

Abstract
Domains are basic evolutionary units of proteins and most proteins have more than one domain. Advances in domain modeling and collection are making it possible to annotate a large fraction of known protein sequences by a linear ordering of their domains, yielding their architecture. Protein domain architectures link evolutionarily related proteins and underscore their shared functions. Here, we attempt to better understand this association by identifying the evolutionary pathways by which extant architectures may have evolved. We propose a model of evolution in which architectures arise through rearrangements of inferred precursor architectures and acquisition of new domains. These pathways are ranked using a parsimony principle, whereby scenarios requiring the fewest number of independent recombination events, namely fission and fusion operations, are assumed to be more likely. Using a data set of domain architectures present in 159 proteomes that represent all three major branches of the tree of life allows us to estimate the history of over 85% of all architectures in the sequence database. We find that the distribution of rearrangement classes is robust with respect to alternative parsimony rules for inferring the presence of precursor architectures in ancestral species. Analyzing the most parsimonious pathways, we find 87% of architectures to gain complexity over time through simple changes, among which fusion events account for 5.6 times as many architectures as fission. Our results may be used to compute domain architecture similarities, for example, based on the number of historical recombination events separating them. Domain architecture “neighbors” identified in this way may lead to new insights about the evolution of protein function.
Keywords: protein domain architecture, multi-domain proteins, domain recombination, domain evolution, protein family
Introduction
Proteins are composed of evolutionarily conserved units called domains, often corresponding to subunits of the 3-D structure of a protein, that have distinct molecular function and structure.1 The sequential order of domains in a protein sequence is known as its protein domain architecture. Architectures are useful for classifying evolutionarily related proteins, in particular to detect evolutionarily distant homologs based on shared domains rather than on pairwise sequence similarity. Large collections of protein domains and families of domains have been gathered into databases such as the CDD,2,3 SCOP,4 Pfam,5 SMART,6 COG,7 and TIGRFAMs.8 Search algorithms such as RPS-BLAST2 and HMMer9 use these domain definitions to identify conserved domains in protein sequences while domain architectures can be identified using the CDART10 and Superfamily11 algorithms, for example.
Novel yet specific combinations of domains are essential for creating diversity in proteins. Two-thirds of all prokaryotic proteins and 80% of eukaryotic proteins have more than one domain.12–14 Many earlier studies analyzed domain combinations to better understand how domains work together to promote the function of a protein.15–17 They established that domains combine under selection rather than by chance.18,19 Specifically, some combinations appear more frequently than others, and the distribution of the number of domain neighbors follows a power law.20 Analysis of protein domain pairs showed that pairs of domains that are close neighbors on a protein sequence tend to appear in the same order in different proteins and their relative spatial orientation might be as well conserved.20 Due to conservation, the domain content of whole genomes can be used to partially reconstruct their phylogeny.21
To study protein evolution, we will consider domain architectures, which unlike domain combinations fully specify the sequential organization of conserved units in entire proteins. Changes to architectures indicate divergence of protein sequence and structure that may affect the function of the protein. Domain architectures of contemporary proteins emerged over time as their respective genes underwent such events as fusion and fission, by which two genes are combined into one or a gene is split into two or more separate genes.17 Proteins that are related through gene fusions and fissions include Rosetta stone proteins and their split forms; their relationship has been used to infer protein function and physical protein–protein interactions.22–26 Fusion and fission have been shown to play a major role in the evolution of multi-domain bacterial proteins.27 Further, it has been shown that in multi-domain proteins, fusions occur more frequently than fissions.28,29 It is also believed that proteins with the same domain architecture are close homologs30 while more evolutionarily distant proteins may differ in their domain architectures. Therefore, the comparison of protein domain architectures can be used for inferring evolutionary relationships between different proteins and protein families.28,31 Recently, a graph theoretical approach based on Dollo parsimony was used to explore the evolution of multi-domain proteins.32
Here, we identify the pathways by which known domain architectures may have evolved. These pathways describe the rearrangements leading to each architecture and their chronological order. In order to develop a large-scale, comprehensive model, we use a data set of all known domain architectures from 159 complete proteomes, representing over 85% of architectures in the sequence database. We consider alternative recombination histories under the constraint that precursor architectures must be inferred to be present in the ancestral species of organisms whose genomes contain a given architecture. These pathways are then ranked using a parsimony principle, by which rearrangements that require the fewest number of independent fission and fusion operations are assumed to be most likely. We find the proposed pathways to be consistent with previous studies of domain recombination, which focused primarily on statistics of co-occurrences of different types of domains with one another, but the inferred most likely pathways differ because of the taxonomic constraints.
Analyzing these proposed pathways, we find evidence that architectures gain complexity over time through simple changes. While showing that fusions and fissions play a large role in the development of new architectures, we also take into account new domains and complex rearrangements to accommodate the diversity and evolutionary distance of architectures. We find that single-domain architectures usually appear as new domains rather than through the breakdown of multi-domain proteins, and the majority of multi-domain architectures evolve through only fusions or only fissions. Among the most parsimonious pathways, 5.6 times as many architectures arise from fusion as from fission. We validate our results by showing that the selection of rearrangement classes is robust over various rules for inferring the presence of precursor architectures and that the most likely pathways require a small number of rearrangement operations. Finally, we demonstrate that evolution only realizes a few of the many possible ways by which each architecture could have evolved.
Results and Discussion
Searching for rearrangements
Each new architecture may be formed by one or more combinations of existing architectures and new domains. We infer presence or absence of domains in ancestor nodes representing non-extant species using maximum parsimony (MP) as described in Materials and Methods. More precisely, referring to nodes from the NCBI Taxonomy tree, an architecture is presumed to be gained at node N if it is present at N but not its parent. At node N, we call it a new architecture. Every architecture can be gained in at least one node. We postulate that each new architecture evolved from existing architectures, that is, architectures present in the parent node, called parent architectures, when all of its domains can be accounted for among parent architectures. If the new architecture contains a domain that is not seen in any parent architecture, new domains necessarily play a role in the evolutionary event. Rearrangements of architectures and new domains are carried out through fission and fusion operations. In accordance with parsimony, we highlight the rearrangements with lowest cost, which we define to be the total number of fission and fusion operations. We denote these lowest cost series of fusions and fissions as putative rearrangement solutions. A dynamic programming procedure, described in Materials and Methods, is used to identify rearrangement solutions with lowest cost.
Our model classifies potential solutions into rearrangement classes based on their use of fission and fusion operations (Table 1). Simple rearrangement classes include New Domain, Fission, and Fusion. These correspond to new architectures with a single novel domain, and rearrangements that use only fission operations and only fusion operations, respectively. We also distinguish rearrangements that involve new domains. The Fusion class is partitioned into three sub-cases to indicate whether each architecture is constructed from only new domains, parent architectures, or both. In contrast with the simple rearrangement classes, complex rearrangement classes require both fusion and fission operations. They include Deletion and Insertion, which correspond to gene deletion and gene insertion, both of which are known to occur in nature. Other combinations of fusion and fission operations are possible as well. When we do not identify a solution in any of the above classes, we compute more general complex solutions that we label class Other. Complicated rearrangements from this last class are less likely to be correct.
Table 1.
Rearrangement classes for architecture creation
| Rearrangement class | Description | Cost | Examples |
|---|---|---|---|
| Simple | |||
| New domain | Single new domain. No operations | 0 | →d1 |
| Fission | Parent architecture is split to produce new architecture | 1–2 | ABC → AB |
| Fusion (3 subclasses) | (a) Fusion of new domains | ≥1 | d1+d2 → d1d2 |
| (b) Fusion of parent architectures | A+BC → ABC | ||
| (c) Fusion of parent architecture(s) and new domain(s) | A+d1 → Ad1 | ||
| Complex | |||
| Deletion | New architecture is non-consecutive sub-list of parent architecture | ≥3 | ABC → AC |
| Insertion (2 subclasses) | (a) New domain is inserted into parent architecture | 3 | AC+d1 → Ad1C |
| (b) One parent architecture is inserted into another | AC+B → ABC | ||
| Other | Other fusion–fission combination | ≥2 | A+BC → AB |
Cost is the total number of fusion and fission operations required for each class. The examples denote architectures as letters A, B, C and new domains as d1 and d2.
Example of evolutionary pathway
We illustrate one evolutionary pathway computed by our method by identifying the fusion and fission rearrangements that may have produced the architectures containing the C2, WW, or HECTc domains (Figure 1). These domains are present in several eukaryotic protein families. In particular, the C2-WW-HECTc architecture characterizes over 160 eukaryotic proteins in Entrez including Smad ubiquitination regulatory factor proteins (Smurf1 and Smurf2) and E3 ubiquitin protein ligase. Using a eukaryotic species tree, we pinpoint the nodes at which each architecture is believed to originate and the rearrangements that take place.
Figure 1.
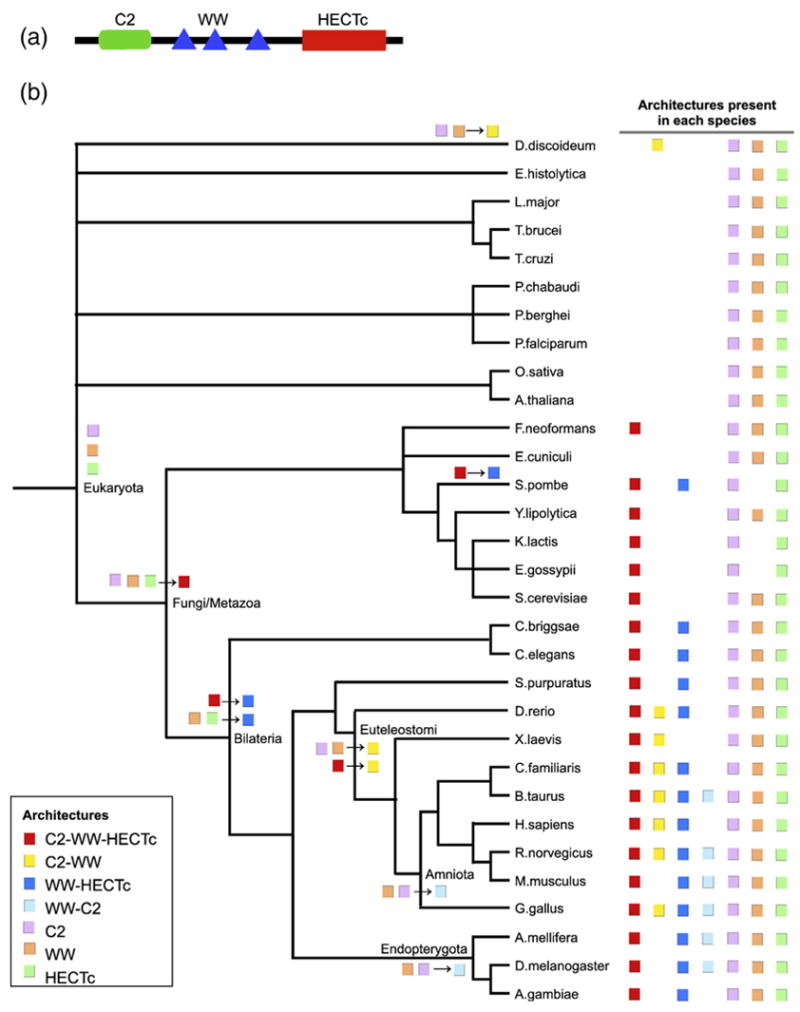
C2-WW-HECTc architecture. (a) Schematic architecture diagram from CDART. (b) Rearrangement tree for architectures containing the C2, WW, or HECTc domains and no other domains. Architectures shown here include C2-WW-HECTc (red), C2-WW (yellow), WW-HECTc (blue), WW-C2 (light blue), C2 (purple), WW (orange), and HECTc (green). The presence of each architecture in each species is indicated at the right. Each line of boxes on the tree corresponds to a potential rearrangement event that produces a new architecture at the closest labeled node. C2, WW, and HECTc single-domain architectures appear in Eukaryota as rearrangement class New Domain. C2-WW-HECTc appears at the Fungi/Metazoa node as a fusion of three architectures. The emergence of WW-HECTc and C2-WW can be attributed to the fission of C2-WW-HECTc or fusion of the respective one-domain architectures; the potential solutions differ for each of their new occurrences. The C2 and WW domains also appear in the other order, as WW-C2 architecture, which comes about through the respective fusions.
Analysis
Analyzing our proposed pathways, we find that 87% of architectures most likely evolve by simple rearrangements. The cases are distributed among a large number of single-domain new architectures, fusion events, and relatively few fission events, supporting the hypothesis that architectures gain complexity over time. We tally the number of architectures attributed to each rearrangement class using a few different measures (Table 2). The distribution of architectures into rearrangement classes is stable under these measures. We define the majority class for each architecture to be the class (from Table 1) of the rearrangement solutions proposed most frequently for that architecture, i.e. for the largest number of new occurrences of that architecture (column C of Table 2). If the most frequent rearrangement solutions come from more than one rearrangement class, the architecture’s majority class is designated Tie. We will refer to this measure in the analysis below.
Table 2.
The number of architectures described by each rearrangement class (Table 1), under three natural measures
| Rearrangement class | (A) Allsolution | (B) Common solutions | (C) Majority class |
|---|---|---|---|
| Simple | |||
| New domain | 4646 | 4483 | 4387 |
| Fission | 1991 | 1387 | 875 |
| Fusion of new domains | 730 | 586 | 489 |
| Fusion of parent architectures | 3734 | 3409 | 2825 |
| Fusion of both | 2266 | 1921 | 1610 |
| Complex | |||
| Deletion | 108 | 80 | 55 |
| Insertion of new domain | 27 | 19 | 13 |
| Insertion of parent architecture | 61 | 48 | 30 |
| Other | 914 | 690 | 473 |
| Tie | 895 | ||
| Total | 14,477 | 12,623 | 11,652 |
A, The number of architectures with any solution of that class. B, The number of architectures whose most common solutions, i.e. the solutions that were proposed for the largest number of new occurrences, include that class. C, The number of architectures whose most common solution(s) are of exactly that rearrangement class. In A and B, an architecture can be attributed to more than one class. In C, architectures whose most common solutions describe more than one class are labeled Tie.
Breaking down the number of architectures in each majority class by the number of domains in each architecture shows the relationship between type of rearrangement and number of domains (Table 3). Nearly all single-domain architectures, which comprise over 42% of all architectures, appear as a new one-domain architecture rather than the fission product of a multi-domain architecture. This accounts for the large number of architectures with case New Domain. Few multi-domain architectures contain all new domains, showing that multi-domain architectures usually evolve from existing architectures. Multi-domain architectures overwhelmingly develop through fusions. Overall, 5.6 times as many architectures come about through fusion (4924 total) than through fission. In contrast with recent work by Kummerfeld & Teichmann who trace gene fusions and fissions only for architectures that are known to exist in fused and split forms,29 we survey possible evolutionary events for every known architecture. Under our model, these events may involve fusion, fission, both, or neither of these operations. While our fusion-to-fission ratio and that by Kummerfeld & Teichmann are not directly comparable, both corroborate that fusion is biologically preferable over fission. Indeed, it is possible that protein domains emerged in evolution from the fusion of individually optimized shorter modules; this recombination provided an efficient way to gain properties not present in the individual modules.33,34 A similar trend apparently occurs in the evolution of multi-domain proteins. In this case, genes encoding for different proteins which participate in the same biological process and physically interact can be fused into one gene to optimize their co-expression and co-regulation.23 Less is known about the genetic mechanisms of fission. It has been proposed that fission can be attributed to point mutations that introduce new start and stop codons, evidenced by more frequent loss of domains at the sequence termini,35 or to a high rate of frameshift sequencing errors that favors independent coding of the components in a protein complex, evidenced by a higher gene fission rate in thermophiles.28
Table 3.
Breakdown of architectures for each majority class (column C of Table 2) by number of domains in each architecture
| Domains in architecture | 1 | 2 | 3 | 4 | 5 | 6+ | Total |
|---|---|---|---|---|---|---|---|
| Simple | |||||||
| New domain | 4387 | 0 | 0 | 0 | 0 | 0 | 4387 |
| Fission | 474 | 219 | 103 | 45 | 26 | 8 | 875 |
| Fusion of new domains | 0 | 425 | 49 | 12 | 2 | 1 | 489 |
| Fusion of parent architectures | 0 | 1887 | 606 | 213 | 80 | 39 | 2825 |
| Fusion of both | 0 | 1071 | 370 | 116 | 36 | 17 | 1610 |
| Complex | |||||||
| Deletion | 0 | 24 | 17 | 9 | 2 | 3 | 55 |
| Insertion of new domain | 0 | 0 | 9 | 2 | 2 | 0 | 13 |
| Insertion of parent architecture | 0 | 0 | 17 | 7 | 4 | 2 | 30 |
| Other | 0 | 215 | 130 | 70 | 36 | 22 | 473 |
| Tie | 96 | 457 | 220 | 75 | 28 | 19 | 895 |
| Total | 4957 | 4298 | 1521 | 549 | 216 | 111 | |
We also identify a small number of complex rearrangements, which may occur in reality. Deletions and insertions combined account for less than 1% of architectures, a negligible fraction, yet when they are used the suggested rearrangements often appear to be natural solutions. Figure 2 illustrates a proposed insertion of the WW single-domain architecture into the CH-RasGAP-RasGAP_C architecture, which yields proteins that are a variant of the IQGAP3 family. The tenability of this rearrangement is supported by the WW domain’s presence in a large variety of proteins and its general protein-binding function, multiple occurrences of this architecture in our data set, and scarcity of other arrangements between these domains that might suggest alternative rearrangement possibilities. The relatively small number of complex cases strengthens the case that in general, architecture evolution proceeds with simple steps.
Figure 2.
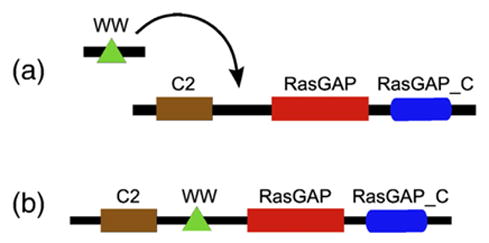
(a) Insertion rearrangement of WW into architecture CH-RasGAP-RasGAP_C takes place at the Amniota ancient species to produce (b) CH-WW-RasGAP-RasGAP_C. WW is present in most Eukaryotes, CH-RasGAP-RasGAP_C in many Fungi/Metazoa, and CH-WW-RasGAP-RasGAP_C in exactly four of the six Amniota species in our data set.
To probe the robustness of our method against alternative rules for inferring the presence of architectures in ancestral species, we compare our MP setting with other intuitive parsimony schemes: Dollo parsimony, a commonly assumed model which assumes that a feature may be gained only once in evolution and may be lost in multiple species; and variations of MP that allow a parent node to be labeled present for an architecture with more or fewer children containing that architecture (see Supplementary Data). Except for Dollo parsimony, the alternative parsimony rules do not substantially affect the assignment of architectures to nodes or the relative proportions of rearrangement classes among the solutions. Using Dollo parsimony, fusion (5678 architectures) is seen 10.8 times as often as fission (525 architectures) and 93% of architectures are described best by simple rearrangement classes. Because Dollo assumes that every architecture originates at the earliest possible node, this protocol assigns more architectures to be present in the most ancient nodes. This assignment results in a larger number of new architectures attributed to fusion of parent architectures only or new domains only and a reduction in all other types of rearrangements.
Our model is substantiated by the low cost of most solutions and the small number of solutions detected for each architecture. Low cost signifies that our proposed evolutionary events can be achieved through simple evolutionary mechanisms. We find that 95.9% of architectures have cost at most three (Figure 3). Additionally, 98.8% of Fusion solutions and 58.2% of Complex-Other solutions, whose cost varies by the number of domains, have cost at most three. Many complex solutions may be attributed to incomplete domain annotation or other sources of error. Nevertheless, the low cost of several complex solutions, including Deletion and Insertion cases plus complex rearrangements in the Other category that consist of a fission operation and one or two fusion operations, suggests that rearrangements that require both fission and fusion operations are realistic. The small number of proposed rearrangements for many architectures minimizes ambiguity in identifying optimal solutions, thus increasing confidence in them. Our method consistently identifies a small number of solutions for each architecture even though MP allows the architecture to be gained in multiple nodes with presumably different sets of parent architectures. A total of 89.2% of architectures have one solution proposed for the largest number of new occurrences of that architecture and 99.1% at most three solutions (Figure 4). In the next section, we show that the small number of solutions cannot be fully attributed to a small search space of feasible solutions.
Figure 3.
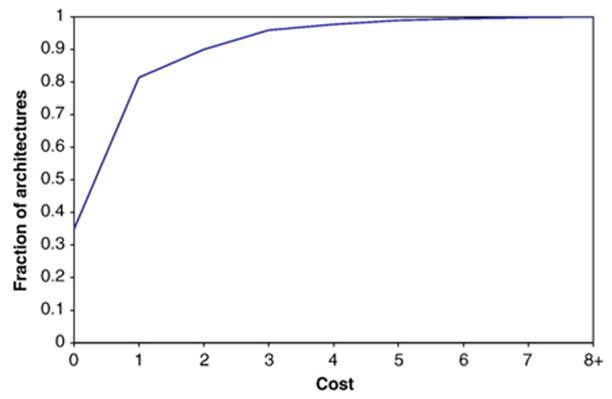
Fraction of architectures with cost at most i.
Figure 4.
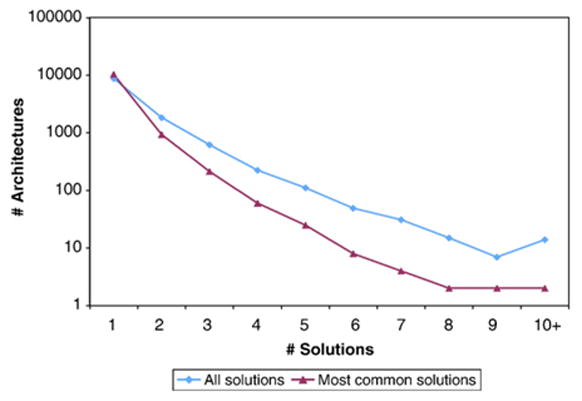
Number of architectures for every number of solutions, including all solutions (blue) and only the most-common solutions (pink).
Actual source–target network is sparse and scale-free
In identifying the most likely pathways, our protocol eliminates a large fraction of possible pathways, consequently indicating the difficulty of predicting rearrangements from domain content. We devise a source–target network model to quantify this difference. A new architecture, the target, is formed by parent architectures and new domains, its sources. We consider two graphs with nodes corresponding to architectures. The first graph describes the source–target network based on our proposed rearrangements, that is, actual sources and targets. In this graph, directed edges go from every source to each of its targets using all of our rearrangement solutions. The second graph connects all potential sources and targets, defined here to mean that the first architecture could be a source for the other through a Fusion or Fission rearrangement. In practice, any two architectures that share a domain could form a potential source–target pair but we consider only potential sources and targets for rearrangements of the two most common rearrangement classes to accurately estimate the number of likely pairs. We find that taking into account inferred ancestral architectures constrains the choice of solutions considerably. On average, for architectures included in the potential source–target network, the ratio of actual targets to potential targets is 19.1%. Further analysis of these networks reveals that only 29.8% of architectures are actual sources. Another 48.9% of architectures have potential but no actual targets. The remaining 21.3% of architectures, most of which have one domain, are unconnected nodes in either graph.
Figure 5 shows that like many other types of biological networks, the target–source network follows a power law. This extends previous analyses showing that the distribution of domain combinations is scale free for those domains that co-exist or are found to be neighbors in the same protein.20,36 The architectures with the largest number of targets are listed in Table 4. All of these architectures have one domain and are known to repeat, e.g. ANK and TPR, participate in cell signaling, or form binding sites. Some of these architectures encapsulate large or general groups of domains and proteins, e.g. ATP-binding site, which increases the frequency of their appearances.
Figure 5.
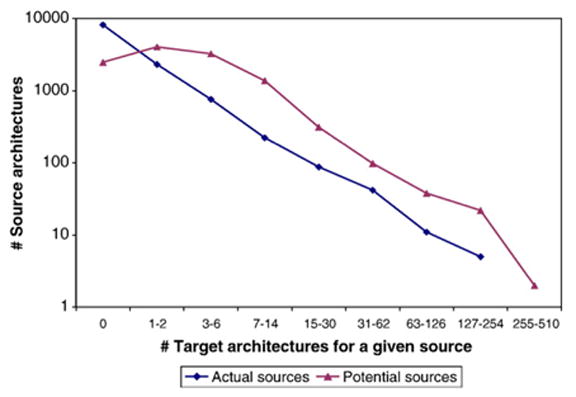
Number of source architectures plotted against the number of target architectures, using a version of a log-log graph in which the y-axis uses a logarithmic scale while the x-axis represents values grouped into bins. Bin i includes 2i targets starting with i=0 and 0 targets.
Table 4.
List of architectures that evolve into the largest number of children architectures (actual targets)
| Domain/architecture name | Targets |
|---|---|
| ATP-binding site | 260 |
| Protein kinases | 221 |
| Tar and SH3 signal transduction, tropomyosin, pre-folding, and more | 177 |
| Ankyrin | 141 |
| Tetratricopeptide repeat domain | 132 |
| PH domain | 124 |
| REC signal receiver domain | 115 |
| RING-finger domain | 112 |
| WD40 domain | 107 |
| SH3 domain | 104 |
| Signal transduction histidine kinase | 104 |
Summary
We have used taxonomic and parsimony constraints to construct recombination pathways by which present-day architectures may have evolved. These pathways describe likely rearrangements of precursor architectures and new domains as well as their chronological order. Analysis of the most likely pathways reveals that simple architecture fusions and fissions, plus the introduction of new domains over time, are sufficient to explain the evolution of 87% of architectures. In particular, most architectures appear with a new single domain or as fusions of existing architectures or new domains. Far fewer architectures originate through a fission event. These observations support the hypothesis that architectures and proteins have gained complexity over time through simple steps. Further, our proposed rearrangements are constrained to form a small fraction of all rearrangements that might be deduced from domain content. Since the accuracy and sensitivity of our results depend on the available data, our method is expected to produce more precise evolutionary pathways as additional domains are uncovered, proteomes are more fully derived from sequenced organisms, and relationships between species are better specified. Our protocol may be used to compute domain architecture similarities, for example, based on the number of historical recombination events separating them. Linking domain architecture “neighbors” identified in this way may lead to new insights about the evolution of protein function. Our results thus provide a basis for closer inspection of the evolution of particular architectures and their corresponding protein families.
Materials and Methods
Genomic and domain architecture assignment data
We compile a list of 111 bacteria, 17 archaea, and 31 eukaryotes to represent a diversity of lineages (Supplementary Data, Table 1). The list includes many contemporary organisms from the complete genomes at NCBI Entrez Genomes. To balance the lineages, we retain only one species from each bacterial genus and add other fully sequenced eukaryotes. We assume taxonomic relationships from the NCBI Taxonomy†. The selected organisms constitute the leaf nodes of the tree and their 11,652 domain architectures are taken from the NCBI CDART database‡. The procedure used to create CDART computes architectures for proteins in the NCBI non-redundant database (nr) by locating all significant matches to CDD domain profiles (e-value<0.01) and then identifying non-overlapping clusters of similar domains in each protein. This procedure allows all significant matches to be considered without discriminating against short domains. The domain definitions are imported from Pfam, SMART, and CDD and represent a wide variety of proteins, avoiding the bias towards globular and structurally determined proteins within the SCOP database.
Inferring ancestral architectures
Architectures are assigned to leaf nodes based on proteins belonging to a given organism and to internal nodes using maximum parsimony. We implement a modification of Fitch’s algorithm,37 usually defined for a binary tree, to allow nodes to have any number of children. The first step of this algorithm applies the following rule recursively, starting from the leaves, to label every internal node: if an architecture is present in more than, less than, or exactly half of labeled children, label the parent “present,” “absent,” or “unknown,” respectively. A second traversal of the tree from root to leaf removes unknown labels by assigning each node the same label as its parent. We break ties at balanced trees, i.e. trees with unknown root, by setting the root to present. The resulting labeling is most parsimonious although it may not be the only labeling that produces the fewest gains and losses.
Computing low-cost rearrangement solutions
For each new architecture we search for rearrangement solutions with lowest cost, using dynamic programming to compute lowest-cost Fusion and Other solutions. An architecture may be gained at more than one node so, to demonstrate that costs are low, we take the cost of producing each architecture to be the maximum cost over all nodes. The algorithms are straightforward (see Supplementary Data). For each new architecture we search for a solution among the simple rearrangement classes. If no simple solution is found, we look for complex solutions. There can be many complex solutions of equal cost, so we consider the number of source architectures in order to minimize the number of solutions. We first search for a Deletion solution because it requires one source. If there is none, we search for Insertion and Other solutions of lowest cost and report the solutions that use fewest parent architectures.
We include architectures with tandem repeats of a single domain by collapsing the repeats into one instance in the architecture definition. We do not compute rearrangements leading to architectures with tandem repeats of two or more different domains because these architectures likely arose through specific domain duplication events and they are present in fewer than 2% of all architectures.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Library of Medicine, and the NIH Undergraduate Scholarship Program.
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/ j.jmb.2006.11.017
Edited by M. Sternberg
References
- 1.Bork P. Mobile modules and motifs. Curr Opin Struct Biol. 1992;2:413–421. [Google Scholar]
- 2.Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucl Acids Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, et al. CDD: a conserved domain database for protein classification. Nucl Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 5.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, et al. The Pfam protein families database. Nucl Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, et al. Recent improvements to the SMART domain-based sequence annotation resource. Nucl Acids Res. 2002;30:242–244. doi: 10.1093/nar/30.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucl Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucl Acids Res. 2003;31:371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 10.Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12:1619–1623. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gough J, Chothia C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucl Acids Res. 2002;30:268–272. doi: 10.1093/nar/30.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teichmann SA, Park J, Chothia C. Structural assignments to the Mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc Natl Acad Sci USA. 1998;95:14658–14663. doi: 10.1073/pnas.95.25.14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerstein M. How representative are the known structures of the proteins in a complete genome? A comprehensive structural census. Fold Des. 1998;3:497–512. doi: 10.1016/S1359-0278(98)00066-2. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Rost B. CHOP proteins into structural domain-like fragments. Proteins: Struct Funct Bioinformatics. 2004;55:678–688. doi: 10.1002/prot.20095. [DOI] [PubMed] [Google Scholar]
- 15.Bornberg-Bauer E, Beaussart F, Kummerfeld SK, Teichmann SA, Weiner J., 3rd The evolution of domain arrangements in proteins and interaction networks. Cell Mol Life Sci. 2005;62:435–445. doi: 10.1007/s00018-004-4416-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA. Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol. 2004;14:208–216. doi: 10.1016/j.sbi.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Chothia C, Gough J, Vogel C, Teichmann SA. Evolution of the protein repertoire. Science. 2003;300:1701–1703. doi: 10.1126/science.1085371. [DOI] [PubMed] [Google Scholar]
- 18.Apic G, Huber W, Teichmann SA. Multi-domain protein families and domain pairs: comparison with known structures and a random model of domain recombination. J Struct Funct Genomics. 2003;4:67–78. doi: 10.1023/a:1026113408773. [DOI] [PubMed] [Google Scholar]
- 19.Vogel C, Teichmann SA, Pereira-Leal J. The relationship between domain duplication and recombination. J Mol Biol. 2005;346:355–365. doi: 10.1016/j.jmb.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 20.Apic G, Gough J, Teichmann SA. An insight into domain combinations. Bioinformatics. 2001;17 (suppl 1):S83–S89. doi: 10.1093/bioinformatics/17.suppl_1.s83. [DOI] [PubMed] [Google Scholar]
- 21.Yang S, Doolittle RF, Bourne PE. Phylogeny determined by protein domain content. Proc Natl Acad Sci USA. 2005;102:373–378. doi: 10.1073/pnas.0408810102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 23.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 24.Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D. A census of protein repeats. J Mol Biol. 1999;293:151–160. doi: 10.1006/jmbi.1999.3136. [DOI] [PubMed] [Google Scholar]
- 25.Enright AJ, Ouzounis CA. Functional associations of proteins in entire genomes by means of exhaustive detection of gene fusions. Genome Biol. 2001;2:research0034.1–research0034.7. doi: 10.1186/gb-2001-2-9-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasek S, Risler JL, Brezellec P. Gene fusion/fission is a major contributor to evolution of multi-domain bacterial proteins. Bioinformatics. 2006;22:1418–1423. doi: 10.1093/bioinformatics/btl135. [DOI] [PubMed] [Google Scholar]
- 28.Snel B, Bork P, Huynen M. Genome evolution. Gene fusion versus gene fission. Trends Genet. 2000;16:9–11. doi: 10.1016/s0168-9525(99)01924-1. [DOI] [PubMed] [Google Scholar]
- 29.Kummerfeld SK, Teichmann SA. Relative rates of gene fusion and fission in multi-domain proteins. Trends Genet. 2005;21:25–30. doi: 10.1016/j.tig.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Bashton M, Chothia C. The geometry of domain combination in proteins. J Mol Biol. 2002;315:927–939. doi: 10.1006/jmbi.2001.5288. [DOI] [PubMed] [Google Scholar]
- 31.Jordan IK, Henze K, Fedorova ND, Koonin EV, Galperin MY. Phylogenomic analysis of the Giardia intestinalis transcarboxylase reveals multiple instances of domain fusion and fission in the evolution of biotin-dependent enzymes. J Mol Microbiol Biotechnol. 2003;5:172–189. doi: 10.1159/000070268. [DOI] [PubMed] [Google Scholar]
- 32.Przytycka T, Davis G, Song N, Durand D. Graph theoretical insights into evolution of multi-domain proteins. J Comput Biol. 2006;13:351–363. doi: 10.1089/cmb.2006.13.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panchenko AR, Luthey-Schulten Z, Wolynes PG. Foldons, protein structural modules, and exons. Proc Natl Acad Sci USA. 1996;93:2008–2013. doi: 10.1073/pnas.93.5.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soding J, Lupas AN. More than the sum of their parts: on the evolution of proteins from peptides. Bioessays. 2003;25:837–846. doi: 10.1002/bies.10321. [DOI] [PubMed] [Google Scholar]
- 35.Weiner J, 3rd, Beaussart F, Bornberg-Bauer E. Domain deletions and substitutions in the modular protein evolution. FEBS J. 2006;273:2037–2047. doi: 10.1111/j.1742-4658.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- 36.Wuchty S. Scale-free behavior in protein domain networks. Mol Biol Evol. 2001;18:1694–1702. doi: 10.1093/oxfordjournals.molbev.a003957. [DOI] [PubMed] [Google Scholar]
- 37.Fitch WM. Toward defining the course of evolution: minimum change for a specified tree topology. System Zoo. 1971;20:406–416. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.