

Abstract
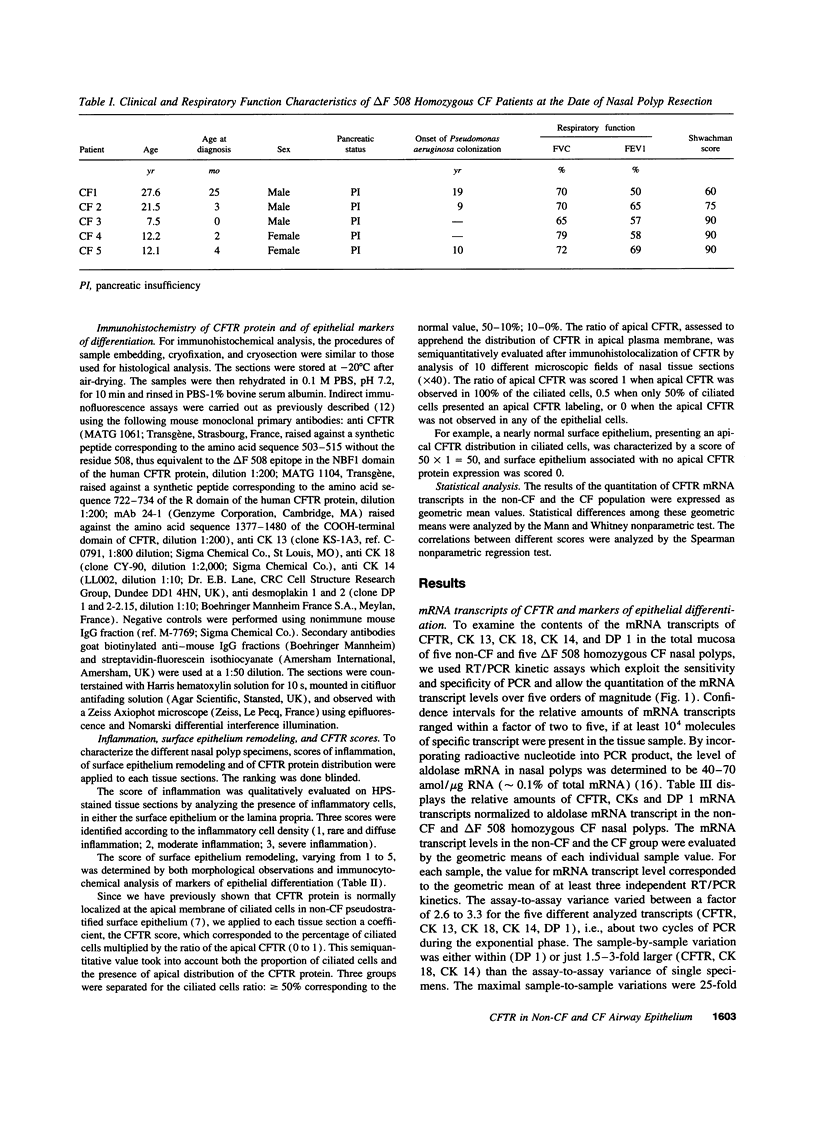
Human nasal polyps from non-CF and delta F 508 homozygous CF patients were used to compare the expression of CFTR and markers epithelial differentiation, such as cytokeratins (CK) and desmoplakins (DP), at the transcriptional and translational levels. mRNA expression was assessed by semiquantitative RT/PCR kinetic assays while the expression and distribution of proteins were evaluated by immunofluorescence analysis. In parallel, for each nasal tissue specimen, the importance of surface epithelium remodeling and inflammation was estimated after histological observations. Our results show that the steady-state levels of CFTR, CK13, CK18, CK18, CK14, or DP 1 mRNA transcripts in delta F 508 CF nasal polyps were not significantly different from those of non-CF tissues. A variability in the CFTR mRNA transcript level and in the pattern of CFTR immunolabeling has been observed between the different tissue samples. However, no relationship was found between the level of CFTR mRNA transcripts and the CFTR protein expression and distribution, either in the non-CF or in the CF group. The histological observations of non-CF and CF nasal polyp tissue indicated that the huge variations in the expression and distribution of the CFTR protein were associated with the variations in the degree of surface epithelium remodeling and inflammation in the lamina propria. A surface epithelium, showing a slight basal cell hyperplasia phenotype associated with diffuse inflammation, was mainly characterized by a CFTR protein distribution at the apex of ciliated cells in both non-CF and CF specimens. In contrast, in a remodeled surface epithelium associated with severe inflammation, CFTR protein presented either a diffuse distribution in the cytoplasm of ciliated cells, or was absent. These results suggest that abnormal expression and distribution of the CFTR protein of CF airways is not only caused by CFTR mutations. Airway surface epithelium remodeling and inflammation could play a critical role in the posttranscriptional and/or the posttranslational regulation of the CFTR protein expression in non-CF and CF airways.
Full text
PDF








Images in this article






Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Birrer P., McElvaney N. G., Rüdeberg A., Sommer C. W., Liechti-Gallati S., Kraemer R., Hubbard R., Crystal R. G. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994 Jul;150(1):207–213. doi: 10.1164/ajrccm.150.1.7912987. [DOI] [PubMed] [Google Scholar]
- Bremer S., Hoof T., Wilke M., Busche R., Scholte B., Riordan J. R., Maass G., Tümmler B. Quantitative expression patterns of multidrug-resistance P-glycoprotein (MDR1) and differentially spliced cystic-fibrosis transmembrane-conductance regulator mRNA transcripts in human epithelia. Eur J Biochem. 1992 May 15;206(1):137–149. doi: 10.1111/j.1432-1033.1992.tb16911.x. [DOI] [PubMed] [Google Scholar]
- Breuer W., Glickstein H., Kartner N., Riordan J. R., Ausiello D. A., Cabantchik I. Z. Protein kinase C mediates down-regulation of cystic fibrosis transmembrane conductance regulator levels in epithelial cells. J Biol Chem. 1993 Jul 5;268(19):13935–13939. [PubMed] [Google Scholar]
- Brezillon S., Dupuit F., Hinnrasky J., Marchand V., Kälin N., Tümmler B., Puchelle E. Decreased expression of the CFTR protein in remodeled human nasal epithelium from non-cystic fibrosis patients. Lab Invest. 1995 Feb;72(2):191–200. [PubMed] [Google Scholar]
- Champigny G., Imler J. L., Puchelle E., Dalemans W., Gribkoff V., Hinnrasky J., Dott K., Barbry P., Pavirani A., Lazdunski M. A change in gating mode leading to increased intrinsic Cl- channel activity compensates for defective processing in a cystic fibrosis mutant corresponding to a mild form of the disease. EMBO J. 1995 Jun 1;14(11):2417–2423. doi: 10.1002/j.1460-2075.1995.tb07239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990 Nov 16;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Chirgwin J. M., Przybyla A. E., MacDonald R. J., Rutter W. J. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979 Nov 27;18(24):5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Dalemans W., Barbry P., Champigny G., Jallat S., Dott K., Dreyer D., Crystal R. G., Pavirani A., Lecocq J. P., Lazdunski M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature. 1991 Dec 19;354(6354):526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- Dalemans W., Hinnrasky J., Slos P., Dreyer D., Fuchey C., Pavirani A., Puchelle E. Immunocytochemical analysis reveals differences between the subcellular localization of normal and delta Phe508 recombinant cystic fibrosis transmembrane conductance regulator. Exp Cell Res. 1992 Jul;201(1):235–240. doi: 10.1016/0014-4827(92)90368-i. [DOI] [PubMed] [Google Scholar]
- Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992 Aug 27;358(6389):761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Denning G. M., Ostedgaard L. S., Cheng S. H., Smith A. E., Welsh M. J. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J Clin Invest. 1992 Jan;89(1):339–349. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning G. M., Ostedgaard L. S., Welsh M. J. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol. 1992 Aug;118(3):551–559. doi: 10.1083/jcb.118.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drumm M. L., Wilkinson D. J., Smit L. S., Worrell R. T., Strong T. V., Frizzell R. A., Dawson D. C., Collins F. S. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991 Dec 20;254(5039):1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Engelhardt J. F., Yankaskas J. R., Ernst S. A., Yang Y., Marino C. R., Boucher R. C., Cohn J. A., Wilson J. M. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet. 1992 Nov;2(3):240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- Gaillard D., Ruocco S., Lallemand A., Dalemans W., Hinnrasky J., Puchelle E. Immunohistochemical localization of cystic fibrosis transmembrane conductance regulator in human fetal airway and digestive mucosa. Pediatr Res. 1994 Aug;36(2):137–143. doi: 10.1203/00006450-199408000-00002. [DOI] [PubMed] [Google Scholar]
- Hiraide F., Kakoi H. Histochemical study on innervation of glands and blood vessels in nasal polyps. Acta Otolaryngol Suppl. 1986;430:5–11. [PubMed] [Google Scholar]
- Hoof T., Riordan J. R., Tümmler B. Quantitation of mRNA by the kinetic polymerase chain reaction assay: a tool for monitoring P-glycoprotein gene expression. Anal Biochem. 1991 Jul;196(1):161–169. doi: 10.1016/0003-2697(91)90133-e. [DOI] [PubMed] [Google Scholar]
- Kanai N., Denburg J., Jordana M., Dolovich J. Nasal polyp inflammation. Effect of topical nasal steroid. Am J Respir Crit Care Med. 1994 Oct;150(4):1094–1100. doi: 10.1164/ajrccm.150.4.7921442. [DOI] [PubMed] [Google Scholar]
- Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat Genet. 1992 Aug;1(5):321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Li C., Ramjeesingh M., Reyes E., Jensen T., Chang X., Rommens J. M., Bear C. E. The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet. 1993 Apr;3(4):311–316. doi: 10.1038/ng0493-311. [DOI] [PubMed] [Google Scholar]
- Li M., McCann J. D., Liedtke C. M., Nairn A. C., Greengard P., Welsh M. J. Cyclic AMP-dependent protein kinase opens chloride channels in normal but not cystic fibrosis airway epithelium. Nature. 1988 Jan 28;331(6154):358–360. doi: 10.1038/331358a0. [DOI] [PubMed] [Google Scholar]
- Lukacs G. L., Chang X. B., Bear C., Kartner N., Mohamed A., Riordan J. R., Grinstein S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993 Oct 15;268(29):21592–21598. [PubMed] [Google Scholar]
- Massion P. P., Inoue H., Richman-Eisenstat J., Grunberger D., Jorens P. G., Housset B., Pittet J. F., Wiener-Kronish J. P., Nadel J. A. Novel Pseudomonas product stimulates interleukin-8 production in airway epithelial cells in vitro. J Clin Invest. 1994 Jan;93(1):26–32. doi: 10.1172/JCI116954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C., Guggino W. B., Montrose M. H. Cellular differentiation regulates expression of Cl- transport and cystic fibrosis transmembrane conductance regulator mRNA in human intestinal cells. J Biol Chem. 1991 Mar 5;266(7):4495–4499. [PubMed] [Google Scholar]
- Morris A. P., Cunningham S. A., Benos D. J., Frizzell R. A. Cellular differentiation is required for cAMP but not Ca(2+)-dependent Cl- secretion in colonic epithelial cells expressing high levels of cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1992 Mar 15;267(8):5575–5583. [PubMed] [Google Scholar]
- Morris A. P., Cunningham S. A., Tousson A., Benos D. J., Frizzell R. A. Polarization-dependent apical membrane CFTR targeting underlies cAMP-stimulated Cl- secretion in epithelial cells. Am J Physiol. 1994 Jan;266(1 Pt 1):C254–C268. doi: 10.1152/ajpcell.1994.266.1.C254. [DOI] [PubMed] [Google Scholar]
- Nakamura H., Yoshimura K., Bajocchi G., Trapnell B. C., Pavirani A., Crystal R. G. Tumor necrosis factor modulation of expression of the cystic fibrosis transmembrane conductance regulator gene. FEBS Lett. 1992 Dec 21;314(3):366–370. doi: 10.1016/0014-5793(92)81507-i. [DOI] [PubMed] [Google Scholar]
- Puchelle E., Gaillard D., Ploton D., Hinnrasky J., Fuchey C., Boutterin M. C., Jacquot J., Dreyer D., Pavirani A., Dalemans W. Differential localization of the cystic fibrosis transmembrane conductance regulator in normal and cystic fibrosis airway epithelium. Am J Respir Cell Mol Biol. 1992 Nov;7(5):485–491. doi: 10.1165/ajrcmb/7.5.485. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 Sep 8;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rommens J. M., Iannuzzi M. C., Kerem B., Drumm M. L., Melmer G., Dean M., Rozmahel R., Cole J. L., Kennedy D., Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 Sep 8;245(4922):1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Rommens J., Kerem B. S., Greer W., Chang P., Tsui L. C., Ray P. Rapid nonradioactive detection of the major cystic fibrosis mutation. Am J Hum Genet. 1990 Feb;46(2):395–396. [PMC free article] [PubMed] [Google Scholar]
- Sood R., Bear C., Auerbach W., Reyes E., Jensen T., Kartner N., Riordan J. R., Buchwald M. Regulation of CFTR expression and function during differentiation of intestinal epithelial cells. EMBO J. 1992 Jul;11(7):2487–2494. doi: 10.1002/j.1460-2075.1992.tb05313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tizzano E. F., Chitayat D., Buchwald M. Cell-specific localization of CFTR mRNA shows developmentally regulated expression in human fetal tissues. Hum Mol Genet. 1993 Mar;2(3):219–224. doi: 10.1093/hmg/2.3.219. [DOI] [PubMed] [Google Scholar]
- Tran-Paterson R., Davin D., Krauss R. D., Rado T. A., Miller D. M. Expression and regulation of the cystic fibrosis gene during rat liver regeneration. Am J Physiol. 1992 Jul;263(1 Pt 1):C55–C60. doi: 10.1152/ajpcell.1992.263.1.C55. [DOI] [PubMed] [Google Scholar]
- Trapnell B. C., Chu C. S., Paakko P. K., Banks T. C., Yoshimura K., Ferrans V. J., Chernick M. S., Crystal R. G. Expression of the cystic fibrosis transmembrane conductance regulator gene in the respiratory tract of normal individuals and individuals with cystic fibrosis. Proc Natl Acad Sci U S A. 1991 Aug 1;88(15):6565–6569. doi: 10.1073/pnas.88.15.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell B. C., Zeitlin P. L., Chu C. S., Yoshimura K., Nakamura H., Guggino W. B., Bargon J., Banks T. C., Dalemans W., Pavirani A. Down-regulation of cystic fibrosis gene mRNA transcript levels and induction of the cystic fibrosis chloride secretory phenotype in epithelial cells by phorbol ester. J Biol Chem. 1991 Jun 5;266(16):10319–10323. [PubMed] [Google Scholar]
- Veeze H. J., Halley D. J., Bijman J., de Jongste J. C., de Jonge H. R., Sinaasappel M. Determinants of mild clinical symptoms in cystic fibrosis patients. Residual chloride secretion measured in rectal biopsies in relation to the genotype. J Clin Invest. 1994 Feb;93(2):461–466. doi: 10.1172/JCI116993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C. L., Kopito R. R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994 Oct 14;269(41):25710–25718. [PubMed] [Google Scholar]
- Yang Y., Devor D. C., Engelhardt J. F., Ernst S. A., Strong T. V., Collins F. S., Cohn J. A., Frizzell R. A., Wilson J. M. Molecular basis of defective anion transport in L cells expressing recombinant forms of CFTR. Hum Mol Genet. 1993 Aug;2(8):1253–1261. doi: 10.1093/hmg/2.8.1253. [DOI] [PubMed] [Google Scholar]
- Yoshimura K., Nakamura H., Trapnell B. C., Dalemans W., Pavirani A., Lecocq J. P., Crystal R. G. The cystic fibrosis gene has a "housekeeping"-type promoter and is expressed at low levels in cells of epithelial origin. J Biol Chem. 1991 May 15;266(14):9140–9144. [PubMed] [Google Scholar]