept is also applied to some eukaryotic microbial groups, e.g. the Protists, by use of microscopic identification. However, even microscopic examination is unsuitable as a sole criterion, with rare exceptions, for the identification of species of Bacteria and Archaea.
Bacterial taxonomists must first of all obtain a pure culture of an organism before it can be officially named according to the International Code of Nomenclature of Bacteria (Sneath 1992). Then, it must be grown in the laboratory and compared to the most closely related strains using phenotypic and genotypic properties. Not only are these tests time consuming and expensive, but some bacteria grow very poorly in the laboratory. For these reasons, many bacteria have not yet been studied and described.
Furthermore, many bacterial species have not yet been grown in pure culture. Typical cultivation procedures used for the recovery of bacteria from natural habitats are poor in that they recover less than 1% of the organisms that are present. This has been termed the ‘great plate count anomaly’ and is thought to be due primarily to the difficulty microbiologists have encountered in designing conditions that are favourable for the growth of many of these bacteria (Staley & Konopka 1985). Therefore, many novel phyla of microbial life that have been identified in environments by the use of molecular approaches (Hugenholz et al. 1998) remain unstudied in pure culture and therefore unnamed.
It is also noteworthy that bacteria can be named as candidatus organisms if it is not possible to grow them in the laboratory as long as sufficient information is known about them, including their mol% G+C content, their 16S rRNA gene sequence, cell size and shape, and other phenotypic properties. This enables the provisional naming of organisms, such as pathogens that cannot be isolated from their hosts, or micro-organisms like the anammox (anaerobic ammonia oxidizing) bacteria of the Planctomycetes that can be grown in highly enriched conditions sufficient to enable their characterization.
(c) Horizontal gene transfer
Another major issue that influences bacterial taxonomy and evolution is lateral or horizontal gene transfer. Evidence indicates that it is a confounding factor in the understanding of the relatedness among bacteria at all taxonomic levels. At one extreme, some have argued that it has highly muddled up evolutionary trees, that it has made it virtually impossible to understand microbial evolution. Others point out that our lack of understanding of bacterial speciation precludes any final decision on its significance.
3. Bacterial speciation and the genomic–phylogenetic species concept
The twenty-first century holds great promise as the time during which microbiologists will truly answer the question, ‘How do bacteria speciate?’ Already, genomic analyses of members of the same species are providing insight to this question (Konstantinidis & Tiedje 2005a,b) as well as multilocus sequence typing (Feil & Spratt 2001) and multilocus sequence analyses (MLSA). Only genomic and MLSA approaches appear to be useful for the classification of bacteria from domain to species (table 1).
Bacteriologists have not yet developed a concept for a species (Rosselló-Mora & Amann 2001). However, with the accumulating evidence from molecular sequencing and genomic studies, the time seems ripe for a species concept. Indeed, at the highest taxonomic levels, the sequence of the ribosomal RNA in the small ribosome subunit, i.e. 16S rRNA for Bacteria and Archaea and 18S rRNA for the Eucarya, relies on the phylogenetic concept and has been shown to be very useful in the classification of all organisms on Earth into a single Tree of Life (Woese et al. 1990). Although the Tree of Life is imperfect, it has nonetheless been immensely useful for the classification of the Bacteria and Archaea at higher taxonomic levels (e.g. Garrity & Holt 2001). However, as mentioned previously, because 16S rRNA gene is such a highly conserved molecule, it is inadequate for the resolution of species of Bacteria and Archaea as well as other organisms (table 1).
A phylogenetic species concept has been proposed previously for Bacteria and Archaea (Staley 2004). Species are considered to be an ‘irreducible cluster of organisms diagnosably different from other such clusters and within which there is a parental pattern of ancestry and descent’ (Craycraft 1989).
Genome analyses open the way for the implementation of array technology, and studies of gene expression, as well as providing the complete DNA sequence of the organism. When combined with genomic analysis, the phylogenic approaches are greatly strengthened. However, because genomic sequences require extensive labour and materials costs, they are not practicable as a requirement for a species concept. Their main role for the immediate future is to provide guidelines in our understanding of bacterial speciation. Thus, intensive genomic sequencing of closely related organisms from communities at varying geographical separation will provide a rich base for understanding speciation. This information will provide a means for assessing the clustering of strains within a ‘species’. Information from genomic studies can be used to identify the specific group of genes that will be useful in phylogenetic analysis for that particular group of organisms. In addition, genomic approaches will provide additional information about gene regulation and expression. For this reason, a combined genomic–phylogenetic species concept is proposed.
The advantages of the genomic–phylogenetic species concept (GPSC) are as follows:
The GPSC is a methodological species concept that relies heavily on macromolecular sequences (e.g. protein, RNA and DNA) that are not subject to biases of interpretation. Interestingly, most other prokaryotic species concepts use phylogeny to assess speciation events indicating the potential universality of the GPSC.
-
The organism's ecology is encompassed by the GPSC in that adaptive radiations will be reflected in the alteration of gene and protein sequences in response to the organism's niche.
The niche of an organism is a, if not the, primary cohesive feature of a species. By definition, no two species may occupy the same niche at the same time. The niche encompasses the habitat and how the organism makes its livelihood in its environment. The molecular sequences of the functional proteins and enzymes that are unique to the species are a reflection of the niche, as are the genes that reveal genetic drift and adaptive radiation associated with biogeography. Furthermore, the core housekeeping genes of the species will also probably reflect this same evolutionary pattern.
The biogeography of an organism is considered by the GPSC in that genetic drift will result in a change in the gene and protein sequences of core genes. The ecological species concept is more limited in that it does not consider genetic drift that may result solely from physical or geographical separation.
The evolutionary history of the organism is considered through the phylogeny of gene and protein sequences. In contrast, the evolutionary species concept is based exclusively on the evolution of an organism. The GPSC is more pragmatic in that it uses sequence information to infer the evolutionary pattern with the recognition that the true evolutionary pathway of an organism cannot be thoroughly understood.
Sequence-based information from genomic–phylogenetic analyses is archival.
Sequence information is readily portable and globally accessible.
Taxa can be identified rapidly once the set of relevant genes for a particular taxonomic group have been identified.
The GPSC has the potential for universality inasmuch as the GPSC has already been used in the Tree of Life and is being used by some for the taxonomy of plants and animals.
A potential though unlikely disadvantage of the GPSC is that phenotypic tests would have to become less significant in describing species, simply because it is difficult to phenotypically distinguish organisms that are very closely related to each other. Even now, if no phenotypic differences can be found, the organism is termed a genomospecies, i.e. an organism whose description is based entirely upon the results of the DNA–DNA hybridization data (Ursing et al. 1995). Such examples cannot be presently described as species although they would be very likely using the GPSC. Of course with complete genome sequences, inferred pathways, enzymes and other proteins can be determined and proteomics can be used to assess phenotype.
The primary reason that the phenotype was highly important for the bacterial species definition was the pragmatic need for the rapid identification of pathogens in clinical laboratories. This was because DNA–DNA hybridization was highly laborious and time-consuming for routine testing and identification of pathogens. However, now that molecular sequencing procedures are available, including commercial ‘kits’ for the rapid diagnosis of pathogenic species, this is no longer a serious issue. Moreover, molecular sequence information can be designed to be applicable across the taxonomic hierarchy, including the subspecies category for which phenotypic tests are not practical (table 1).
In order for a phylogenetic concept to be practicable, it must rely on a set of core genes from an organism as well as those that reflect its niche. To the extent that typical housekeeping genes parallel this evolutionary pattern, they will be useful for this purpose. The identification of these core genes and their sequence analysis could serve as a basis for assessing species in that particular taxonomic group. Thus, the MLSA approach could be used to determine what the irreducible clusters are that comprise a species.
4. Application of the genomic–phylogenetic species concept to bacterial taxonomy
(a) Co-speciation symbioses
The GPSC could be applied to bacterial taxonomy rather quickly, at least for some taxa. To illustrate this, consider the situation in which bacteria have been shown to co-evolve with host animals. Buchnera aphidicola is an excellent example. Strong evidence supports that this bacterium, which lives in the bacteriome, a special compartment in the aphid's body cavity, began to co-evolve with its aphid hosts 150–250 Myr ago (Moran et al. 1993). As a result, trees based on 16S rRNA gene sequencing of B. aphidicola and 18S rRNA gene and mitochondrial DNA sequencing of the host, combined with fossil evidence, produce a tree for B. aphidicola that is a mirror image of the aphid phylogeny (Moran et al. 1993; Baumann 2005).
However, at this time, the taxonomy of the bacterium does not mirror the taxonomy of its hosts. Thus, a single bacterial genus and species has co-evolved and perhaps co-speciated with an aphid host which comprises several suborders and families, many genera and potentially some 4000 species (Paul Baumann 2006, personal communication).
This present bacterial taxonomic treatment must be challenged owing to the following rationale: if symbiotic Bacteria or Archaea have been shown to co-evolve with their host species, then they may also co-speciate with them. Therefore, for each aphid host taxon (species, genus, family, suborder and order), there may be a comparable, separate and parallel bacterial taxon (table 2). I term this the ‘co-speciation quid pro quo tenet’ in bacterial taxonomy.
Table 2.
Present and hypothetical GPSC taxonomy of Buchnera aphidicola.
| taxon | present taxonomya | hypothetical GPSC taxonomy |
|---|---|---|
| domain | Bacteria | Bacteria |
| kingdom | NA | Proteobacteria |
| phylum | Proteobacteria | Gammaproteobacteria |
| class | Gammaproteobacteria | ? |
| order | Enterobacteriales | Buchnerales |
| family | Enterobacteriaceae | Buchneraceae+others |
| genus | Buchnera | Buchnera+many others |
| species | aphidicola | aphidicola+4000 others |
This illustrative example for B. aphidicola should not be interpreted as a blanket policy that for each aphid species there is a separate and parallel species of bacteria. Instead, it would be necessary to demonstrate empirically that each purported species pair has truly co-speciated. For this, appropriate MLSA studies would be necessary for the bacteria accompanied by similar analyses for the aphid host.
Interestingly, some controversy exists about the coevolution/co-speciation of Buchnera–aphid symbiosis based upon phylogenetic analyses of two bacterial genes (Martinez-Torres et al. 2001), the results of which do not show complete parallelism. However, the authors do not attribute the explanation to the false Buchnera phylogeny, but to the questionable phylogeny of the aphid hosts. It is important to recognize in Buchnera that its rate of evolution based upon 16S rRNA gene divergence is considerably higher than that of the aphid, making the Buchnera phylogeny more robust.
This proposed classification of Buchnera based on co-speciation would potentially increase the number of bacterial species by about 4000 (Paul Baumann 2006, personal communication), which when combined with the official listing of 5000 bacterial species brings the total to about 9000. Therefore, this single example of co-speciation would almost double the present number of bacterial species. Considering that most animal species, including the highly speciated insects, harbour bacterial symbionts, many of which may co-speciate, this would probably increase the number of bacterial species many fold.
(b) Commensal co-speciation1
Another group of organisms in which co-speciation may occur are the commensal bacteria. Of course, this situation differs markedly from that of the organism pairs in which coevolution occurs, as discussed for Buchnera. In the case of Buchnera, there is virtually no possibility of exchanging genes with other bacteria because the cells are isolated from other bacteria as well as phage in the aphid's bacteriome. Therefore, the possibility of HGT with other bacteria is largely, if not entirely, precluded. Furthermore, Buchnera has undergone reductive evolution in which many genes that are unnecessary for an independent existence have been lost. Therefore, the genome size has decreased from that of the ancestral type by at least fivefold, from about 3 to 4 Mb to about 650 kb.
In the case of commensal bacteria, many are still capable of independent growth on rich media in the laboratory. One example of such a bacterium is the genus Simonsiella belonging to the Betaproteobacteria (Hedlund & Staley 2002). This organism resides in the oral cavity of mammalian hosts where it lives on the epithelial cells. There is no evidence that it is pathogenic. The unique morphology of this bacterium makes it readily identifiable (figure 1). Interestingly, this filamentous bacterium is a gliding organism that shows dorsal–ventral asymmetry. It can be readily cultivated in the laboratory on a medium to which mammalian serum is added.
Figure 1.
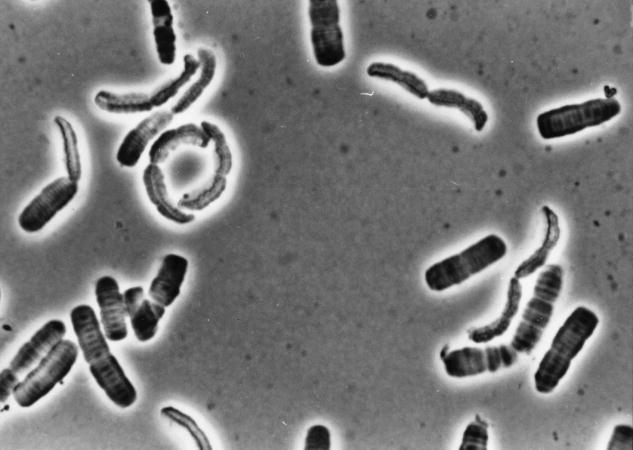
A phase photomicrograph showing a strain of the mammalian oral bacterium, Simonsiella. Note the filamentous cells and dorsal–ventral asymmetry. Cell diameter is about 2 μm.
Studies of the 16S rRNA gene sequence of this bacterium show that four strains from various mammalian species, humans, sheep, dogs and cats, form separate clades (Hedlund & Staley 2002). When the 16S rRNA gene sequences are compared with the most closely related known sequences from GenBank, the resulting tree has the following features. Each host animal species harbours a separate clade of Simonsiella strains that represent a species. For the carnivore commensals, i.e. the dog and cat strains, the tree is monophyletic, supporting the view that there has been no HGT. However, the primate host (Homo sapiens) and ruminant host (sheep) are separated from each other and the carnivore strains by polytomies. This indicates that there has been HGT and recombination with other organisms. These recombination events may have been initiated by the unidirectional transfer of Simonsiella DNA to highly competent species of the Betaproteobacteria including Neisseria. At this time, there is no evidence that Simonsiella has received genes from other members of the Betaproteobacteria.
If a tree is constructed based on 16S rRNA gene of the Simonsiella strains without the inclusion of other bacteria, the result is similar to that found for Buchnera. That is, the Simonsiella strains show a phylogeny that is a mirror image of the phylogeny of the host animal species (figure 2). Of course, in order for this tree to reflect ‘co-speciation’, it would be necessary to demonstrate that Simonsiella does not receive DNA from other members of the Betaproteobacteria in the oral cavity and to reconcile evolutionary timelines of host and symbiont. This could be assessed by looking at MLSA patterns of the genes in Simonsiella versus those in Neisseria, Kingella and Eikenella which account for the polytomies of the published 16S rDNA tree (Hedlund & Staley 2002).
Figure 2.

A 16S rDNA tree of Simonsiella strains from four different mammalian species, human, sheep, cat and dog. Note that the four strains from each animal host form clades and there is a pattern typical of coevolution illustrated with the evolution of the animal hosts. See text for details on the phylogeny. Courtesy of Brian Hedlund.
If it could be shown that Simonsiella strains do indeed co-evolve with their mammalian hosts, then their phylogeny would be similar to that described for Buchnera and would parallel that of their host mammals. As a consequence, since there are about 5000 mammalian species, this, as with Buchnera, could also result in almost doubling the present number of bacterial species.
(c) Free-living genomic–phylogenetic species concept ecospecies?
The situation concerning the evolution of free-living bacterial and archaeal species is less well understood. A consideration of the Prochlorococcus genomes (Rocap et al. 2003) may help illustrate the application of the GPSC with respect to such free-living bacteria. Some strains of this photosynthetic genus of marine cyanobacteria, which accounts for a major percentage of global primary productivity, are adapted to high light intensity, whereas others are adapted to low light intensity. The MED4 strain is adapted to high light, whereas the MIT9312 strain is a low light strain that underlies the high light group in the marine water column. The 16S rDNA tree indicates that they are descended from a common low light ancestor and have diverged through time to form the two separate ecotypes (GPSC ecospecies?). Surprisingly, the size of the MED4 genome is only 1.65 Mb, whereas the low light strain MIT9313 is 2.41 Mb and the mol% G+C content is 30.8 versus 50.7 for the two strains, respectively. These are remarkable differences not expected within a single species or perhaps even a genus! This difference is explained in part by reductive evolution and loss of certain genes, including mutY which removes incorrectly paired adenosine. The loss of this gene therefore may have led to an increase in adenine mispairing, leading to GC-TA transversions, with a resultant lower GC content.
These two genomes share 1350 genes in common. MED4 has 366 additional genes and MIT9313 has 925. Evidence is provided that these genes have been either differentially retained from the common ancestor, duplicated, or acquired via HGT. The upshot is that Prochlorococcus has a core set of genes that can be used to understand their phylogeny. Therefore, the phylogenetic species concept could be applied to the strains in this genus despite the fact that there is clear evidence that multiple HGT events have occurred particularly at ‘hot spots’ on the genomes.
A follow-up study of two high light-adapted strains of Prochlorococcus, MED4 and MIT9312, which are also separate ecotypes (GPSC ecospecies?), indicate that they are different from each other as well (Coleman et al. 2006). Most interesting from the standpoint of the GPSC is that, although these strains differ from each other by 0.8% in 16S rDNA sequence, the median sequence identity of the 1574 shared genes is only 78%. Again, it is evident that the phylogenetic species concept could be applied to these strains as well. These ecotypes have an abundance of shared genes that can, at least in theory (no one to my knowledge has yet undertaken the phylogenetic analyses), be used for their classification by the GPSC. This example based on genome sequences illustrates that the GPSC can be readily applied to ecotypes.
(d) Free-living genomic–phylogenetic species concept geospecies?
In the past several years, considerable interest has been shown in bacterial biogeography (Staley 1999; Staley & Gosink 1999; Martiny et al. 2006). Increasingly evidence is accumulating that geovars of bacteria occur, particularly in extreme environments. For example, the island communities of hot springs provide evidence that entire genera and species may differ from one location on Earth to another, despite strong similarities in the physical and the chemical properties of the different habitats (Papke et al. 2003).
One of the most striking examples of geovars is reported with Sulfolobus icelandicus strains in which separate clades are found in hot springs from Iceland, North America and Russia (Whitaker et al. 2003). Based on the concatenated sequence information reported in the paper, a proposal could be made that each of these locations and the probable sublocations within these areas (such as the Uzon and the Mutnovsky geyser field locations at Kamchatka) would appear to be ideal locations to harbour phylospecies. However, additional data indicate that some genetic loci used for the concatenated analyses from strains at one location are more similar to those from another location (Whitaker 2006). Therefore, HGT events appear to have occurred between locations. From a taxonomic standpoint, it was concluded that, without further information, these organisms should be considered geovars of S. icelandicus, not phylogenetic geospecies (R. Whitaker 2006, personal communication).
(e) Pathogens and phylogeography
Several human pathogens have provided evidence of geographical distribution. Helicobacter pylori has been shown to follow the migratory pathways of H. sapiens as they dispersed from Africa about 40 000 years ago (Falush et al. 2003). Evidence from MLSA shows that the H. pylori strains from different human racial groups, Asians, Europeans and Africans, produce separate patterns in support of the continental distributions and migrational patterns of their hosts.
A similar pattern of phylogeography has been found with Mycobacterium tuberculosis (Gagneux et al. 2006). In both species, the distributional patterns follow the migratory routes of human populations that became separated over time by geographical distances. However, unlike M. tuberculosis, the subspecies of H. pylori undergo extensive recombination with strains within the human racial group in which they reside, resulting in clusters that separate them from the pathogenic strains found in other human populations.
From a taxonomic standpoint, H. pylori and M. tuberculosis have been treated as pathovars. Some microbiologists believe that these are examples of speciation in progress. If the separation of the human races were to persist for many additional millennia, these would probably become true species in concert with their human racial counterparts. However, with the rapidity at which humans are mixing geographically and racially, this consequence is unlikely.
The substance of these examples is that these phylogeographic patterns point to the probability that these types of associations can lead to divergence and subsequent speciation events in microbial symbionts in concert with their plant and animal host species, leading to co-speciation in specific regions on Earth.
In many of the examples cited previously, the assumption that provides for clear examples of speciation are dependent either on a reduction in the extent of recombination or its absence altogether. However, in the real world, the degree to which microbial taxa are free from confounding recombinant events is unclear. The following section discusses the effects of HGT, recombination and expression on speciation.
5. Highly recombinant bacteria
Gene transfer between and among Bacteria and Archaea can occur through a variety of mechanisms, including transformation, transduction and conjugation. It is estimated that about 1% of bacteria are able to undergo natural transformation in which reasonably large segments of the DNA can be potentially available for recombination (Thomas & Nielsen 2005). Since all organisms studied have been found to harbour phages, it is probable that transduction is one of the most widespread mechanisms known to effect gene transfer. The transfer of plasmids, which may involve large segments of the DNA, appears to be less common.
Conceptually, the bacterial species is in a state of dynamic genetic flux. First and foremost to these organisms is that they must, on the one hand, adapt to their niche, while on the other hand, be prepared to move into another. Therefore, genes may enter or leave the organism as long as they do not result in rapidly displacing the organism from its niche, which could lead to their untimely extinction. Indeed, recombinant mobile genetic elements may provide the recipient organism with some advantage in a changing environment, as the new genes may proffer a selective advantage that enables the organism to evolve to adjust its niche and improve its fitness.
For example, two polycyclic aromatic hydrocarbon (PAH)-degrading organisms of the Gammaproteobacteria, Neptunomonas naphthovorans and a Pseudoalteromonas sp. that are located at the same marine site, have been shown to harbour a dioxygenase whose sequence indicates that they are clearly related to each other (Hedlund & Staley 2006). Since this creosote-contaminated, US Environmental Protection Agency Superfund Site contains PAH compounds, the logical inference is that these bacteria have been able to incorporate and express the genes involved in PAH degradation, most probably via plasmid transfer from one to the other or from another PAH-degrading organism. However, the housekeeping genes of the organism have probably remained unchanged and will continue to be useful for classification by the GPSC as ecospecies or, in this example, ‘ecogenera’ using the MLSA approach. Eventually, however, the strong selective pressure of this environment may result in the evolution of the capabilities of these organisms towards novel species that are better suited to this recently changed environment.
Even interdomain gene transfers have been reported in the literature. One notable example of this was the discovery of six formaldehyde oxidation genes in two groups, the methanogens of the Archaea and the methanotrophs of the Proteobacteria. Recently, however, another phylum of the Bacteria, the Planctomycetes were found to harbour these same genes and furthermore, all six of the Planctomycetes proteins involved in formaldehyde oxidation exhibited an intermediate phylogeny between that of the Proteobacteria and the Archaea (Chistoserdova et al. 2004). This could be interpreted in a variety of ways, some of which do not involve interdomain HGT. Therefore, simply because some evidence may suggest interdomain HGT, it does not necessarily mean that it is the actual explanation, inasmuch as the simple vertical transfer of these genes with loss from some taxa could explain the data equally well.
Although the mechanisms by which DNA can be transferred from one bacterium to another are known, their effect at great distances is poorly understood. How probable is it that a bacterium wafted in the air or carried by water currents thousands of kilometres around Earth will serve as a vector to transfer genes through transduction, transformation or conjugation? Is it possible that phages are the principal agents involved in long-distance transfers?
Recent evidence indicates that homologous recombination (HR) of small segments of the genome containing less than 1000 bp is occurring across considerable geographical distances. Thus, evidence from the hot springs genera, Thermotoga (Nesbo et al. 2006) and Sulfolobus (Whitaker 2006) indicates that this process is significant. Perhaps, phages are responsible for the transfer. This is consistent with what has been reported for genomes, also, such as Prochlorococcus species (genera?) that reside at higher and lower depths in the marine water column (Coleman et al. 2006). For Thermotoga, evidence was also provided for HR of large gene fragments. However, as the authors state, it is uncertain whether these came from organisms at global scales or rather from the local environments owing to inadequate local sampling (Nesbo et al. 2006).
Although the data are not extensive at this time, these patterns suggest that the genomes of resident bacteria at one locale may be markedly affected by genes arising from a distant location. If so, this could be a major driving force for bacterial evolution in that the genomes of bacteria endemic to one area can be impacted by a distant genetic source. As long as these transfers are not extensive, they should not override the core genome of a bacterium. In that case, the phylogeographic pattern of the endemic bacterium can be discerned and the taxonomy of the organism can be determined by the GPSC. Clearly, however, much more extensive work will be needed to understand the genetic diversity of species within and between locations.
Recent MLSA work on 770 strains of the highly recombinant genus, Neisseria indicates that the individual species, N. meningitidis, N. lactamica and N. gonorrhoeae can all be identified using concatenated sequences of seven housekeeping loci (Hanage et al. 2005). A few strains arose from a branch separating N. meningitidis from N. lactamica. These strains were regarded as ‘fuzzy species’ or incipient species.
Interestingly, when highly recombinant bacteria are considered, it could be argued that they fulfil the definition of a pseudo-biological species concept in which sexuality plays the major role in speciation. However, it must be recognized that within animal species all the genes are transferred; the transfer is only intraspecies, it is the sole means of reproduction and fertile progeny are produced. None of these criteria fully apply to the Bacteria and Archaea (table 3).
Table 3.
Differences in sexual exchange between Bacteria and Archaea versus animal species according to the biological species concept (BSC).
| sexual exchange effects | Bacteria and Archaea | animal species (BSC) |
|---|---|---|
| genes of organism | some | all |
| taxa only | intraspecies, interspecies to domain? | intraspecies |
| reproduction | binary transverse fission and other mechanisms | sole mechanism |
| product progeny | merodiploid | fertile |
Therefore, the primary challenge of a GPSC for Bacteria and Archaea is that it cannot apply to bacteria which are so highly recombinant that the phylogeny cannot be discerned. The highly recombinant types that cannot be phylogenetically resolvable would need to be considered as sub-specific varieties. On the other hand, if further evidence indicates that recombination is the driving as well as cohesive force in the speciation of some groups of highly recombinant bacteria, then a recombinant species concept for these Bacteria and Archaea may be more appropriate.
6. Future directions for a genomic–phylogenetic species concept
Much of the work that is being carried out by bacteriologists is based on an assumption that phylogeny is an acceptable approach to bacterial classification. This is one of the reasons that the 16S rRNA gene sequence has acquired such significance. Thus, it seems reasonable that a GPSC for Bacteria and Archaea would be acceptable to a majority of bacteriologists, providing experimental evidence supports this approach. Clearly, however, the adoption of the GPSC for Bacteria and Archaea will entail further research. What criteria are necessary for identification of a phylogenetic species? Evidence from phylogenetic analyses of gene or protein sequences that supports the evolution of a novel cluster or clade. A sufficient number of sequences as well as a sufficient number of strains would be needed.
Of course, the genomic approach is the quintessential way to examine the speciation process in bacteria. Indeed, even the expense of doing this has dropped considerably in the past few years. Some present procedures indicate that a typical 3–4 Mb genome can be sequenced for less than US $10 000 (Margulies et al. 2005). With complete genomes of several strains of a closely related group (genus or species), it is possible to identify core genes. The phylogeny of the core genes could be readily extended to a larger collection of strains by MLSA to see whether the strains form phylogenetic clusters. Furthermore, approaches such as the average nucleotide index of the strains would also be beneficial in identifying similarities among the strains (Konstantinidis & Tiedje 2005a,b) and RT-PCR and expression arrays could be used to determine if the recombinant loci are actually being expressed.
Once the genomes of a given taxonomic group have been analysed, it would be possible to assess which genes would be most useful for MLSA. This would result in a less time-consuming and expensive method for the taxonomy of that group. Genomes of some 500 bacterial strains are available at this time and several have already proven to be useful to study the GPSC.
Even without complete genome sequences, the MLSA approach has been very useful in assessing the phylogeny of bacteria (Feil & Spratt 2001). In general, its findings have been very consistent with the presently accepted taxonomy of Bacteria. Some clusters representing new species have been discovered (Brian Spratt 2006, personal communication). If the MLSA approach as it is conducted presently were accepted as an alternative manner to describe Bacteria and Archaea, it could be implemented very soon for some microbial groups. Furthermore, it would be a welcome improvement in the taxonomy of Bacteria and Archaea because it would greatly expedite their identification.
An interim period is proposed to allow for research on the GPSC for bacteria and the utility of phylospecies. The primary goal of this interim period would be to identify model groups of organism for analysis. Many human pathogens have already been studied in considerable depth (Feil & Spratt 2001). Likewise, Buchnera and commensal bacteria such as Simonsiella would appear to be ideal models in which rapid progress could be made to investigate the GPSC. In addition, it would be desirable to have further work performed on environmental organisms. For example, some of the hot springs cyanobacteria have been brought into culture and are amenable to MLSA (D. Ward 2006, personal communication).
With sufficient information available from a wide variety of Bacteria and Archaea, it should be possible to extend the GPSC more broadly for the taxonomy of Bacteria and Archaea. Most importantly, studies of the microbial speciation process are fundamental to understanding how these fascinating and successful forms of life on Earth have evolved.
Acknowledgments
I wish to thank Paul Baumann for information on Buchnera and Brian Hedlund for providing permission to use figure 2. I also appreciate the helpful suggestions and comments of Brian Oakley, Brian Spratt & Rachel Whitaker. In addition, I wish to acknowledge the National Science Foundation as well as the NASA University of Washington Astrobiology Institute program for supporting my laboratory's research.
Endnote
The quotation marks used for ‘commensal co-speciation’ indicate that, for commensal bacteria, it is uncertain and perhaps doubtful that the symbiont is directing the host's evolution, although some have argued that the development of the immune systems of hosts is probably affected by commensal organisms. In this example, commensal co-speciation is identified as a somewhat different process from the classical description of co-speciation in the organisms, such as Buchnera and the aphids in which both the partners are clearly co-dependent for their livelihood.
One contribution of 15 to a Discussion Meeting Issue ‘Species and speciation in micro-organisms’.
References
- Barghorn E.S, Tyler S.A. Microorganisms from the gunflint chert. Science. 1965;147:563–577. doi: 10.1126/science.147.3658.563. [DOI] [PubMed] [Google Scholar]
- Baumann P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005;59:155–189. doi: 10.1146/annurev.micro.59.030804.121041. doi:10.1146/annurev.micro.59.030804.121041 [DOI] [PubMed] [Google Scholar]
- Baumann P, Baumann L, Moran N.A. Genus V Buchnera. In: Brenner D.J, Staley J.T, Krieg N.R, Garrity G, editors. Bergey's manual of systematic bacteriology. 2nd edn. vol. 2. Springer; New York, NY: 2005. pp. 633–638. [Google Scholar]
- Breed R.S, Murray E.G.D, Smith N.R, editors. Bergey's manual of determinative bacteriology. Williams and Wilkins; Baltimore, MD: 1957. [Google Scholar]
- Brenner D.J, McWhorter A.C, Leete Knutson J.K, Steigerwalt A.G. Escherichia vulneris, a new species of Enterobacteriaceae associated with human wounds. J. Clin. Microbiol. 1982;15:1133–1140. doi: 10.1128/jcm.15.6.1133-1140.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D.J, Staley J.T, Krieg N.R. Classification of prokaryotic organisms and the concept of speciation. In: Brenner D.J, Staley J.T, Krieg N.R, Garrity G, editors. Bergey's manual of systematic bacteriology. 2nd edn. vol. 2. Springer; New York, NY: 2005. pp. 27–32. [Google Scholar]
- Chistoserdova L, Jenkins C, Kalyuzhnaya M, Marx C.J, Lapidus A, Vorholt J.A, Staley J.T, Lidstrom M.E. The enigmatic Planctomycetes may hold a key to the origins of methanogenesis and methylotrophy. Mol. Biol. Evol. 2004;21:1234–1241. doi: 10.1093/molbev/msh113. doi:10.1093/molbev/msh113 [DOI] [PubMed] [Google Scholar]
- Cohan F.M. What are bacterial species? Annu. Rev. Microbiol. 2002;56:457–487. doi: 10.1146/annurev.micro.56.012302.160634. doi:10.1146/annurev.micro.56.012302.160634 [DOI] [PubMed] [Google Scholar]
- Coleman M.L, Sullivan M.B, Martiny A.C, Steglich C, Barry K, DeLong E.F, Chisholm S.W. Genomic islands and the ecology and evolution of Prochlorococcus. Science. 2006;311:1768–1770. doi: 10.1126/science.1122050. doi:10.1126/science.1122050 [DOI] [PubMed] [Google Scholar]
- Craycraft J. Speciation and ontology: the empirical consequences of alternative species concepts for understanding patterns and processes of differentiation. In: Otte D, Endler J.A, editors. Speciation and its consequences. Sinauer Associates, Inc; Sunderland, MA: 1989. pp. 28–59. [Google Scholar]
- Falush D, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. doi:10.1126/science.1080857 [DOI] [PubMed] [Google Scholar]
- Feil E.J, Spratt B.G. Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol. 2001;55:561–590. doi: 10.1146/annurev.micro.55.1.561. doi:10.1146/annurev.micro.55.1.561 [DOI] [PubMed] [Google Scholar]
- Fox G.E, Wisotzkey J.D, Jurtshuk P. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int. J. Syst. Bacteriol. 1992;42:166–170. doi: 10.1099/00207713-42-1-166. [DOI] [PubMed] [Google Scholar]
- Gagneux S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. doi:10.1073/pnas.0511240103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity G.M, Holt J.G. In: Bergey's manual of systematic bacteriology. 2nd edn. Boone D, Castenholz R, Garrity G, editors. vol. 1. Springer; New York, NY: 2001. pp. 119–166. [Google Scholar]
- Gevers D, et al. Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 2005;3:733–739. doi: 10.1038/nrmicro1236. doi:10.1038/nrmicro1236 [DOI] [PubMed] [Google Scholar]
- Hanage W.P, Fraser C, Spratt B. Fuzzy species among recombinogenic bacteria. BMC Biol. 2005;3:6–13. doi: 10.1186/1741-7007-3-6. doi:10.1186/1741-7007-3-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund B.P, Staley J.T. Phylogeny of the genus Simonsiella and other members of the Neisseriaceae. Int. J. Syst. Evol. Microbiol. 2002;52:1377–1382. doi: 10.1099/00207713-52-4-1377. doi:10.1099/ijs.0.01952-0 [DOI] [PubMed] [Google Scholar]
- Hedlund B.P, Staley J.T. Microbial endemism and biogeography. In: Bull A.T, editor. Microbial diversity and bioprospecting. ASM Press; Washington, DC: 2004. pp. 225–231. [Google Scholar]
- Hedlund B.P, Staley J.T. Isolation and characterization of Pseudoalteromonas strains with divergent polycyclic aromatic hydrocarbon (PAH) catabolic properties. Environ. Microbiol. 2006;8:178–182. doi: 10.1111/j.1462-2920.2005.00871.x. doi:10.1111/j.1462-2920.2005.00871.x [DOI] [PubMed] [Google Scholar]
- Hugenholz P, Goegel B.M, Pace N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.L. Nuleic acids in bacterial classification. In: Krieg N.R, Holt J.G, editors. Bergey's manual of systematic bacteriology. 1st edn. vol. 1. Williams and Wilkins; Baltimore, MD: 1984. pp. 8–11. [Google Scholar]
- Konstantinidis K.T, Tiedje J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl Acad. Sci. USA. 2005a;102:2567–2572. doi: 10.1073/pnas.0409727102. doi:10.1073/pnas.0409727102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis K.T, Tiedje J.M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005b;187:6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005. doi:10.1128/JB.187.18.6258-6264.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:378–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Torres D, Buades C, Latorre A, Moya A. Molecular systematics of aphids and their primary endosymbionts. Mol. Phylogenet. Evol. 2001;20:437–449. doi: 10.1006/mpev.2001.0983. doi:10.1006/mpev.2001.0983 [DOI] [PubMed] [Google Scholar]
- Martiny J.G.H, et al. Microbial biogeography: putting microbes on the map. Nat. Rev. Microbiol. 2006;4:102–112. doi: 10.1038/nrmicro1341. doi:10.1038/nrmicro1341 [DOI] [PubMed] [Google Scholar]
- May R.M. How many species are there on earth? Science. 1988;241:179–181. doi: 10.1126/science.241.4872.1441. [DOI] [PubMed] [Google Scholar]
- Mayr E. Two empires or three? Proc. Natl Acad. Sci. USA. 1998;95:9720–9723. doi: 10.1073/pnas.95.17.9720. doi:10.1073/pnas.95.17.9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N.A, Munson M.A, Baumann P, Ishikawa H. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc. R. Soc. B. 1993;253:167–171. [Google Scholar]
- Nesbo C.L, Dlutek M, Doolittle W.F. Recombination in Thermotoga: implications for species concepts and biogeography. Genetics. 2006;172:759–769. doi: 10.1534/genetics.105.049312. doi:10.1534/genetics.105.049312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke R.T, Ramsing N.B, Bateson M.M, Ward D.M. Geographical isolation in hot spring. Environ. Microbiol. 2003;5:650–659. doi: 10.1046/j.1462-2920.2003.00460.x. doi:10.1046/j.1462-2920.2003.00460.x [DOI] [PubMed] [Google Scholar]
- Rocap G, et al. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature. 2003;424:1042–1046. doi: 10.1038/nature01947. doi:10.1038/nature01947 [DOI] [PubMed] [Google Scholar]
- Rosselló-Mora R, Amann R. The species concept for prokaryotes. FEMS Microbiol. Rev. 2001;25:39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x. doi:10.1016/S0168-6445(00)00040-1 [DOI] [PubMed] [Google Scholar]
- Skerman V.B.D, McGowan V, Sneath P.H.A. Approved lists of bacterial names. Int. J. Syst. Bacteriol. 1980;30:225–420. [Google Scholar]
- Sneath P.H.A, editor. International code of nomenclature of bacteria (bacteriological code 1990 revision) American Society of Microbiology; Washington, DC: 1992. [PubMed] [Google Scholar]
- Stackebrandt E, Goebel B.M. Taxonomic note: a place for DNA–DNA reassociation and 16S rDNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 1994;44:846–849. [Google Scholar]
- Stackebrandt E, et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002;52:1043–1047. doi: 10.1099/00207713-52-3-1043. doi:10.1099/ijs.0.02360-0 [DOI] [PubMed] [Google Scholar]
- Staley J.T. Biodiversity: are microbial species threatened? Curr. Opin. Biotechnol. 1997;8:340–345. doi: 10.1016/s0958-1669(97)80014-6. doi:10.1016/S0958-1669(97)80014-6 [DOI] [PubMed] [Google Scholar]
- Staley J.T. Bacterial biodiversity: a time for place. ASM News. 1999;10:1–6. [Google Scholar]
- Staley J.T. A microbiological perspective of biodiversity. In: Staley J.T, Reysenbach A.-L, editors. Biodiversity of microbial life: foundation of Earth's biosphere. Wiley; New York, NY: 2002. pp. 3–23. [Google Scholar]
- Staley J.T. Speciation and bacterial phylospecies. In: Bull A.T, editor. Microbial diversity and bioprospecting. ASM Press; Washington, DC: 2004. pp. 40–48. [Google Scholar]
- Staley J.T, Gosink J.J. Poles apart: biodiversity and biogeography of polar sea ice bacteria. Annu. Rev. Microbiol. 1999;53:189–215. doi: 10.1146/annurev.micro.53.1.189. doi:10.1146/annurev.micro.53.1.189 [DOI] [PubMed] [Google Scholar]
- Staley J.T, Konopka A.E. Measurement of in situ activities of heterotrophic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. doi:10.1146/annurev.mi.39.100185.001541 [DOI] [PubMed] [Google Scholar]
- Thomas C.M, Nielsen K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005;3:711–721. doi: 10.1038/nrmicro1234. doi:10.1038/nrmicro1234 [DOI] [PubMed] [Google Scholar]
- Torsvik V, Goksoyr J, Daae F.L. High diversity in DNA of soil bacteria. Appl. Environ. Microbiol. 1990;56:782–787. doi: 10.1128/aem.56.3.782-787.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursing J.B, Rosselló-Mora R, Garcia-Valdes E, Lalucat J. Taxonomic note: a pragmatic approach to the nomenclature of phenotypically similar genomic groups. Int. J. Syst. Evol. Microbiol. 1995;45:604. [Google Scholar]
- Ward D.M. A natural species concept for prokaryotes. Curr. Opin. Microbiol. 1998;1:271–277. doi: 10.1016/s1369-5274(98)80029-5. doi:10.1016/S1369-5274(98)80029-5 [DOI] [PubMed] [Google Scholar]
- Wayne L.G, et al. Report of the ad hoc committee on reconcilliation of approaches to bacterial systematics. Int. J. Syst. Bacteriol. 1987;37:463–464. [Google Scholar]
- Whitaker R.J. Allopatric origins of microbial species. Phil. Trans. R. Soc. B. 2006;361:1975–1984. doi: 10.1098/rstb.2006.1927. doi:10.1098/rstb.2006.1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker R.J, Grogan D.W, Taylor J.W. Geographic barriers isolate endemic populations of hyperthermophilic Archaea. Science. 2003;301:976–978. doi: 10.1126/science.1086909. doi:10.1126/science.1086909 [DOI] [PubMed] [Google Scholar]
- Woese C.R, Kandler O, Wheelis M.C. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria and Eucarya. Proc. Natl Acad. Sci. USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. doi:10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, Pauling L. Molecules as documents of evolutionary history. J. Theor. Biol. 1965;8:357–366. doi: 10.1016/0022-5193(65)90083-4. doi:10.1016/0022-5193(65)90083-4 [DOI] [PubMed] [Google Scholar]