

Abstract
The cAMP and ERK/MAP kinase (MAPK) signal transduction pathways are critical for hippocampus-dependent memory, a process that depends on CREB-mediated transcription. However, the extent of crosstalk between these pathways and the downstream CREB kinase activated during memory formation have not been elucidated. Here we report that PKA, MAPK, and MSK1, a CREB kinase, are co-activated in a subset of hippocampal CA1 pyramidal neurons following contextual fear conditioning. Activation of PKA, MAPK, MSK1, and CREB is absolutely dependent on Ca2+-stimulated adenylyl cyclase activity. We conclude that adenylyl cyclase activity supports the activation of MAPK, and that MSK1 is the major CREB kinase activated during training for contextual memory.
Introduction
A critical step in the generation of long-term memory (LTM) is memory consolidation, a process that depends on de novo transcription and translation (Davis and Squire, 1984). One of the transcriptional pathways strongly implicated in memory consolidation is the CREB/CRE-mediated transcriptional pathway (for reviews see (Impey et al., 1999; Silva et al., 1998; Tully et al., 2003). In mice, contextual fear conditioning stimulates phosphorylation of CREB at serine 133 and CRE-mediated transcription in the hippocampus (Impey et al., 1998b; Taubenfeld et al., 1999). CREB activity is essential for long-term facilitation in Aplysia (Dash et al., 1990; Martin et al., 1997), as well as LTM in Drosophila (Yin et al., 1995; Yin et al., 1994) and mice (Bourtchuladze et al., 1994; Pittenger et al., 2002). Furthermore, administration of CRE oligonucleotide decoys to the hippocampus, which inhibits CRE-mediated transcription in vivo, blocks contextual memory formation (Athos et al., 2002).
Although several signaling pathways can mediate activity-dependent phosphorylation of CREB in hippocampal neurons, the MAPK pathway is particularly interesting because it is implicated in stimulation of CRE-mediated transcription and LTM (for reviews see Impey et al., 1999; Mazzucchelli et al., 2002; Sweatt, 2004). MAPK activation is required for formation of hippocampus-dependent memory (Atkins et al., 1998; Blum et al., 1999) and training-induced increases in CREB phosphorylation are blocked by inhibitors of MAPK signaling in vivo (Athos et al., 2002). Furthermore, regulation of protein synthesis by MAPK may also play a critical role in long-lasting synaptic plasticity and memory (Kelleher et al., 2004). Another major signal transduction system important for synaptic plasticity and hippocampus-dependent memory is the cAMP pathway. For example, reduction of cAMP-dependent protein kinase (PKA) activity in transgenic mice causes defects in L-LTP (long-lasting LTP) and spatial memory (Abel et al., 1997). Mice deficient in type 1 adenylyl cyclase (AC1) exhibit impaired spatial memory (Wu et al., 1995), mossy fiber LTP (Villacres et al., 1998) and cerebellar LTP (Storm et al., 1998). Furthermore, double-knockouts in AC1 and AC8 (DKO), in which Ca2+/calmodulin-stimulated cAMP production is greatly reduced, lack L-LTP and LTM for passive avoidance and contextual fear (Wong et al., 1999). Although it is not known why cAMP signaling is required for hippocampus-dependent memory, cAMP regulates ion channel activity and trafficking, gene expression, and neurotransmitter release, all of which can influence synaptic plasticity.
There are a number of major unanswered questions concerning the roles of MAPK and cAMP in memory formation. Why is cAMP required for LTM? Does crosstalk between cAMP and MAPK contribute to memory? Because cAMP stimulates MAPK activity in cultured neurons (Villalba et al., 1997; Vossler et al., 1997) and is required for its nuclear translocation (Impey et al., 1998a), increases in PKA activity may be necessary to support the activation and nuclear translocation of MAPK during memory formation. However, it has not been established that fear conditioning activates hippocampal PKA, nor is there any evidence that PKA and MAPK are co-activated in the same neurons. There is also no evidence that adenylyl cyclase activity is required for stimulation of MAPK when animals are trained for hippocampus-dependent memory. Moreover, there are a number of CREB kinases downstream from MAPK. Which of these CREB kinases is activated during memory formation? To address these issues, we used confocal imaging to identify individual hippocampal neurons that are biochemically activated following contextual fear conditioning. We discovered that PKA, MAPK, and MSK1 are activated in the same subset of CA1 pyramidal neurons, and that Ca2+-stimulated adenylyl cyclase activity is indispensable for the training-induced activation of MAPK, MSK1 and CREB.
Results
Cellular and subcellular analysis of MAPK activation
When mice are trained for contextual fear conditioning, there is a transient increase in MAPK activity in the hippocampus that can be monitored by Western analysis for dually phosphorylated ERK1/ERK2 (referred to here as pERK) (Atkins et al., 1998). However, Western analysis does not allow for the identification of the cellular and subcellular localization of this signal nor can it be used to determine if MAPK is co-activated with other proteins in the same cells. Therefore, we used immunohistochemical methods to identify individual hippocampal cells in which MAPK activity is elevated following fear conditioning (Figure 1). Mice were trained by placing them in a novel context and delivering a footshock two minutes later (paired mice). Unpaired controls, which were shocked immediately after being placed in context, or unshocked controls, did not develop contextual fear memory (n = 9–12 mice per group; effect of treatment on percent freezing, H(2, 33) = 21.8; p < 0.0001) (Figure 1B). Paired mice exhibited increased numbers of cells immunopositive for pERK (pERK+) in the CA1 pyramidal layer compared to naïve, unshocked or unpaired mice (n = 8–14 mice per group; effect of treatment, H(3, 40) = 14.42; p < 0.0001) (Figures 1A, 1C). In contrast, no significant increases were observed in areas CA2, CA3, or dentate gyrus. Activation of MAPK in CA1 was rapid and transient, reaching a maximum 30 min after training and returning to baseline within 60 min (n = 6–12 mice per group; effect of time, H(2, 28) = 18.42; p < 0.0001) (Figure 1D).
Figure 1.

MAPK is selectively and transiently activated in the CA1 subregion of the hippocampus after contextual fear conditioning (A) Immunohistochemistry for pERK in CA1, CA3 and dentate gyrus (DG) from sections representative of naïve, unpaired, context and paired mice 30 minute after training. Neuropil staining was heaviest in the CA3 mossy fibers (MF), moderate in the inner (iml) and outer (oml) layers of DG, and mild to moderate in the dendritic layers of Ammon’s horn. Only pERK staining in the pyramidal cell layer (sp) and stratum radiatum (sr) in CA1 increased with training. Scale bar, 100 μm. (B) Effect of training on 6 hour memory for contextual fear conditioning. n = 9–12 mice per group. (C) Effect of fear conditioning on the number of pERK+ cells in the main hippocampal subregions (30 min). (D) Time course of pERK+ cell numbers in CA1, normalized to unpaired animals.
Training-induced pERK increases co-localized with the neuronal marker NeuN, and the pyramidal cell markers CaMKIIα or MAP2, indicating that MAPK activation was confined to CA1 pyramidal neurons (Figure 2A, first and second rows). We found no evidence of training-induced MAPK activation in interneurons or astrocytes (Figure 2A, second row). pERK+ neurons could be readily differentiated from pERK negative (pERK-) neurons due to the pronounced increase in labeling intensity (grayscale level in soma: 106 ± 15 in pERK+ neurons, 14 ± 1 in pERK- neurons; p = 0.0004). This allowed us to unambiguously quantify the percentage of neurons undergoing MAPK activation after training. MAPK was activated in 10.9 ± 1.1 % (141 of 1,281) of CA1 pyramidal neurons in paired mice compared to 1.7 ± 0.5 % (13 of 720) in unpaired mice (mean ± SD; n = 7 mice per group; p = 0.00005) (Figure 2B).
Figure 2. Training for contextual memory triggers MAPK activation selectively in CA1 pyramidal neurons.
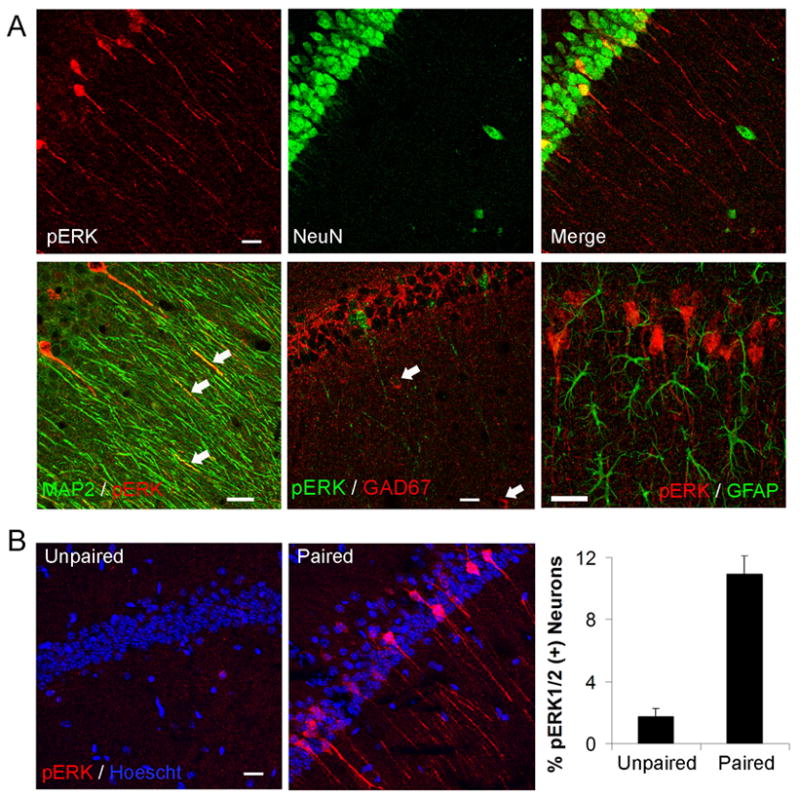
Images are three dimensional reconstructions of confocal z-stacks. (A) First row, pERK+ cell bodies co-localized with the neuronal marker NeuN. Second row, pERK labeling was found in cell bodies and apical dendrites immunoreactive to MAP2, but not in GABAergic interneurons stained with GAD67, or in astrocytes stained with GFAP. (B) Fear conditioning increased the percentage of CA1 pyramidal neurons stained for pERK. Scale bars, 20 μm.
To identify subcellular pools of MAPK activated during memory formation, we used line scan analysis to assess the intensity profile of the pERK immunosignal across the cytoplasm and nucleus in optical sections from CA1. pERK substantially increased in the perinuclear region of the cytoplasm and peaked inside the nucleus in activated neurons of trained animals (Figure 3A). Line scan profiles were similar at 10 and 30 min after training (data not shown). In addition, activation of MAPK in dendrites was found in proximal and distal domains, and appeared limited to a few (mainly first- and second-order) branches in pERK+ neurons (Figure 3B).
Figure 3. Nuclear and synaptic activation of MAPK.
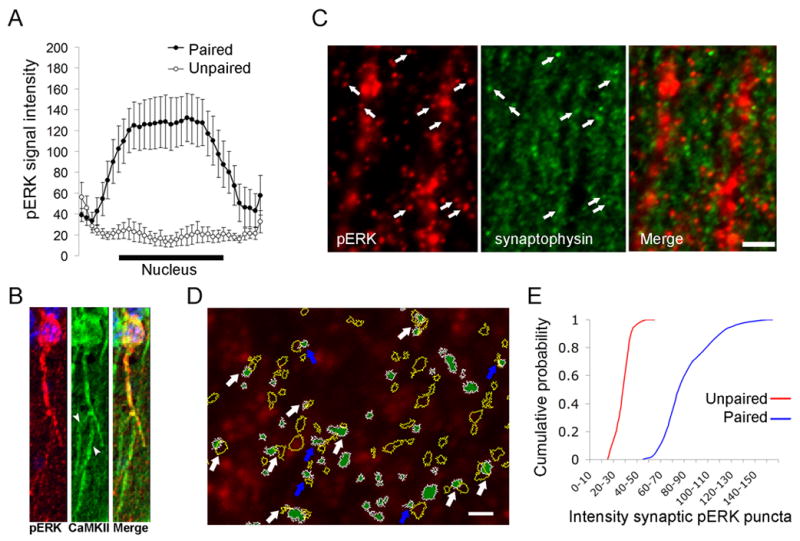
(A) Average line scans of pERK staining in paired (n = 42 neurons, pERK+ subset) and unpaired (n = 36 neurons) mice (30 min); x-axis represents relative position across the soma. (B) Double-labeling for pERK and CaMKIIα in a CA1 pyramidal neuron after fear conditioning. Arrowheads indicate dendritic branches of the neuron shown in which pERK staining was absent. (C) Apical dendritic branches (occupying areas devoid of synaptophysin labeling) were decorated with pERK+ puncta, which were often juxtaposed to synaptophysin boutons (arrows). Scale bar, 5 μm. (D) Example of a single confocal section subfield included in the formal quantitative analysis. Green regions with white margins represent pERK+ puncta that met criteria of size and labeling intensity after thresholds were applied. Raw image is in red. Yellow-outlined objects represent overlaid synaptophysin-labeled puncta obtained after similar processing. pERK+ and synaptophysin+ loci showed partial overlap (white arrows) or were juxtaposed to one another (blue arrows). Scale bar, 1 μm. (E) Quantification of the gray scale signal intensity of pERK+ puncta associated with synaptophysin in unpaired and paired mice.
We also examined MAPK activation in apical CA1 synapses (> 100 μm away from the pyramidal layer). Synaptophysin-labeled puncta (corresponding to individual presynaptic boutons) typically were found in close apposition to or partially overlapping with pERK+ puncta, which often emanated from dendritic shafts (Figures 3C, 3D). Presynaptic boutons in contact with pERK+ puncta (i.e. pERK+ synapses) showed a larger average size than those forming pERK- synapses (bouton area at pERK+ synapses 0.34 ± 0.01 μm2, n = 911; bouton area at pERK- synapses 0.3 ± 0.005 μm2, n = 2,625; p = 0.0006). Neither the frequency (unpaired 20 ± 6 %, paired 20 ± 5 %; p = 0.9) nor the size (p = 0.71) of pERK+ synapses changed within 10–30 min after training. However, the intensity of pERK staining increased in 80 ± 16 % of pERK+ synapses with training (i.e. 16 ± 4 % of all CA1 synapses) (p = 0.02) (Figure 3E). Together, these data indicate that MAPK activation in the hippocampus following contextual fear conditioning is restricted to approximately 10 % of CA1 pyramidal neurons and involves two distinct intracellular pools of MAPK, namely, a nuclear pool, and a pool associated with synapses.
Co-activation of MAPK and PKA in CA1 neurons
Although cAMP signaling is required for hippocampus-dependent memory (Abel et al., 1997; Wong et al., 1999; Wu et al., 1995), it is not known whether contextual fear conditioning actually activates PKA, which would suggest an instructive, rather than permissive, role for PKA in LTM. Therefore, we used an antibody that recognizes proteins phosphorylated at a conserved PKA phosphorylation motif to monitor PKA activation following training (Maas et al., 2005; Vanhoose et al., 2006; Zhang et al., 2005). Control experiments showed that the immunostaining for phosphorylated PKA substrates (pPKA-s) significantly increased upon infusion of the cAMP analogue Sp-cAMP into area CA1 of conscious mice compared to the vehicle-infused, contralateral CA1 (H(2, 14) = 10.15; p < 0.01) (Figure 4A). The effect of Sp-cAMP was fully blocked by co-infusion of the PKA antagonist Rp-cAMP, but not by prior injection of the MEK1/2 inhibitor SL327, thus confirming the ability of the antibody to specifically detect PKA activation in vivo. This pPKA-s antibody was used to monitor PKA activity in mice trained for contextual fear. Random sampling of CA1 pyramidal somata revealed a subset of neurons with elevated pPKA-s immunostaining associated with training (n = 357 neurons, 5–7 mice per group) (Figure 4B). Based on the similar size of this subpopulation (~15 % of neurons) and that of pERK+ neurons, we asked whether the MAPK and PKA pathways are co-activated in the same subset of neurons. The mean level of pPKA-s staining was significantly higher in pERK+ neurons compared to pERK- neurons in all trained mice examined (n = 192 neurons, 7 mice at 30 min, p < 0.00005; n = 96 neurons, 5 mice at 10 min; p = 0.015) (Figure 4C). Up to 82 ± 5% of pERK+ neurons showed labeling intensities for pPKA-s more than two standard deviations above that in unpaired mice, whereas only 25 ± 7% of pERK- neurons met the same criterion. Line scan analysis showed a two-fold increase in pPKA-s staining in pERK+ neurons compared to pERK- neurons or unpaired mice (p < 0.0001) (Figures 4D, 4E). Collectively, these data indicate that contextual fear conditioning results in a highly co-localized activation of MAPK and PKA in individual CA1 pyramidal neurons.
Figure 4. Co-localized activation of MAPK and PKA signaling in CA1 pyramidal neurons.
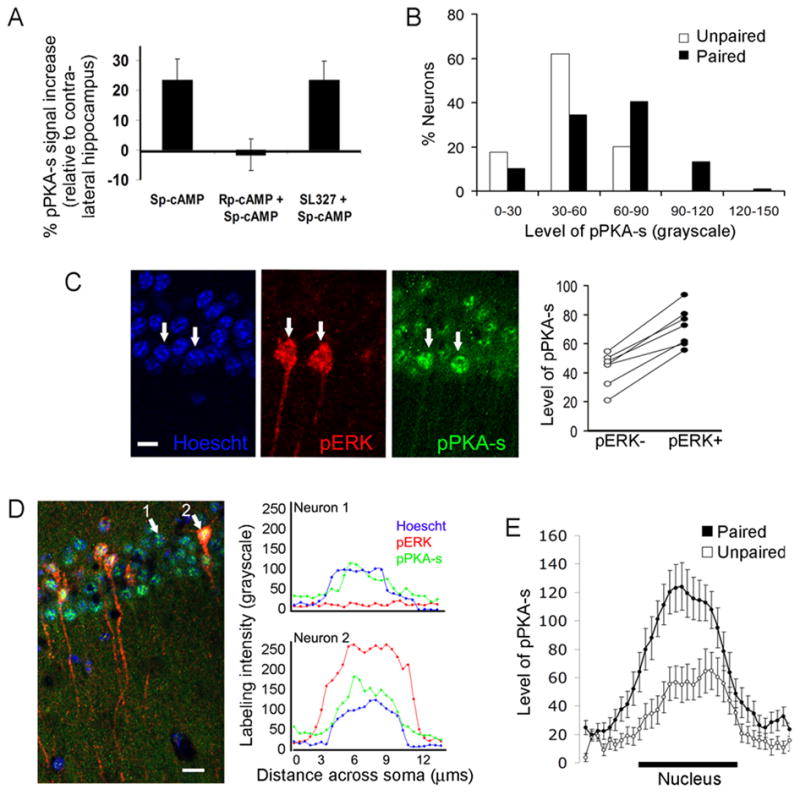
(A) Acute infusion of Sp-cAMP into the CA1 region increased the immunoreactivity for phosphorylated PKA substrates (pPKA-s), which was blocked by Rp-cAMP but not by SL327. (B) Frequency distribution of the levels of pPKA-s labeling in CA1 somata in paired and unpaired mice (30 min after training). (C) A representative optical section from CA1 showing higher levels of pPKA-s labeling in pERK+ neurons (arrows). Scale bar, 10 μm. Right, summary data. Each dot pair represents the mean gray scale intensity of pPKA-s in pERK+ and pERK- somata for a given mouse. (D) Single-cell line scans showing the correlation between the nuclear levels of pERK and pPKA-s from two neurons that were centered at the same plane along the z-axis. Scale bar, 20 μm. (E) Average line scans of pPKA-s staining in paired (n = 42 cells, pERK+ subset) and unpaired (n = 36 cells) mice;x-axis represents relative position across the soma.
MAPK activation depends on Ca2+-stimulated adenylyl cyclase activity
The above data indicate that the co-activation of MAPK and PKA may be necessary for memory consolidation. To analyze the extent of crosstalk between these two pathways, we first verified that cAMP stimulates MAPK in the hippocampus in vivo. Acute infusion of Sp-cAMP greatly increased staining for pERK in dendrites and somata of CA1 pyramidal neurons (Figure S1). MAPK activation depended on MEK1/2 and was completely blocked by Rp-cAMP.
To assess the interaction between the MAPK and cAMP signaling pathways during fear conditioning, we used mice lacking the type 1 and type 8 adenylyl cyclases (DKO mice, Wong et al., 1999). DKO mice are selectively impaired in LTM, but not STM, and show no Ca2+-stimulated adenylyl cyclase activity in hippocampal membranes (Wong et al., 1999). Contextual fear conditioning did not elicit MAPK activation in DKO mice: pERK levels increased with training in wild type, but not DKO, hippocampi (n = 6 mice per group, significant interaction between genotype and treatment, H(3, 23) = 12.51; p = 0.002) (Figure 5A). Accordingly, the number of pERK+ cells remained unchanged in paired DKO mice compared to naïve or unpaired DKO mice (n = 7–9 mice per group; no effect of treatment, H(2, 22) = 0.62; p = 0.54) (Figure 5B). Basal MAPK expression in DKO mice appeared increased compared to wild type mice (n = 8 mice per group, p = 0.01) but was not activated by training (Figure 5C).
Figure 5. Impaired activation of MAPK and PKA following contextual fear conditioning in DKO mice.

(A) Western blot analysis of hippocampal pERK in WT and DKO mice after training (5–10 min). Blot shows pooled samples; the graph shows mean data from individually run samples. U, unpaired; P, paired. (B) Immunohistochemical analysis of pERK in DKO mice after training (30 min). Representative images of the CA1 region. Scale bar, 50 μm. (C) Western blot analysis of total ERK expression in WT and DKO hippocampi. (D) Average intensity levels of pPKA-s in pERK- and pERK+ CA1 somata from WT and DKO hippocampi after training (30 min). (E) Ratio of the pERK labeling intensity in the nucleus relative to the cytoplasm in WT and DKO mice.
To determine whether the deficit in Ca2+-stimulated adenylyl cyclase activity prevented the activation of PKA after fear conditioning, we assessed the levels of pPKA-s in DKO mice. There was no increase in pPKA-s levels associated with training (n = 116 neurons, 5 mice per group; levels in paired vs. unpaired neurons, p = 0.37) or MAPK activation (n = 142 neurons, 5 mice; levels in pERK+ vs. pERK- neurons, p = 0.61) in DKO mice (Figure 5D). Moreover, consistent with a role for PKA in the nuclear translocation of MAPK, the nuclear-to-cytoplasmic ratio of pERK was increased by training in wild type, but not DKO, mice (significant interaction between genotype and treatment, H(3, 135) = 50.5; p < 0.0001) (Figure 5E). These data support the hypothesis that Ca2+-stimulation of adenylyl cyclases is required for MAPK activation following fear conditioning and provide one of the first clues concerning the role of cAMP in memory formation.
Co-activation of MAPK, PKA, MSK1 and CREB in single neurons
The mitogen and stress-activated kinase 1 (MSK1) and the ribosomal S6 kinase 1/2 (RSK1/2) are major neuronal nuclear substrates of ERK that can activate the transcription factor CREB (Arthur et al., 2004). However, it has not been shown that either CREB kinase is activated in contextual fear conditioning. Phosphorylation of MSK1 at Thr581 and of RSK1/2 at Ser221/227 is crucial for its kinase activities (McCoy, et al., 2005). The percentage of CA1 pyramidal neurons showing nuclear phospho-MSK1 increased from 6.9 ± 1.1 % (77 of 1,095) in unpaired mice to 17.2 ± 3.9 % (174 of 1,023) in paired mice (n = 5–7 mice per group; p = 0.003), an increase of 2.5 fold. Double labeling for pMSK1 and pERK showed a striking co-localization in pyramidal neurons (Figure 6A, first row). The intensity of pMSK1 staining was significantly higher in pERK+ neurons compared to pERK- neurons in paired mice (58 ± 9 % nuclear increase; n = 96 neurons, 6 mice; p = 0.0004). Because MSK1 can be phosphorylated by p38 MAP kinase as well, we examined the nuclear levels of phospho-activated p38 in paired mice, and found no difference between pMSK1+ and pMSK1- neurons (n = 74 neurons, 4 mice; p = 0.33) (data not shown).
Figure 6. Activation of MAPK/PKA and downstream CREB kinases following contextual fear conditioning.
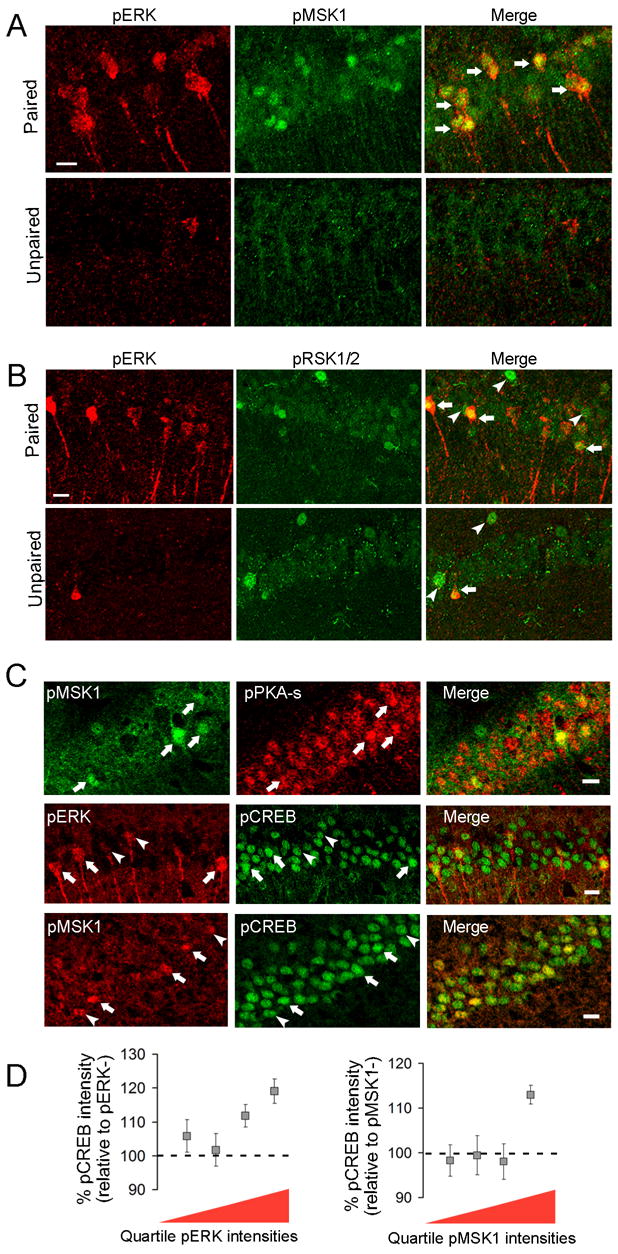
Images are three dimensional reconstructions of confocal z-stacks. (A, B) Immunodetection of pERK and pMSK1 (A) or pERK and pRSK1/2 (B) in the CA1 layer of paired and unpaired mice (30 min). Arrows indicate co-localized signals, arrowheads indicate segregated signals. Scale bar, 20 μm. (C) First row, immunodetection of pPKA-s and pMSK1. Arrows indicate neurons heavily labeled for both kinases. Second row, immunodetection of pCREB and pERK. Third row, immunodetection of pCREB and pMSK1. Low-to-moderate pERK or pMSK1 staining (indicated by arrowheads) was not associated with high pCREB staining, whereas high pERK or pMSK1 staining (indicated arrows) was. Images were taken from the CA1 layer of paired mice (30 min). Scale bar, 20 μm. (D) Summary data of the relative pCREB levels as a function of the quartile intensity for pERK or pMSK1.
Nuclear levels of pRSK1/2 increased modestly in pERK+ neurons compared to pERK- neurons in paired mice (30 ± 4 % increase; n = 114 neurons, 6 mice; p = 0.002) (Figure 6B). However, the cellular activation patterns of pERK and pRSK1/2 in CA1 only partially overlapped.
Consistent with the co-activation of MAPK and PKA, training-induced increases in nuclear phospho-MSK1 also correlated with higher levels of pPKA-s in single neurons (40 ± 11 % increase; n = 110 neurons, 5 mice; p = 0.006) (Figure 6C, upper row).
We next asked whether activation of MAPK and MSK1 was accompanied by enhanced CREB phosphorylation at Ser133 in the same subset of neurons after training. A basal level of CREB phosphorylation was evident in CA1 neurons of control mice (data not shown). After training, however, those neurons with the highest degree of nuclear MAPK or MSK1 phosphorylation (i.e. with labeling intensities in the upper quartile) showed significantly increased levels of phospho-CREB compared to pERK- or pMSK1- neurons, respectively (p = 0.008 for pERK, n = 161 neurons and 5 mice; p = 0.0009 for pMSK1, n = 146 neurons and 7 mice) (Figures 6C, D).
The PKA/MAPK pathway regulates training-induced MSK1 activation
To determine whether cAMP-dependent MAPK activation couples to CREB phosphorylation via MSK1, we first confirmed that infusion of Sp-cAMP increases the levels of phospho-MSK1 in CA1 (Figure 7A). Increased levels of pMSK1 were blocked by co-infusion of Sp-cAMP and Rp-cAMP, as well as by previous administration of a MEK1/2 inhibitor (significant effect of treatment, H(2, 14) = 8.23; p = 0.016). Next, wild type mice were injected with SL327 (50 mg/Kg, i.p) or vehicle (DMSO) immediately after training to assess the ERK-dependence of MSK1 activation in fear conditioning (Figure 7B). SL327 prevented the increase in pERK 30 min after training, whereas DMSO did not (significant interaction between treatment and drug, H(3, 18) = 10.44; p = 0.015). In addition, SL327 completely blocked the training-induced phosphorylation of MSK1 (significant interaction between treatment and drug, H(3, 18) = 11.12; p = 0.004). Pair-wise comparisons showed an effect of training on the percentage of pMSK1+ nuclei in CA1 between DMSO groups (p = 0.012), but not between SL327 groups (p = 0.21). Total levels of nuclear MSK1 were unaffected by drug treatment or training, supporting a specific effect on the activation of MSK1.
Figure 7.

Calcium-stimulated adenylyl cyclase and MAPK regulate MSK1 and CREB activation following contextual fear conditioning. (A) Infusion of Sp-cAMP into the CA1 region increased the immunoreactivity for phosphorylated MSK1, which was blocked by Rp-cAMP and SL327. (B) Post-training inhibition of MEK1/2 prevents the co-localized phosphorylation of MSK1 and MAPK in CA1 nuclei. Images are three dimensional reconstructions of confocal z-stacks. Scale bar, 20 μm. Summary data is shown on the right. (C) Nuclear extracts from wild type and DKO hippocampi were prepared from context-exposed or paired mice and analyzed for relative levels of pCREB, pMSK1 or pRSK2. Blots show individually run samples. Summary data is shown on the right.
Based on the absence of MAPK stimulation in DKO mice, and the need of MAPK for MSK1 activation, we then assessed the phosphorylation states of CREB and MSK1 in DKO mice (Fig. 7C). In contrast to wild type mice, relative levels of pCREB and pMSK1 failed to increase after training in nuclear extracts of DKO hippocampi (p = 0.005 and p = 0.025 for wild type mice, respectively; p = 0.26 and p = 0.16 for DKO mice, respectively). Moreover, the differential induction of MSK1 in wild type and DKO mice was confirmed with a second antibody recognizing phosphorylated MSK1/2 at Ser360/376 (p = 0.014 for wild type; p = 0.84 for DKO). On the other hand, phosphorylation of nuclear RSK2 was not affected by training in wild type (p = 0.63) or DKO mice (p = 0.9). Collectively, these data demonstrate that calcium-sensitive adenylyl cyclase activity is required for ERK-mediated activation of MSK1 and CREB during memory formation.
Discussion
One of the major objectives of this study was to identify which MAPK-activated CREB kinase is stimulated during memory formation. Furthermore, it was important to define the relationship between MAPK and cAMP signaling following training for contextual fear conditioning, and to determine why Ca2+-stimulated adenylyl cyclase activity is required for contextual memory. There are several mechanisms by which cAMP could contribute to memory, including regulation of AMPA receptor trafficking (Esteban et al., 2003) and MAPK activation (Vossler et al., 1997). We have been unable to observe increased PKA phosphorylation of AMPA receptors following contextual fear conditioning (Scheiner and Storm, unpublished data). Consequently, we focused on the role of cAMP signaling for MAPK activation because of the central role played by MAPK during memory formation. We used confocal imaging to identify individual hippocampal cells in which PKA, MAPK and CREB kinases are activated after contextual fear conditioning. Prior to this study, it had not been shown that contextual fear conditioning activates PKA, nor was it known that PKA and MAPK are activated in the same neurons in the hippocampus. Furthermore, there was no evidence for activation of specific CREB kinases following fear conditioning.
Training for contextual memory caused a five to six fold increase in MAPK activation in approximately 10% of CA1 pyramidal neurons in two distinct intracellular pools: a nuclear pool and a postsynaptic pool. Furthermore, PKA was activated in the same subset of neurons as MAPK, and both showed increased nuclear activities after training. MAPK activation strongly correlated with activation of MSK1, a CREB kinase. Most importantly, the training-induced increases in MAPK, PKA and MSK1 activities were ablated in mice lacking Ca2+-stimulated adenylyl cyclase activity. We conclude that one of the major reasons cAMP signaling is required for memory is to support the activation and nuclear translocation of MAPK in CA1 pyramidal neurons.
Signal transduction pathways are usually implicated in memory formation because they are activated in specific areas of the brain by training, and inhibition of the pathway blocks memory. For example, MAPK activity is stimulated in area CA1 following training for hippocampus-dependent memory and administration of MEK inhibitors blocks both training-induced increases in MAPK and memory formation (Athos et al., 2002; Atkins et al., 1998; Blum et al., 1999). Ca2+-stimulated adenylyl cyclase (Wong et al., 1999a; Wu et al., 1995) and PKA (Abel et al., 1997; Atkins et al., 1998) activities are required for memory formation, suggesting that either basal PKA activity is necessary or that an increment in PKA activity contributes to memory. Using an antibody that recognizes phosphorylated PKA substrates, we discovered that PKA is not only activated in area CA1 following contextual fear conditioning, but there is also a strong correlation between neurons showing MAPK activation and those in which PKA was activated. In keeping with this, we have also observed increased nuclear levels of the PKA catalytic alpha subunit in pERK+ neurons after training (Sindreu and Storm, unpublished data). The increase in pPKA-s was readily blocked by inhibitors of PKA and lost in mice lacking Ca2+-stimulated adenylyl cyclase activity, thus validating the use of the pPKA-s antibody to monitor PKA activation.
Our observation that fear conditioning activates MAPK selectively in area CA1 agrees with other evidence that stimulation of transcription in this area of the hippocampus is particularly important for contextual memory formation (Athos et al., 2002; Impey et al., 1998b). Much less was known, however, about the identity and size of the cellular population activated during training for contextual memory, and in which intracellular compartments MAPK is stimulated. Although our analysis focused on the role of MAPK in the nucleus because of its importance for CREB-mediated transcription, MAPK was simultaneously activated in dendrites and at distal synapses following fear conditioning. It is noteworthy that MAPK regulates a number of other proteins, including dendritic K+ channels (Yuan et al., 2002), glutamate receptors (Zhu et al., 2002), and may also control dendritic protein synthesis (Kelleher et al., 2004). Thus, the parallel activation of synaptodendritic and somatonuclear pools of MAPK supports the general hypothesis that memory formation depends on several MAPK-regulated events including synaptic activity, dendritic protein synthesis, and transcription (for a review see Sweatt, 2004).
Although CREB-mediated transcription is necessary for memory formation and depends on MAPK signaling (Athos et al., 2002; Bourtchuladze et al., 1994; Pittenger et al., 2002), the CREB kinase activated by MAPK following training for contextual memory was not known. We were particularly interested in determining if training for contextual fear activates RSK2 or MSK1 because studies using cultured neurons have implicated both kinases in CREB-mediated transcription through the phosphorylation of transcription factors and histones (Impey et al., 1998a; Arthur et al., 2004; Brami-Cherrier et al., 2005; Soloaga et al., 2003). We discovered that fear conditioning activates MSK1, but not RSK2, in CA1 neurons, and that activation of MAPK and MSK1 was tightly correlated on a cell-by-cell basis. Furthermore, the activation of MSK1 induced by training was abrogated in mice lacking Ca2+-stimulated adenylyl cyclases or by post-training inhibition of MEK1/2. This is the first evidence implicating MSK1 in MAPK-dependent CREB phosphorylation during formation of contextual memory. The identification of MSK1, and not RSK2, as the activated CREB kinase emphasizes that signaling mechanisms inferred from cultured neuron studies do not necessarily apply in vivo. Definitive evidence as to the relative importance of both CREB kinases during memory formation may come from the use of conditional mutant mice or novel MSK1 antagonists.
In summary, our data indicates that stimulation of MAPK in dendrites and the nucleus following training for contextual memory depends on Ca2+-stimulated adenylyl cyclase activity and leads to the activation of the CREB kinase MSK1. Furthermore, we have discovered that signaling elements for CREB-mediated transcription starting with the initial cAMP signal, PKA, MAPK, MSK1 and CREB are all activated in the same subset of neurons after training. We conclude that one of the major reasons adenylyl cyclase activity is required for memory is to support the activation of MAPK, MSK1 and CREB in hippocampal neurons.
Experimental Procedures
Animals
Experiments were approved and in accordance with the animal care committee's guidelines at the University of Washington. Adult (3–4 months) male C57BL/6 mice and AC1/AC8 double knockout (DKO) mice were used. DKO mice were generated as described (Wong et al., 1999) and bred into the C57BL/6 background for more than nine generations.
Contextual fear conditioning
One week before conditioning, mice were housed individually and handled daily. On the day of training, the mouse was placed into a conditioning chamber made of transparent walls with a metal grid floor (Colbourn) and allowed two minutes to explore. A two-second, 0.7 mA foot shock was then delivered, and after an additional minute in the cage the mouse was returned to its home cage. Unpaired controls were immediately shocked after they were placed in context and then returned to their home cages. Non-shocked mice were placed in the chamber for three minutes but never shocked. Naive mice were neither shocked nor placed in the conditioning chamber.
Tissue preparation and immunohistochemistry
Mice were anesthetized with a ketamine/xylazine cocktail and transcardially perfused with 4% formaldehyde in 0.1 M PB (pH 7.4). Brains were cryoprotected in 30% sucrose, and serial coronal sections (20 μm) were cut in a cryostat. Sections were pretreated with 1% NaBH4 for 15 min, 1.5% H2O2 in 10% ethanol in PB for 20 min, blocked and permeabilized in 10% normal serum, 2% BSA, 50 mM glycine, 0.2% Triton X-100 in PBS for 2 h, and incubated in rabbit anti-phospho-Thr202/Tyr204-ERK1/2 (1:250), rabbit anti-phospho-Thr581-MSK1 (1:400) or rabbit anti-phospho-PKA substrates (1:200) antibodies (Cell Signaling) for 24–36 h at 4°C. Sections were then incubated with biotinylated goat anti-rabbit IgG (1:250) for 2 h, followed by standard ABC (Vectashield), and developed with diaminobenzidine-nickel. Solutions used in steps up to and including the primary antibody contained 50 mM NaF and 1 mM Na3VO4 to block phosphatases. Sections from the different experimental conditions were stained in parallel using common working solutions and developed for identical times.
Intra-hippocampal infusions
Freely moving mice with bilaterally implanted cannula (Plastics One) (coordinates from bregma: AP −1.65, ML 1.5, DV −1.5) received infusions of 40 mM Sp-cAMP or Rp-cAMP (Sigma) dissolved in sterile PBS (0.5 μl/hippocampus; rate 0.25 μl/min) with an automated syringe pump (World Precision Instruments) connected to the infusion cannula via polyethylene tubing. Ten minutes later, mice were anesthetized, perfused and processed for immunohistochemistry as above. SL327 (Calbiochem) was injected at 50 mg/Kg, i.p., 30 minutes before intra-hippocampal infusions.
Quantitative analysis
For cell counts in DAB-stained sections, 6–8 hippocampal sections per mouse—spanning between −1.6 mm and −2.2 mm from bregma—were analyzed under the microscope (200X). Counts per subregion and section were averaged across sections to yield individual means, and these were in turn averaged across mice to yield group means. For optical density measurements, three coronal sections 100 μm apart and encompassing the center of the infusion site were analyzed per mouse. The stratum pyramidale spanning 200 μm laterally from the infusion site was outlined using ImageJ software and the mean grayscale signal intensity within was measured and normalized to the contralateral hippocampus (infused with vehicle). Values were corrected for background, which was taken from sections stained without primary antibody. All quantifications were done blind to condition and genotype.
Immunofluorescent staining
For detection of pERK, the best results were obtained with tyramide signal amplification (TSA). Sections were pretreated as above and primary antibodies visualized by deposition of Cyanine3-tyramide complexes according to manufacturer's instructions (Perkin Elmer). For multiple labeling, sections were first stained for pERK (1:10,000), phospho-Thr180/Tyr182-p38 MAPK (1:4,000; Cell Signaling), or phospho-MSK1 (1:5,000) with TSA, rinsed and blocked again, and incubated in one (or two) of the following antibodies: mouse anti-synaptophysin (1:400), mouse anti-GFAP (1:500), from Sigma; mouse anti-NeuN (1:1,000), mouse anti-CaMKIIα (1;2,000), rabbit anti-MAP2 (1:2,000), from Chemicon; rabbit anti-phospho-PKA substrates (1:200), rabbit anti-phospho-Msk1 (1:400), from Cell Signaling; rabbit anti-phospho-CREB (1:300; Upstate), or rabbit anti-phospho- Ser221/227-RSK1/2 (1:200; R&D Systems). Second primary antibodies were detected with corresponding secondary antibodies conjugated to Alexa Fluor 488 or 647 (1:500). Sections were Hoescht counterstained and mounted in anti-fading medium (Gelmount). The first primary antibody was used at a dilution so low that it consistently failed to be recognized by conventional fluorescent secondary antibodies but it was clearly detected by TSA amplification. Absence of cross-reactivity between the first primary antibody and the second secondary antibody was confirmed in experiments in which the second primary antibody was omitted (data not shown).
Confocal imaging and analysis
Images were captured on a Leica SP1 or SL confocal microscope and analyzed with MetaMorph software using the Integrated Morphometry Analysis and Region Measurements tools (Universal Imaging Corporation, West Chester, PA). Density filter, pinhole aperture, detector gain and offset were initially set to obtain pixel densities within a linear range, and were then kept constant for experimental comparisons. Signals were acquired using sequential line scanning. For analysis of cell bodies, Z-series stacks (z-step size 1 μm) were collected as 512 x 512 pixel images (average of 4 frames per image) with a 40X objective (NA, 1.4). To estimate the percentage of activated neurons, Z-stacks 6–8 μm high of the rostral one-third of the CA1 hippocampus were acquired from at least two sections per mouse. Analysis of average somatic intensity and line scan intensity profiles were performed at the midplane of every neuron based on the nuclear counterstaining. Line scans were randomly oriented from the center of the nucleus. MAP2 or CaMKIIα, and Hoescht were used to define cytoplasmic and nuclear boundaries, respectively. Co-localization of immunosignals was confirmed in x, y and z dimensions. For analysis of synapses, 1024 x 1024 pixel images (average of 8 frames per image, z-step size 0.4 μm) were collected with a 63X objective and zoomed to an x/y pixel size of 58 nm. Synaptic analysis was limited to the first 2 μm from the surface of the section, where synaptophysin puncta were most sharp and bright. Threshold was set to the sum of the average intensity and standard deviation of the field. We defined synapses as those synaptophysin puncta labeled above threshold with a size ranging between 0.1–0.7 μm2. Labeling intensity was represented on a gray scale of 0 to 255.
Immunoblotting
Mice were sacrificed by cervical dislocation and their dorsal hippocampi were quickly dissected and frozen in an ethanol/dry ice bath. For whole-cell fractions, frozen tissue was weighed and sonicated in homogenization buffer (0.32M sucrose, 10mM HEPES, pH 7.4, 2mM EDTA, 1mM DTT, 1mM PMSF, pH 7.4, 1:100 dilutions of Sigma Protease, Phosphatase 1 & Phosphatase 2 Inhibitor Cocktails) using 2.5 μl buffer per μg of tissue. For nuclear extracts, tissue homogenates were centrifuged at 1,000 g, the pellet was resuspended in the presence of 0.5% NP-40, centrifuged again, and the second pellet was incubated in nuclear extraction buffer (10 mM HEPES, 400 mM NaCl, 0.5% NP-40, 10 % glycerol, 2 mM EDTA, 5 mM DTT, with protease and phosphatase cocktails) for 30 min, and centrifuged at 20,000 g for 10 min. The final supernatant was used as the nuclear extract. An equal volume of 4X SDS-PAGE sample buffer was added to each sample and they were heated to 90°C for 10 minutes. Samples were loaded on Tris-HCl resolving gels for SDS-PAGE analysis. Proteins were then transferred to PVDF membranes (Immobilon-P, Millipore) and blocked in 10% nonfat dry milk and 0.1 % Triton X-100 in PBS (PBST). Primary antibodies were used at 1:1,000 dilution overnight at 4°C in 5% BSA in PBST, and included: rabbit anti-phospho-ERK1/2, rabbit anti-phospho-MSK1, rabbit anti-pCREB, and rabbit anti-CREB, from Cell Signaling; goat anti-ERK1/2, and goat anti-RSK2, from Santa Cruz; mouse anti-actin, from Chemicon; rabbit anti-phospho-MSK1/2, rabbit anti-phospho-RSK1/2, and goat anti-MSK1, from R&D Systems. Alkaline phosphatase- or HRP-conjugated secondary antibodies (1:2,000; Cappel) were used at room temperature for one hour in 5% BSA in PBST. Immunoblots were developed using CDP-Star (Tropix) or ECL Plus (Amersham). When necessary, immunoblots were stripped in 25mM glycine, pH 2, 1% SDS for 30 minutes and washed in PBST. Bands were visualized with Kodak X-Omat Blue film and digitized with a scanner calibrated to measure optical density. ImageJ was then used to measure the integrated optical density of bands. Care was taken to expose film so that band intensities fell within the linear range of the calibration curve.
Statistical tests
The Wilcoxon Sum-of-Ranks test and the Kruskal-Wallis test were used for two-sample and three- (or more) sample comparisons, respectively. Nevertheless, most data appeared to follow a normal distribution, according to Shapiro-Wilk test, and similar statistical conclusions were reached when data sets were subjected to parametric analysis (T test, or ANOVA followed by Tukey pair-wise comparisons; data not shown). Unless otherwise stated, measures are expressed as mean ± standard error of the mean. Significance was set at 0.05.
Acknowledgments
We thank Greg Martin for expert technical assistance with confocal microscopy, Mike Garelick for help with mouse cannulations, and members of the Storm lab for discussions. This work was supported by National Institutes of Health grants NS 20498 and MH 073601. C.B.S was supported by a postdoctoral Ruth L. Kirschstein research award. Z.S. was supported in part by grant T32 GM07108.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel T, Nguyen PV, Barad M, Deuel TAS, Kandel ER. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Arthur JS, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, Impey S. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24:4324–4332. doi: 10.1523/JNEUROSCI.5227-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athos J, Impey S, Pineda V, Chen X, Storm D. Hippocampal CRE-mediated gene expression is required for contextual memory formation. Nature Neuroscience. 2002;5:1119–1120. doi: 10.1038/nn951. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, Simon AJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR. Making new connections: role of Erk/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998a;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nature Neurosci. 1998b;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Maas JW, Jr, Vogt SK, Chan GC, Pineda VV, Storm DR, Muglia LJ. Calcium-stimulated adenylyl cyclases are critical modulators of neuronal ethanol sensitivity. J Neurosci. 2005;25:4118–4126. doi: 10.1523/JNEUROSCI.4273-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Casadio A, Zhu HX, E YP, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-form facilitation of Aplysia sensory to motor synapses: A function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Brambilla R. Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell Mol Life Sci. 2000;57:604–611. doi: 10.1007/PL00000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JS. MSK1 activity is controlled by multiple phosphorylation sites. Biochem J. 2005;387:507–517. doi: 10.1042/BJ20041501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. Embo J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar LTP in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Wiig KA, Bear MF, Alberini CM. A molecular correlate of memory and amnesia in the hippocampus. Nat Neurosci. 1999;2:309–310. doi: 10.1038/7217. [DOI] [PubMed] [Google Scholar]
- Tully T, Bourtchouladze R, Scott R, Tallman J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov. 2003;2:267–277. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- Vanhoose AM, Clements JM, Winder DG. Novel blockade of protein kinase A-mediated phosphorylation of AMPA receptors. J Neurosci. 2006;26:1138–1145. doi: 10.1523/JNEUROSCI.3572-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR. Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci. 1998;18:3186–3194. doi: 10.1523/JNEUROSCI.18-09-03186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen- activated protein kinase (MAP kinase) pathway. J Neurosci Res. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci USA. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Del Vecchio M, Zhou H, Tully T. CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell. 1995;81:107–115. doi: 10.1016/0092-8674(95)90375-5. [DOI] [PubMed] [Google Scholar]
- Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437:569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]