

Abstract
In situ X-ray data collection has the potential to eliminate the challenging task of mounting and cryocooling often fragile protein crystals, reducing a major bottleneck in the structure determination process. An apparatus used to grow protein crystals in capillaries and to compare the background X-ray scattering of the components, including thin-walled glass capillaries against Teflon, and various fluorocarbon oils against each other, is described. Using thaumatin as a test case at 1.8 Å resolution, this study demonstrates that high-resolution electron density maps and refined models can be obtained from in situ diffraction of crystals grown in microcapillaries.
1. Introduction
Automating and miniaturizing the experimental aspects of macromolecular structure determination is greatly aiding investigators working on the elucidation of all types of biomolecules and their complexes (Weselak et al., 2003; Chayen, 2004; Pusey et al., 2005). The in situ collection of X-ray diffraction data from macromolecular crystals could significantly decrease the time and effort with which structures are obtained. Furthermore, by eliminating loop handling, the gene-to-structure pathway is streamlined, and crystals are less prone to damage caused by mechanical and environmental shock. While crystals from microcapillaries can be easily extracted for cryocooling (Zheng et al., 2004), the in situ method is not amenable to flash-cryocooling in general, making radiation damage a significant disadvantage. However, data sets from multiple isomorphic crystals can be used to overcome this drawback. With the advent of improved data processing software and faster CCD detectors (Yagi et al., 2004), this approach is now more feasible.
The advantages of in situ data collection have been demonstrated before (McPherson, 2000; López-Jaramillo et al., 2001; Ng et al., 2003; Luft et al., 1999), and there are undoubtedly much older unpublished accounts, when most crystal diffraction data were collected in capillaries. Commercial products (Watanabe, 2005) are now offered that can be used for plate-based X-ray exposures, for predicting resolution limit and space group. These products are impractical for complete data collection because of crystal dehydration and obstruction of the X-ray beam by the crystal container. In this paper, in situ collection is used in conjunction with a modern crystallization screening technique, where thin glass capillaries are filled with approximately 100 (replicate or unique) 20 nl microbatch trials (naming such capillaries as ‘microcapillaries’) via poly(dimethylsiloxane) (PDMS) stamp-based microfluidics (Chen et al., 2005; Song et al., 2003; Tice et al., 2003). The aqueous ‘plugs’, containing protein and precipitant, are separated by immiscible fluorocarbon oil at both ends (Fig. 1). This system has been used previously to obtain space-group and unit-cell information from crystal diffraction at room temperature (Zheng et al., 2004). In the present study, microcapillaries containing plugs which have crystals are mounted directly in a coldstream set to 277 K, and multiple frames of diffraction data are collected using synchrotron radiation. Despite the lack of cryocooling, here we demonstrate that sub-2.0 Å data sets and refined models are achievable, and that the collection of data from multiple crystals is convenient, given the large number of microbatch trials in a single capillary.
Figure 1.
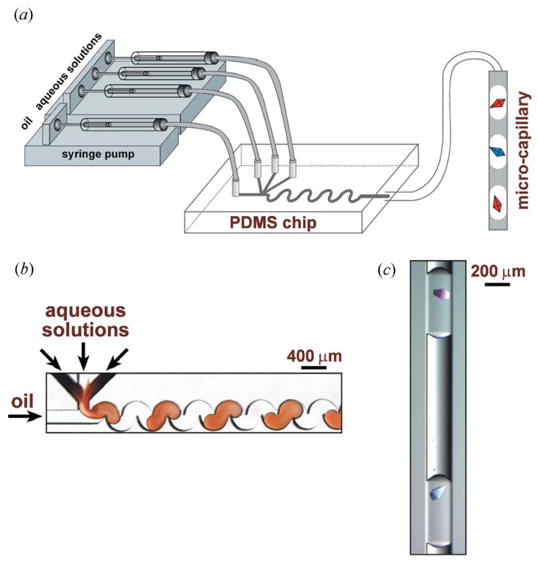
Microfluidic system for in situ diffraction data collection. (a) A schematic of the PDMS device used to form the crystallization trials. (b) A microphotograph of the process of plug formation in a PDMS chip. (c) A microphotograph of two thaumatin crystals inside two separate plugs in the microcapillary.
Although microfluidics-based free-interface diffusion chips (Hansen et al., 2002) have reached the market, our demonstration of microcapillary-based microbatch screening offers an alternative method to increase the efficiency of small volumes, and has the advantage of convenient in situ data collection and a potentially significant reduced cost of production.
2. Experimental methods
The apparatus used to fill microcapillaries with oil-spaced aqueous plugs was constructed as previously described (Chen et al., 2005; Song et al., 2003; Tice et al., 2003; Fig. 1) using PHD 2000 syringe pumps (Harvard Apparatus) and 10 and 50 μl Hamilton Gastight syringes (1700 series, TLL). Microchannels were fabricated using rapid prototyping in PDMS (Dow Corning Sylgard Brand 184 Silicone Elastomer). Devices were sealed using a Plasma Prep II (SPI Suppliers).
Thaumatin (Sigma Chemicals) was chosen as our initial model protein to demonstrate that the overall approach from crystal growth to data collection was feasible. Crystals were successfully grown (in each capillary material) using microfluidics by filling a single capillary with 100 replicate plugs that contained a 1:1 mixture, by volume, of 2.0 M sodium potassium tartrate and a thaumatin concentration of 25.0 mg ml−1. After generating the plugs containing tartrate and thaumatin, the first single crystals (25–50 μm) could be seen within hours. Pictures of microcapillaries were taken using a SPOT Insight color camera mounted on a Leica MZ 12 5 stereoscope.
Before data collection at the synchrotron, we tested the background X-ray scattering/diffraction of capillaries made from thin glass (Hampton) and Teflon in combination with a set of three fluorocarbon oils, FC-(40, 70, 3283) (3M) (subset of data shown in Fig. 2). Capillaries were attached to the stems of pins using HoldFast epoxy aquarium sealant (Marineland) and mounted on the goniometer. Diffraction data collection was performed on a Rigaku MSC R-AXIS IV with 10 min exposures. For each combination of oil and capillary, an exposure was taken both at an aqueous plug and at an oil spacer centered in the beam in order to gain a practical understanding of the background intensity, given a typical 300 μm effective radius of a home-source X-ray beam.
Figure 2.

Background intensities of scattering/diffraction for (a) a water plug centered in the beam using FC-70 as an oil spacer in a thin glass capillary, (b) a water plug centered in the beam using FC-40 as an oil spacer in a thin glass capillary, (c) a water plug centered in the beam using FC-40 as an oil spacer in a Teflon capillary, and (d) a thaumatin crystal centered in the beam using FC-40 as an oil spacer in a Teflon capillary. Each image with the exception of (d) was thresholded to identical bounds (200, 2000) in CrystalClear (Molecular Structure Corporation). FC-3283 plugs had similar intensity profiles to FC-70.
Subsequently, thaumatin crystals grown in thin glass capillaries underwent diffraction data collection at the GM/CA-CAT beamline at the Advanced Photon Source (APS) at 12 000 eV (1.03 Å). Capillaries were attached to pins as previously described and mounted on the goniometer under a coldstream (CryoJet XL, Oxford Diffraction) set to 277 K.
Two data sets were collected (Table 1) from two crystals in adjacent plugs with 1.0° oscillation and 1.2 s exposure per frame, with the unfocused beam attenuated by 50%. Data were integrated and scaled in HKL2000 (Otwinowski & Minor, 1997) and the merged data set was produced using XPREP (Bruker AXS). Molecular replacement (using PDB ID 1thw as a model) was carried out using MOLREP, refinement using REFMAC5 (Murshudov et al., 1997) and solvent building using ARP/wARP (Perrakis et al., 1997) of the CCP4 (Collaborative Computational Project, Number 4, 1994) suite of programs.
Table 1. Diffraction data statistics for two thaumatin crystals in the same capillary.
Refined unit-cell parameters were virtually identical in the two crystals, with a = b = 58.6 Å and c = 151.93 Å in P41212. Rint of merging data sets 1 and 2 was 0.04.
| Data set 1 | Data set 2 | Merged | |
|---|---|---|---|
| Resolution range (Å) | 27.94–1.86 | 36.34–1.90 | 36.34–1.86 |
| Number of reflections | 20 717 | 18 350 | 21 731 |
| Completeness (%) | 94.1 | 99.4 | 98.7 |
| Completeness, outer (%) | 94 | 99.2 | 94 |
| Redundancy | 7.9 | 7.3 | |
| Initial frame I/σ(I) | 9.11 | 7.94 | |
| Final frame I/σ(I) | 6.48 | 5.89 | |
| Initial frame I/σouter(I) | 2.96 | 2.64 | |
| Final frame I/σouter(I) | 1.38 | 1.28 | |
| I/σ(I) | 24.07 | 22.0 | 30.56 |
| I/σouter(I) | 10.69 | 10.05 | 10.69 |
| Rsym | 0.086 | 0.094 | |
| R | 0.145 | 0.146 | 0.148 |
| Rfree | 0.187 | 0.195 | 0.188 |
| Initial frame outer resolution shell (Å) | 1.75–1.61 | 1.75–1.61 | |
| Final frame outer resolution shell (Å) | 2.10–2.0 | 1.80–1.69 | |
| Number of frames | 100 | 100 |
3. Analysis of data
Previous reports of in situ data collection without cryocooling suggest the method is only suitable for weaker ‘home’ X-ray sources (López-Jaramillo et al., 2001), but examples of data collections at synchrotrons exist (Lacy et al., 1998; Reinisch et al., 2000). We demonstrate that single crystals can provide high-resolution (Fig. 3) refinable data sets under the intensity of synchrotron radiation. In the case of acute decay or reflection ‘blind spots’ due to crystal orientation, data sets from multiple crystals can be successfully merged for improved completeness (Table 1). In fact, the higher-intensity synchrotron beam coupled with fast-readout CCD detectors proves advantageous for collecting a maximum amount of data prior to the onset of extensive secondary radiation-induced decay.
Figure 3.
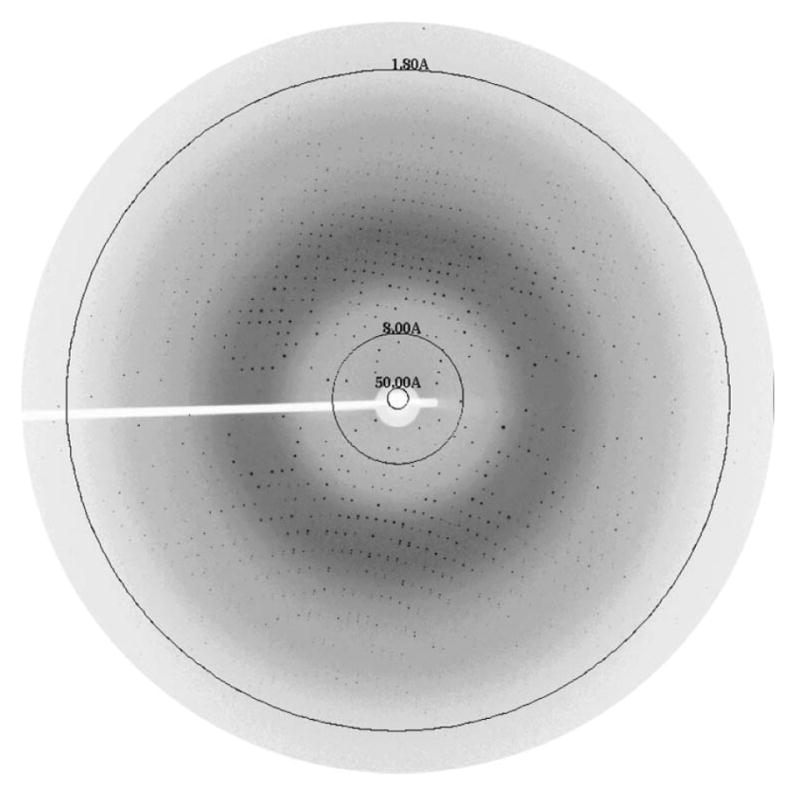
Initial in situ diffraction image from data set 1 of a thaumatin crystal in a thin glass capillary. The frame parameters were 1.0° oscillation, 1.2 s exposure and detector distance of 150 mm.
All of the fluorocarbon oils displayed significant background scattering/diffraction when the respective oil spacer was centered in the X-ray beam, as compared with when the beam was centered on the aqueous plug. The FC-40 oil consistently gave the least intense background from visual observation (Figs. 2c and 2d). Modern synchrotron beams, unlike home sources, match the beam radius to the crystal (via focusing and slitting down). Therefore, we expect that, in a typical situation, the beam could be focused on single crystals in a microcapillary so that the radiation flux through the oil spacer would be minimal.
While thin-walled glass and quartz have long been used for data collection from macromolecular crystals, we considered the use of Teflon-based capillaries as a flexible and cheaper replacement. While about three distinct ‘rings’ are visible in resolution shells under 10 Å (Figs. 2c and 2d), we found that the images obtained were suitable for indexing, suggesting that Teflon might be suited to screening-type diffraction studies, as opposed to those for structure determination (where blind spots could reduce data quality).
Unexpectedly, crystals in microcapillaries appeared visibly stationary under ϕ axis rotation, as confirmed by the observation of minimal variation in the orientation matrix during data reduction. We suggest that this steadfastness may be a consequence of the small 20 nl volumes and small dimensions of the plugs. The inertial effects for fluids become less significant at lower values of the Reynolds number. The Reynolds number decreases as the dimensions of the system decrease, as viscosity increases and as velocity of fluid flow decreases. The confinement of crystals at the water/fluorocarbon or the water/glass interface may provide additional stability. Furthermore, data collection on home sources necessitates the use of much larger crystals, which often dislodge owing to gravity. The intense synchrotron beam allows much smaller samples to be used, which are less likely to be dislodged.
Radiation damage induced by a lack of cryocooling deserves special attention (Blake & Phillips, 1962), as it could limit the applicability of the overall approach we are proposing. As an initial proof of concept, we have biased our experiments towards large crystals of thaumatin (100 μm scale), offsetting primary radiation damage effects which are independent of temperature. For crystals of this size, the total radiation exposures required for minimally complete data sets are well below the Henderson (1990) limit. Secondary damage from the diffusion of reactive radiolytic products is likely to be a much larger effect in the case of non-frozen microcapillaries than it is in flash-cryocooled experiments. We tried to minimize secondary damage and local heating effects by setting the cold stream to 277 K. Despite these measures we can still, as expected, observe weakening of diffraction spots much more rapidly than is typically seen under cryoconditions. This can be observed in both data sets by the decreased overall intensities between the initial and final frames, and in the decreased signal-to-noise ratios of the outer shell of reflections between the initial and final frames (Table 1).
The most obvious indicator of radiation-induced damage is that measured intensities decrease significantly as the X-ray dose accumulates; the total unaveraged I/σ(I) over time can be seen in Fig. 4. The individual Rsym values for the data sets (8.6 and 9.4%) could be considered large but, in addition to decay, could be a result of temperature shifts in the coldstream and/or an expected increased sensitivity to vibrations from low mosaicity (initial mosaicities are 0.056 and 0.049°). Mosaicity has a constant upward drift as a function of frame number (Fig. 5), but variations for other integration parameters are small. The final difference in unit-cell lengths for both data sets is only about 0.1%. There is no clear inflection point or threshold that would indicate a reasonable frame at which to cut off the integration. The two data sets were merged with a low Rint (4%) and are free of the non-isomorphism that can be introduced from flash-cryocooling (Teng & Moffat, 2000).
Figure 4.
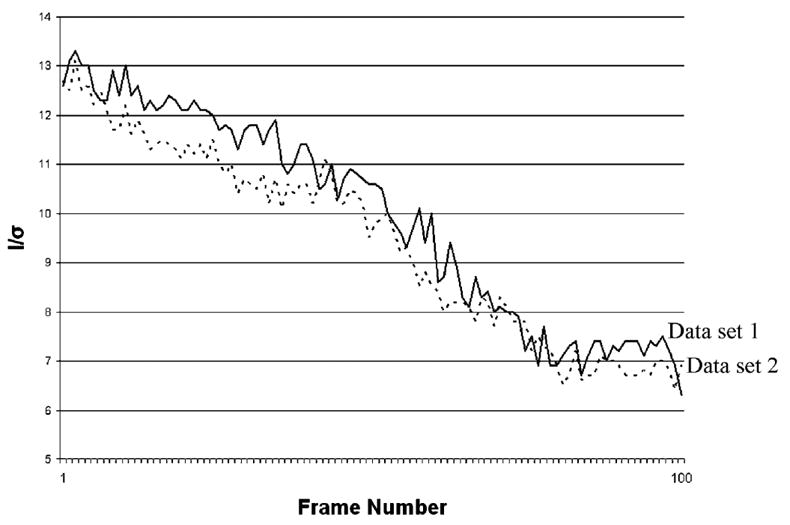
Decreasing intensity signals of both crystals as X-ray dose accumulates.
Figure 5.
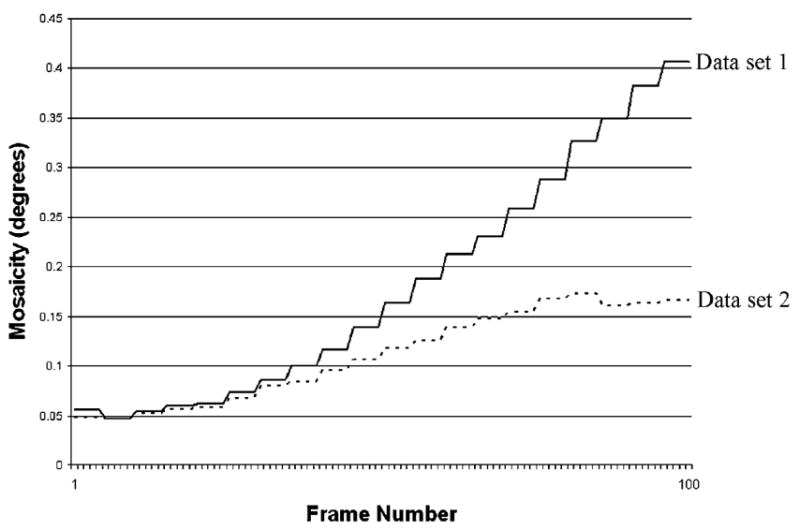
Increasing mosaicities of both crystals as X-ray dose accumulates.
Although thaumatin crystals are hardy and of high symmetry, we describe the collection of two adjacent crystals as a minimal representation of the realistic situation where crystals may be more sensitive to decay and orientation, and multiple samples might need to be employed to obtain a complete high-quality data set. Thaumatin has been refined up to 1.05 Å (PDB ID 1rqw), and crystals grown in agarose gel have provided room-temperature data sets from synchrotron radiation yielding 1.2 Å resolution atomic models (Sauter et al., 2002), but the crystals used in the present study are about a tenth of the scale of those used by Sauter et al. We suggest that our observed diffraction limits of about 2.0 Å are due, at least in part, to short, highly attenuated exposures and progressive weakening of the outer shell of reflections from radiation-induced decay. The I/σ(I) outer values decrease about twofold between the initial and final frames for each data set, and the outer shell limits themselves drop in resolution (Table 1). As a control experiment, several flash-cryocooled thaumatin crystals were prepared using hanging-drop vapor diffusion conditions analogous to the microcapillary conditions using glycerol as a cryoprotectant. We found that no crystal (despite being much larger than those in the microcapillaries) exceeded a visible diffraction limit of 1.7 Å (with similar exposure times).
In order to limit secondary damage, no attempt was made to choose a starting point for data collection by strategy simulations. Instead, 100° of data were collected at random orientation. Data set 1 was processed to 2.0 Å, and we discovered that it had only 94% completeness even though 100° of data were collected from a tetragonal crystal (Table 1). The capillary was then translated along the goniometer z axis and a fresh crystal was recentered in the beam, a process that has become semi-automated at some beamlines (McPhillips et al., 2002). An additional 100 frames were collected on a second crystal (which was processed to 1.9 Å), and the data sets were merged to form a virtually complete data set, with higher redundancy and resolution. The thaumatin model was refined against this data set to achieve R factors about as low as any entry found in the PDB for thaumatin (in the same tetragonal space group and at a comparable resolution). We note that minimally complete data sets (merged and unmerged) also yielded virtually identical R factors, and no striking differences in electron densities (Table 1) are observed. Isomorphic crystals are an important underlying assumption for the microcapillary approach. However, some biological crystals exhibit notorious non-isomorphism (Yonath et al., 1998) and would be impossible to analyze using multiple crystals. The use of multiple isomoprhic crystals does not offer any obvious disadvantage in data quality, but modern protein crystallographers seem to be apprehensive about introducing errors from subtle non-isomorphisms. Virus crystallographers routinely use multiple crystals [see Grimes et al. 1998 for an example of 1000 crystals] in merging complete data sets.
It is difficult to estimate exactly how the decay process affects the quality of the data set, given the low R factors for the refined model. We note that disulfide bonds, known to be susceptible to radiation-induced reduction, have faithful electron density in the refined thaumatin maps (Fig. 6). Some glutamate side chains in each structure demonstrate carboxylate electron density that is less precise than other parts of the model, but it is difficult to determine if this difference is due to decarboxylation.
Figure 6.
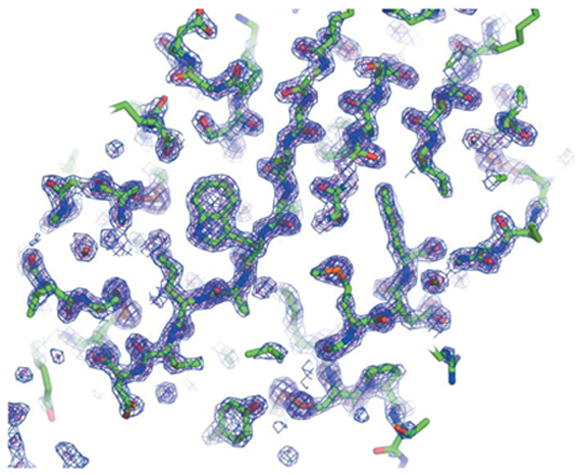
Electron density map of thaumatin from a merged data set (Table 1). Figures generated using PyMOL (DeLano Scientific, San Carlos, CA, http://www.pymol.org).
In order to evaluate the general applicability of the microcapillary approach, we attempted to grow crystals of a protein target from the SARS genome. A specific example for a conserved domain of SARS-CoV protein nsp3 (Saikatendu et al., 2005) is shown in Fig. 7. We were successful in growing single crystals in most cases, which suggests a surprising equivalence between vapor diffusion and microbatch type screening. The crystals grown in the microcapillary apparatus appear to be identical to those grown using traditional methods, under X-ray diffraction. Current efforts are focused on increasing the rate of successful crystal growth via microfluidics-based seeding and on data collection strategies for crystals that suffer acute decay under X-ray exposure.
Figure 7.
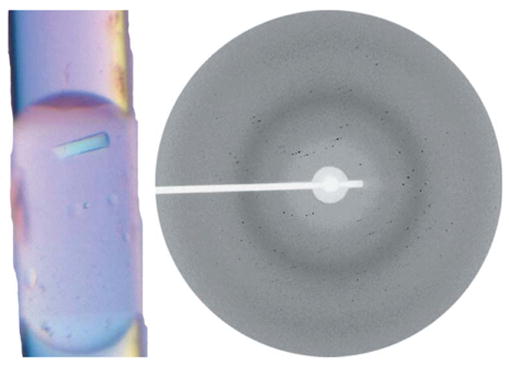
A single crystal of a conserved domain of SARS-CoV protein nsp3 (Saikatendu et al., 2005), with initial diffraction pattern from in situ data collection (3 Å limit). Diffraction image was taken at the GM/CA-CAT beamline at the APS.
4. Results and discussion
We have demonstrated an integrated in situ approach to crystallization screening and X-ray data collection for macromolecules. Plug-based microfluidics using PDMS stamp lithography to load nanovolume crystallization experiments into glass capillaries offers an inexpensive platform to increase volume efficiencies by tenfold over conventional techniques. This method is amenable to automated sparse matrix screening (Zheng & Ismagilov, 2005) and gradient fine screening (Zheng et al., 2003). Microcapillaries provide convenient handling of multiple crystals for data collection, and this technique can be used to offset the shortened lifetime of exposed crystals without the aid of cryocooling. We have validated this approach by using thaumatin as a model system to the point of providing sub-2.0 Å structures. The overall approach is well suited towards the automation of macromolecular structure determination.
We are grateful to Drs Janet Smith and Robert Fischetti for the access to the GM/CA-CAT beamline at the APS during its commissioning time. GM/CA-CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract #W-31-109-Eng-38. This study was supported by NIH/NIAID Contract #HHSN 266200400058C, ‘Functional and Structural Proteomics of the SARS-CoV’ (to PK), NIH Roadmap Award GM073197, ‘Joint Center for Innovative Membrane Protein Technology’ (to RCS), NIH Protein Structure Initiative Specialized Centers Grant GM074961 (to PK, RCS and RFI), and NIH award EB001903 (to RFI). We greatly appreciate the careful reading of and comments on the manuscript by Dr Elspeth Garman.
References
- Blake CCF, Phillips DC. Proceedings of the Symposium on the Biological Effects of Ionizing Radiation at the Molecular Level. Brno, IAEA Symposium; Vienna: 1962. pp. 183–191. [Google Scholar]
- Chayen NE. Curr Opin Struct Biol. 2004;14:577–583. doi: 10.1016/j.sbi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Chen DL, Gerdts CJ, Ismagilov RF. J Am Chem Soc. 2005 doi: 10.1021/ja052279v. In the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Cryst D. 1994;50:760–763. [Google Scholar]
- Grimes JM, Burroughs JN, Gouet P, Diprose JM, Malby R, Zientara S, Mertens PP, Stuart DI. Nature (London) 1998;395:470–478. doi: 10.1038/26694. [DOI] [PubMed] [Google Scholar]
- Hansen CL, Skordalakes E, Berger JM, Quake SR. PNAS. 2002;99:16531–16536. doi: 10.1073/pnas.262485199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. Proc R Soc London Ser B. 1990;241:6–8. [Google Scholar]
- Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- López-Jaramillo FJ, García-Ruiz JM, Gavira JA, Otálora F. J Appl Cryst. 2001;34:365–370. [Google Scholar]
- Luft JR, Rak DM, De Titta GT. J Cryst Growth. 1999;196:450–455. [Google Scholar]
- McPherson A. J Appl Cryst. 2000;33:397–400. [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J Synchrotron Rad. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Ng JD, Gavira JA, Garcia-Ruiz JM. J Struct Biol. 2003;142:218–223. doi: 10.1016/s1047-8477(03)00052-2. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Sixma TK, Wilson KS, Lamzin VS. Acta Cryst D. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- Pusey ML, Liu ZJ, Tempel W, Praissman J, Lin D, Wang BC, Gavira JA, Ng JD. Prog Biophys Mol Biol. 2005;88:359–386. doi: 10.1016/j.pbiomolbio.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Reinisch KM, Nilbert ML, Harrison SC. Nature (London) 2000;404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P. Structure. 2005 doi: 10.1016/j.str.2005.07.022. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter C, Lorber B, Giegé R. Proteins Struct Funct Genet. 2002;48:146–150. doi: 10.1002/prot.10125. [DOI] [PubMed] [Google Scholar]
- Song H, Tice JD, Ismagilov RF. Angew Chem Int Ed. 2003;42:768–772. doi: 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- Teng T, Moffat K. J Sychrotron Rad. 2000;7:313–317. doi: 10.1107/S0909049500008694. [DOI] [PubMed] [Google Scholar]
- Tice JD, Song H, Lyon AD, Ismagilov RF. Langmuir. 2003;19:9127–9133. [Google Scholar]
- Watanabe N. J Appl Cryst. 2005;38:396–397. [Google Scholar]
- Weselak M, Patch MG, Selby TL, Knebel G, Stevens RC. Methods Enzymol. 2003;368:45–47. doi: 10.1016/S0076-6879(03)68004-3. [DOI] [PubMed] [Google Scholar]
- Yagi N, Inoue K, Oka T. J Synchrotron Rad. 2004;11:456–461. doi: 10.1107/S090904950402093X. [DOI] [PubMed] [Google Scholar]
- Yonath A, Harms J, Hansen HA, Bashan A, Schluenzen F, Levin I, Koelln I, Tocilj A, Agmon I, Peretz M, Bartels H, Bennett WS, Krumbholz S, Janell D, Weinstein S, Auerbach T, Avila H, Piolleti M, Morlang S, Franceschi F. Acta Cryst A. 1998;54:945–955. doi: 10.1107/s010876739800991x. [DOI] [PubMed] [Google Scholar]
- Zheng B, Roach LS, Ismagilov RF. J Am Chem Soc. 2003;125:11170–11171. doi: 10.1021/ja037166v. [DOI] [PubMed] [Google Scholar]
- Zheng B, Tice JD, Roach LS, Ismagilov RF. Angew Chem Int Ed. 2004;43:2508–2511. doi: 10.1002/anie.200453974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Ismagilov RF. Angew Chem Int Ed. 2005;117:2576–2579. [Google Scholar]