

Abstract
Aminoglycoside antibiotics target an internal RNA loop within the bacterial ribosomal decoding site. Here, we described the synthesis and SAR of novel 3,5-diamino-piperidine derivatives as aminoglycoside mimetics, and show they act as inhibitors of bacterial translation and growth.
Keywords: Aminoglycosides, Antibiotics, Translation inhibitors, 2-deoxy-streptamine, Ribosome, Decoding site, 3, 5-diamino-piperidine
Bacterial resistance to antibiotics is on the rise and represents a global medical threat. In hospitals in the United States, approximately two million patients per year are infected.1 The majority of these nosocomial pathogens are resistant to at least one antibiotic and result in about 90,000 deaths per year; a number that has increased 7-fold over the last decade. The recent and rapid spread of community acquired methicillin resistant Staphylococcus aureus further highlights the threat of resistance development and illustrates the need for new antibiotics that work by novel mechanisms.2 Given the broad genetic and physiological diversity of bacterial pathogens and the need for empiric therapies that cover a broad panel of organisms, it is not surprising that discovery of new antibiotics has advanced slowly. Central to antibiotic discovery is identifying broadly validated targets. One such proven target is the bacterial ribosome, which is the target for a significant number of clinically important antibiotics that bind at the ribosomal RNA (rRNA).3 Here, we expand on the description of a novel series of antibacterial compounds that target rRNA and blocks bacterial translation and growth.4
Three-dimensional structures of different aminoglycosides bound to the decoding site, or A-site, within the 16S rRNA have been determined by X-ray crystallography.5 Importantly, these studies have shown that 2-deoxystreptamine (2-DOS), a conserved core scaffold among aminoglycosides, binds in a similar manner regardless of the 4,5- or 4,6-disubstitutions found in the neomycin or gentamicin families, respectively (Fig. 1).6 The cis-1,3-configured amino groups of 2-DOS are predominantly involved in base recognition by forming conserved hydrogen bonds with A1493, G1494 and U1495 of the 16S rRNA. These interactions anchor the aminoglycoside scaffold within the A-site internal loop and displace residues A1492 and A1493 from the RNA interior. These two adenine residues act as a molecular switch that is involved in securing the fidelity of translation by interacting with the first two bases of the mRNA-tRNA codon-anticodon hybrid. Thus, aminoglycoside binding impairs the ribosome’s ability to discriminate against near-cognate tRNA-mRNA pairings, which ultimately causes cell death.
Figure 1.
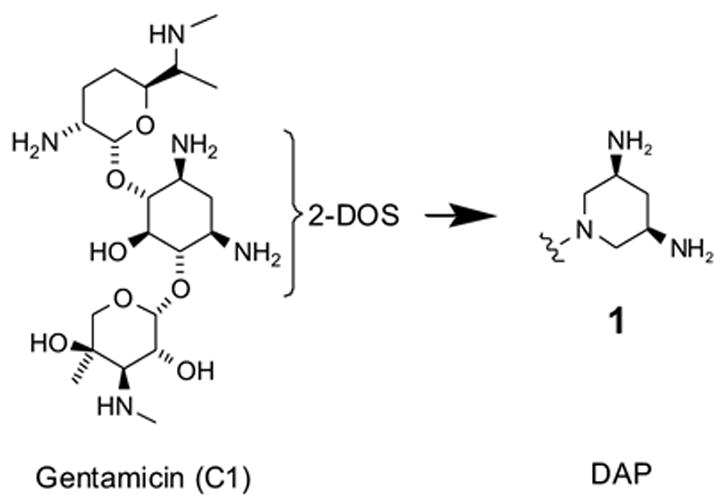
The aminoglycoside gentamicin and the DAP scaffold that mimics 2-DOS of the aminoglycoside.
While 2-DOS is recognized as a key pharmacophore for binding rRNA, its synthesis and modification is challenging due to the five contiguous stereogenic centers.7 Therefore, we had developed synthetic mimetics of 2-DOS, one of which is cis-3,5-diamino-piperidine (DAP) (1) (Fig. 1).4 The DAP ring retains the characteristic cis-1,3-diamine configuration of 2-DOS that is important for RNA recognition. In contrast to aminoglycosides, the reduced chemical complexity renders the DAP series and derivatives amenable to rapid elaboration by parallel synthesis. Here, we focus on the synthesis and structure-activity relationships of DAP and other synthetic mimetics of 2-DOS.
Scheme 1 outlines the synthesis of the Boc-protected DAP (5). In this specific synthesis, the two NO2 groups of 2-chloro-3,5-dinitropyridine (2) was hydrogenated under H2 atmosphere at room temperature in the presence of 10% Pd/C to generate 3,5-diaminopyridine 3. The two amino groups were then protected as a di-Boc derivative 4 which was subsequently hydrogenated at 2200 psi using 5% Rh/C in the presence of acidic acid at 110 °C to yield the piperidine derivative 5.8,9 Since aminoglycoside are relatively large molecules that engage in at least 15 contacts with rRNA,5 a single DAP scaffold would not allow high affinity binding to rRNA and, hence, additional functionality was added. Guided by modeling studies and the availability of a straightforward synthetic route, DAP was linked to a triazine core which resulted in molecules (termed DAPT) that had a high affinity for A-site containing RNA oligonucleotides.4,10 The DAP scaffold and analogs (R1 and R2, termed headpiece) thereof were linked at the 2 and 6 positions of triazine (6) (Fig. 2). The so-called “tailpiece” substituents (R3) were attached at the triazine 4-position via a N-linkage. Representatives of R3 used in this study are shown in Figure 2.4,10
Scheme 1.
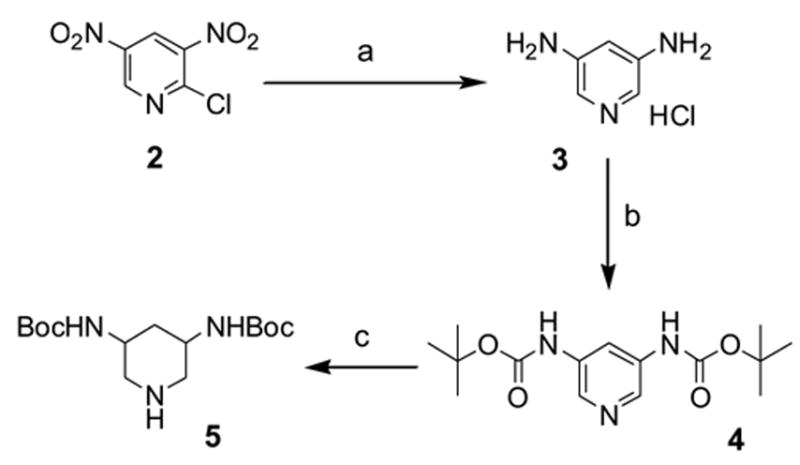
(a) H2, 10% Pd/C, rt. (b) Boc2O, NaHCO3, H2O/MeOH/THF. (c) H2, 2200 psi, 5% Rh/C, CH3COOH, 110 °C.8,9
Figure 2.
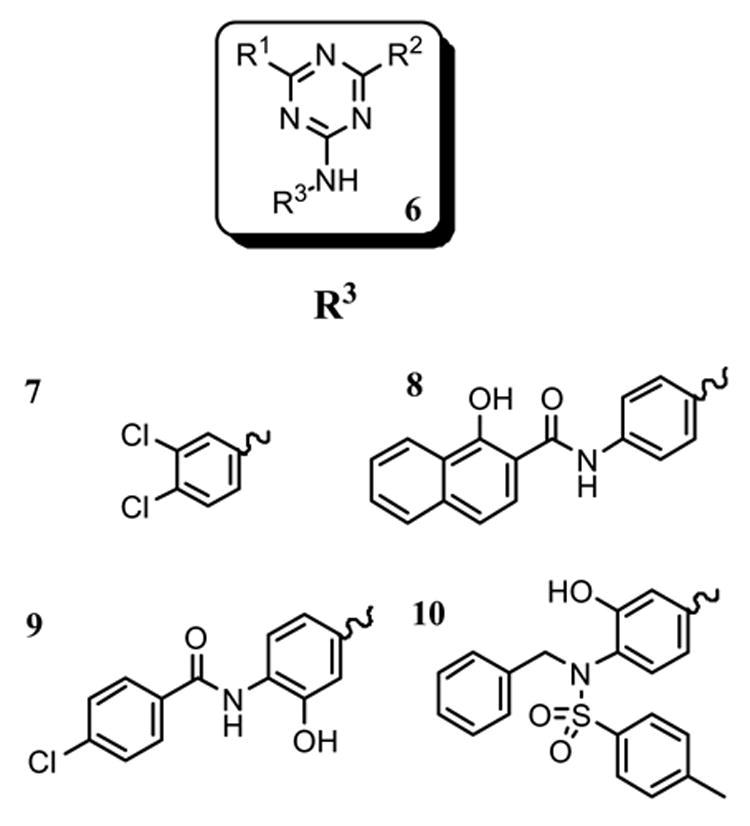
DAPT (6) and tailpiece (R3) structures.
Table 1 highlights the SAR of headpieces where R1 and R2 are identical resulting in a symmetric configuration. The symmetrical arrangement of two DAPs in compounds 11 and 12 resulted in potent translation inhibitors. Importantly, in the symmetric configuration, the acyclic headpiece (13) and a dimethylated DAP (14) retained activity (Table 1). Both of these headpieces allow the amino groups to be located in identical spatial orientation found both in DAP and 2-DOS and, hence, may account for similar hydrogen bonding with target rRNA. Although two amino groups in the five-membered ring headpiece of compound 15 have a slightly different spatial orientation than in DAP, 15 was nevertheless active. In contrast, when one of the amino groups in the headpiece is removed, or the spatial orientation is significantly altered, activity is abolished (compounds 16–18). These results support our hypothesis that DAP can indeed substitute for the 2-DOS pharmacophore. Moreover, these findings show that the signature cis-3,5-diamino fragment of DAP, and by analogy the cis-1,3-diamino fragment of 2-DOS, can tolerate a variety of structure modifications as exemplified in compounds 11–15.
Table 1.
Symmetric headpiece substitutions and protein synthesis inhibitory activity.
| Compounda | R1 and R2 | R3 | IC50b |
|---|---|---|---|
| 11 |
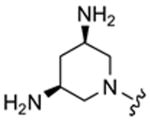
|
7 | 10 |
| 12 | 8 | 7 | |
| 13 |
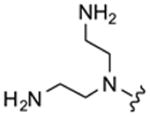
|
8 | 7 |
| 14 |
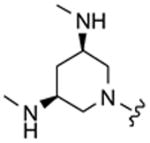
|
7 | 5 |
| 15 |
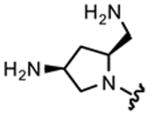
|
8 | 8 |
| 16 |
|
8 | 100 |
| 17 |
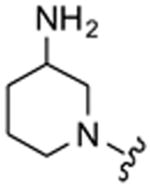
|
7 | >1000 |
| 18 |
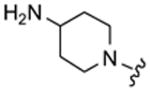
|
7 | 800 |
| Kan | 0.4 | ||
| Tet | 2.8 |
Kan, kanamycin; Tet, tetracycline.
Coupled in vitro transcription-translation assay with Escherichia coli extracts (μM).4
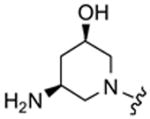
Table 2 summarizes headpiece SAR with asymmetric configurations of R1 and DAP (R2). For most headpiece configurations there are as many as three R3 substitutions on the triazine core (6), which allowed detailed insight into headpiece (R1) SAR. Interestingly, a mono-DAP compound (19) (R1 = H) has similar activity as di-DAP compounds (12, 20 and 21). Consistent with the results shown in Table 1, active headpieces in symmetric configuration (13 and 14) also resulted in active inhibitors when they were asymmetrically combined with DAP (22–24, 25 and 29–31) (Table 2). Conversely, a headpiece with symmetric configuration that resulted in poor activity (Table 1; 16) was found to reduce activity when asymmetrically combined with DAP (38–40). In general, the replacement of an amino group with a hydroxy group on five- (32–34) or six-membered ring headpiece (38–40) slightly reduced activity compared to the di-amino substitutions (29–31 and 12, 20–21, respectively). Similarly, other mono-hydroxy substitutions exhibit moderate to weak activity (35–37, 41–43 and 44–46). An azetidin-3-ylamine substitution was tolerated (52–53), while a pyrrolidine-3,4-diamine substitutions (50–51) resulted in weak translation inhibitors. Di-hydroxy substitutions (47–49) were found to yield particularly weak inhibitors and appeared to destabilize the binding of the asymmetric DAP scaffold to its target. An acetamide substitution on the piperidine abolished activity (54–55). In summary, these results show that a symmetric substitution of two DAP moieties on the triazine core represents an optimal configuration for inhibitors of bacteria translation and growth.4,10 However, this work also shows that there is room to alter the headpiece configuration while retaining antibacterial activity, which may be used to optimize other pharmaceutical properties such as bioavailability.
Table 2.
Asymmetric (R2 is DAP) headpiece substitutions.
| Compda | R1 | R3 | IC50 | Ecb | Sac |
|---|---|---|---|---|---|
| 19 | H | 8 | 16 | 4 | nd |
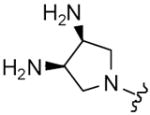
| 12 |
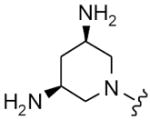
|
8 | 10 | 1 | 2 |
| 20 | 9 | 7 | 2 | 16 | |
| 21 | 10 | 3 | 4 | 8 | |
| 22 |
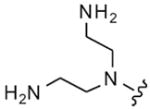
|
8 | 10 | 4 | 8 |
| 23 | 9 | 18 | 16 | 32 | |
| 24 | 10 | 1 | 16 | 8 | |
| 25 |
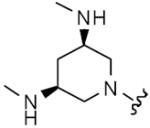
|
8 | 8 | 2 | 8 |
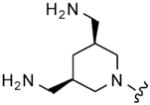
| 26 |
|
8 | 8 | 2 | 4 |
| 27 | 9 | 11 | 8 | 16 | |
| 28 | 10 | 1 | 16 | 8 | |
| 29 |

|
8 | 10 | 4 | 2 |
| 30 | 9 | 12 | 8 | 32 | |
| 31 | 10 | 13 | 32 | 2 | |
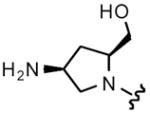
| 32 |
|
8 | 19 | 4 | 8 |
| 33 | 9 | 13 | 16 | 32 | |
| 34 | 10 | 15 | 16 | 8 | |
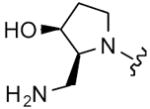
| 35 |
|
8 | 10 | 8 | 16 |
| 36 | 9 | 19 | 16 | 32 | |
| 37 | 10 | 21 | 16 | 8 | |
| 38 |
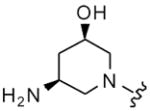
|
8 | 17 | 2 | 8 |
| 39 | 9 | 40 | 8 | 64 | |
| 40 | 10 | 17 | 16 | 16 | |
| 41 |
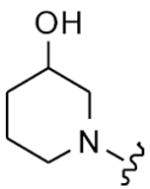
|
8 | 310 | 16 | 16 |
| 42 | 9 | 40 | 64 | 32 | |
| 43 | 10 | 44 | 16 | 8 | |
| 44 |
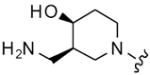
|
8 | 21 | 4 | 16 |
| 45 | 9 | 31 | 32 | 32 | |
| 46 | 10 | 27 | 32 | 16 | |
| 47 |
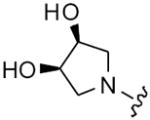
|
8 | 72 | 16 | 32 |
| 48 | 9 | 77 | 32 | 64 | |
| 49 | 10 | 43 | 32 | 32 | |
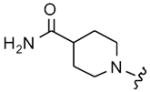
| 50 |
|
9 | 58 | 16 | 16 |
| 51 | 10 | 36 | 32 | 16 | |
| 52 |

|
9 | 14 | 8 | nd |
| 53 | 10 | 11 | 16 | 16 | |
| 54 |
|
9 | >1000 | nd | nd |
| 55 | 10 | >1000 | nd | nd | |
| Cm | 1.5 | 4 | 8 |
nd, not determined.
Cm, chloramphenicol.
E. coli ATCC 25922 minimum inhibitory concentration (μg/mL)4
S. aureus ATCC 25923 (MIC).
Summary and Perspective
This paper focused on headpiece optimization on our novel DAPT translation inhibitors. The active DAP pharmacophore was derived from structural information of the molecular recognition between the conserved 2-DOS scaffold of aminoglycosides and the A-site RNA.5,6 This work expands on our efforts to rationally design 2-DOS mimetics, including the aminoazepane and novel acyclic deoxystreptamine scaffolds.10 After the initial design of DAPT, the series then evolved through an iterative process of parallel synthesis and high throughput testing that led to promising lead compounds. Rapid medicinal chemistry elaboration can now be used to potentially generate a clinical candidate that may improve on the pharmaceutical properties of aminoglycosides, including persisting resistance, poor bioavailability and undesirable toxicity.
In a future communication we will describe SAR around protein binding and the effect of serum on antibacterial potency. In the context of this study, a particularly intriguing observation was found with compound 22 in that the E. coli MIC improved in the presence of serum protein. This synergistic activity with serum was titratable and reached a peak reduction in MIC of 16-fold. Separately, particular core and tailpiece substitutions were found to reduce the serum MIC shift. A striking feature of the DAPT series is their potent antimicrobial activity against Pseudomonas aeruginosa, a pathogen for which there is a unmet clinical need to develop new antibiotics.2 In one study, 53 clinical isolates of P. aeruginosa were tested against a DAPT compound (compound 1a in reference 4) and commercial antibiotics. The MIC90 of the DAPT compound was 8 μg/ml, while the seven reference antibiotics that included amikacin, imipenem, ciprofloxacin, ceftazidime and tazobactam/piperacillin, all had higher MIC90 values. Based on these and other findings we are particularly interested in developing the DAPT compounds as novel anti-P. aeruginosa agents.
Acknowledgments
This work was supported in part by a grant from the National Institute of Allergy and Infectious Disease (AI51105).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.National Institute of Allergy and Infectious Disease fact sheet. Apr, 2006. [Google Scholar]
- 2.(a) DeRyke CA, Maglio D, Nicolau DP. Exp Opin Pharmacother. 2005;6:873. doi: 10.1517/14656566.6.6.873. [DOI] [PubMed] [Google Scholar]; (b) Rice LB. Am J Infect Control. 2006;34:S11. doi: 10.1016/j.ajic.2006.05.220. [DOI] [PubMed] [Google Scholar]
- 3.Sutcliffe JA. Curr Opin Microbiol. 2005;8:1. doi: 10.1016/j.mib.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Y, Gregor VE, Sun Z, Ayida BK, Winters GC, Murphy D, Simonsen KB, Vourloumis D, Fish S, Froelich JM, Wall D, Hermann T. Antimicrob Agents Chemother. 2005;49:4942. doi: 10.1128/AAC.49.12.4942-4949.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]; (b) Francois B, Russell RJM, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. Nucleic Acids Res. 2005;33:5677. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Han Q, Zhao Q, Fish S, Simonsen KB, Vourloumis D, Froelich JM, Wall D, Hermann T. Angew Chem Int Ed Engl. 2005;44:2694. doi: 10.1002/anie.200500028. [DOI] [PubMed] [Google Scholar]
- 6.(a) Pfister P, Hobbie S, Vicens Q, Böttger EC, Westhof E. Chembiochem. 2003;4:1078. doi: 10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]; (b) Simonsen KB, Ayida BK, Vourloumis D, Winters GC, Takahashi M, Shandrick S, Zhao Q, Hermann T. ChemBioChem. 2003;4:886. doi: 10.1002/cbic.200300689. [DOI] [PubMed] [Google Scholar]; (c) Vicens Q, Westhof E. Biopolymers. 2003;70:42. doi: 10.1002/bip.10414. [DOI] [PubMed] [Google Scholar]
- 7.Busscher GF, Rutjes FPJT, van Delft FL. Chem Rev. 2005;105:775. doi: 10.1021/cr0404085. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Y, Vourloumis D, Gregor VE, Winters GC, Hermann T, Ayida BK, Sun Z, Murphy D, Simonsen KB. PCT WO 2005/028467 A1. 2005. [Google Scholar]
- 9.(a) Synthesis of 3,5-diaminopyridine hydrochloride (3): 2-chloro-3,5-dinitro-pyridine (27.36 g, 2) was divided into two equal portions and added into two 2L round bottomed flasks. Methanol (700 mL) was then added to each flask. Nitrogen gas was bubbled through the solution while stirring and 7.15 g of 10% palladium on carbon was divided into two equal portions and added to each flask portion-wise while under nitrogen. The solutions were cooled in an ice-water bath under N2. Hydrogen balloons then replaced the N2 inlets. After evacuation, the solutions were exposed to H2 gas at slightly positive pressure. This vacuum-hydrogen exchange was done at least 5 times. The reactions were then stirred under slight positive pressure of H2 for 2 days. Then the solutions were filtered through Celite under reduced pressure to remove the catalyst. The filtrate was concentrated in vacuo. The crude product (24.27 g, 100% yield) was a dark green solid and was immediately used in the next step without purification. Product 3 was confirmed by LC-MS.(b) Synthesis of di-Boc-protected 3,5-diamino-pyridine (4): Compound 3 (19.56 g, 0.13 mol) was dissolved in 150 mL of methanol in a 2 L reaction vessel. Nitrogen was used to purge the flask and the reaction solution was cooled to 0°C. A solution of 58.67 g (0.27 mol) of di-tert-butyldicarbonate in 85 mL of methanol was added dropwise. 18.9 mL of triethylamine (0.13 mol) was diluted with 20 ml of methanol and added dropwise. Then another 58.67 g of di-tert-butyldicarbonate dissolved in 85 mL of methanol was added dropwise. After that another 29.34 g of di-tert-butyldicarbonate, dissolved in 40 mL of methanol was added dropwise. The reaction mixture was warmed to rt and stirred overnight. TLC analysis showed that no starting material and very little mono-Boc-protected diaminopyridine remained. The methanol was removed in vacuo. The resulting solid was dissolved in ethyl acetate and was washed with brine by extraction. The aqueous layer was extracted three times with ethyl acetate. The combined organic layers were dried with MgSO4 and passed through a layer of silica gel (135 cm x 3 cm) constructed in a fritted funnel. The silica was thoroughly washed with ethyl acetate. The ethyl acetate solution was concentrated in vacuo and the resulting solid was dissolved in a minimal amount of refluxing methylene chloride and then cooled to rt. The crystallized solid was filtered under reduced pressure and washed with a very small amount of CH2Cl2 and hexane and was dried in a vacuum oven at 50–60°C. The first three batches of crystals weighed a total of 26.99 g, and a fourth batch of crystals (from methanol) weighed 3.59 g, yielding a total of 30.58 g of product 4 (73.6% yield).1H NMR (CDCl3): δ 8.22 (d, 2H, J = 2.4 Hz), 8.16 (t, 1H, J = 2.4 Hz), 6.66 (s, 2H, NH), 1.54 (s, 18 H, Boc).(c) Synthesis of (3R, 5S)-3,5-bis-(tert-butoxycarbonylamino)-piperidine (5): Compound 4 (10 g, 32.34 mmol) was dissolved in 350 mL of methanol. Glacial acetic acid (1.85 mL, 32.34 mmol) was added, followed by 5% Rh-C (wet, 6.66 g, 5 mol %), purged with N2 and transferred to a high-pressure hydrogenator. The vessel was flushed twice with H2 gas at 500 psi, the H2 pressure was then increased to 1000 psi and heated to 110°C. The H2 pressure was subsequently increased to 2200 psi and the mixture was stirred for 28 hrs, followed by cooling to rt. The catalyst was filtered off through Celite under reduced pressure. The filtrate was then concentrated in vacuo to remove methanol. Na2CO3 (3.43 g, 32.34 mmol) in 100 mL of H2O was added followed by 100 mL of EtOAc. After stirring for 90 min, a thick white precipitate formed. The mixture was then filtered under reduced pressure and the solid was washed with EtOAc, dried in a vacuum oven overnight at 60°C. The desired product 5 (5.6 g) was obtained as an off-white solid that was confirmed by LC-MS and1H NMR. MS m/e 316 [M+1]+ (exact MS: 315).1H NMR (DMSO-d6): 6.70 (d, 2H, J = 7.2 Hz, 2NHCO), 3.16–3.28 (m, 2H, H3,5ax), 2.76–2.84 (m, 2H, H2,6eq), 2.17 (br, 1H, NH), 1.99 (t, 2H, J = 11.6 Hz, H2,6ax), 1.84–1.90 (m, 1H, H4eq), 1.37 (s, 18H, Boc), 1.10 (q, 1H, J = 11.6 Hz, H4ax). HPLC has a single peak. Melting point: 214.6 °C (decompose). The above process produces the cis-isomer in high purity.
- 9.Zhou Y, Sun Z, Froelich JM, Hermann T, Wall D. Bioorg Med Chem Lett. 2006;16:5451. doi: 10.1016/j.bmcl.2006.07.052. [DOI] [PubMed] [Google Scholar]
- 10.(a) Barluenga S, Simonsen KB, Littlefield ES, Ayida BK, Vourloumis D, Winters GC, Takahashi M, Shandrick S, Zhao Q, Han Q, Hermann T. Bioorg Med Chem Lett. 2004;14:713. doi: 10.1016/j.bmcl.2003.11.028. [DOI] [PubMed] [Google Scholar]; (b) Foloppe N, Chen IJ, Davis B, Hold A, Morley D, Howes R. Bioorg Med Chem. 2004;12:935. doi: 10.1016/j.bmc.2003.12.023. [DOI] [PubMed] [Google Scholar]; (c) Murray JB, Meroueh SO, Russell RJM, Lentzen G, Haddad J, Mobashery S. Chem Bio. 2006;13:129. doi: 10.1016/j.chembiol.2005.11.004. [DOI] [PubMed] [Google Scholar]; (d) Vourloumis D, Winters GC, Takahashi M, Simonsen KB, Ayida BK, Shandrick S, Zhao Q, Hermann T. ChemBioChem. 2003;4:879. doi: 10.1002/cbic.200300688. [DOI] [PubMed] [Google Scholar]