

Summary
Cell division is controlled by cyclin-dependent kinases (CDKs). In metazoans, S-phase onset coincides with activation of Cdk2, whereas Cdk1 triggers mitosis. Both Cdk1 and -2 require cyclin-binding and T-loop phosphorylation for full activity. The only known CDK-activating kinase (CAK) in metazoans is Cdk7, which is also part of the transcription machinery. To test the requirements for Cdk7 in vivo, we replaced wild-type Cdk7 with a version sensitive to bulky ATP analogs in human cancer cells. Selective inhibition of Cdk7 in G1 prevents activation (but not formation) of Cdk2/cyclin complexes and delays S phase. Inhibiting Cdk7 in G2 blocks entry to mitosis, and disrupts Cdk1/cyclin B complex assembly, indicating that the two steps of Cdk1 activation—cyclin-binding and T-loop phosphorylation—are mutually dependent. Therefore, by combining chemical genetics and homologous gene replacement in somatic cells, we reveal different modes of CDK activation by Cdk7 at two distinct execution points in the cell cycle.
Introduction
The cyclin-dependent kinases (CDKs) that promote chromosome duplication in S phase and segregation at mitosis require binding of cyclin and phosphorylation on the activation segment (T-loop) by a CDK-activating kinase (CAK) for full activity (reviewed by Morgan, 1997). Despite universal conservation of this two-step pathway, the organization of the CAK-CDK network—and the identity of CAK—have diverged (reviewed by Fisher, 2005). In metazoans, the only CAK identified to date is the Cdk7/cyclin H/Mat1 complex, which is also a component of the RNA polymerase (Pol) II general transcription factor (TF) IIH. Cdk7 has evolved two distinct recognition mechanisms for its structurally dissimilar substrates—the T-loops of CDKs and the carboxyl-terminal domain (CTD) of the largest subunit of Pol II—to accomplish its dual functions (Larochelle et al., 2006). The ortholog of Cdk7 in the budding yeast Saccharomyces cerevisiae, the Kin28 complex, is a CTD kinase contained in TFIIH (Feaver et al., 1994), but is devoid of CAK activity (Cismowski et al., 1995). CDK activation is instead catalyzed by Cak1, a single-subunit kinase related only distantly to CDKs (Espinoza et al., 1996; Kaldis et al., 1996; Thuret et al., 1996). The fission yeast Schizosaccharomyces pombe has two CAKs: the essential Mcs6 complex and the nonessential Csk1, orthologous to the metazoan and budding yeast enzymes, respectively (Hermand et al., 1998; Lee et al., 1999; Saiz and Fisher, 2002).
In budding yeast, the same CAK is required at both G1/S and G2/M transitions (Sutton and Freiman, 1997). The situation in metazoans is less clear. Cdk7 phosphorylates both Cdk1 and -2 selectively in human cell extracts (Larochelle et al., 2006), and either partial depletion of Cdk7 by RNA interference (RNAi) or near-quantitative immunodepletion with specific antibodies causes a proportional reduction in the Cdk2-activating capacity of a whole-cell extract (Wohlbold et al., 2006). CDK activation appears defective in cdk7 temperature-sensitive mutants of both Drosophila melanogaster (Larochelle et al., 1998) and Caenorhabditis elegans (Wallenfang and Seydoux, 2002), but in each case cell-cycle progression is blocked only at mitosis, not at S phase. T-loop phosphorylation of Cdk2 persists, moreover, in Drosophila cdk7 mutants at restrictive temperature (Larochelle et al., 1998). These observations, and the detection of minor CAK activities in vitro (Kaldis and Solomon, 2000; Liu et al., 2004), left open the possibility that another CAK exists in animal cells. Therefore, absent a genetic test of its function in vivo, the role of Cdk7 as the major or sole CAK in mammalian cells remained unproven (Abbas and Dutta, 2006).
Genetic studies of mammalian Cdk7 have been complicated by the enzyme’s dual roles in cell division and transcription. Mice lacking the Mat1 subunit of the Cdk7 complex die early in embryogenesis, which established that the complex was essential but limited analysis of the accompanying biochemical defects (Rossi et al., 2001). RNAi-mediated depletion of Cdk7 by ~70% in human cells produces no obvious phenotype (Wohlbold et al., 2006). We therefore took a chemical-genetic approach—the introduction into cells of a mutant kinase engineered to accommodate bulky, unnatural ATP analogs in its active site—to discern the functions of human Cdk7 in vivo. Expansion of the ATP-binding pocket by mutation of Phe91 to a glycine residue renders the kinase analog-selective and –sensitive (as) (Larochelle et al., 2006). In a previous study, we identified seven of ~10–15 protein substrates of Cdk7 in HeLa cell nuclear extracts, including Cdk1, Cdk2, Cdk4 and Pol II, by phosphorylation with Cdk7as and a radiolabeled substrate analog (Larochelle et al., 2006). The mutant enzyme was inhibited by a non-hydrolyzable analog with an IC50 ~17 nM, whereas the wild-type kinase was unaffected. The apparent KmATP of Cdk7as was ~sixfold higher than that of wild-type Cdk7, but below the likely intracellular ATP concentration, and the mutation did not affect enzyme turnover, suggesting that the mutant kinase would retain function in vivo (Larochelle et al., 2006).
Here we introduce Cdk7as into human colon cancer cells by homologous gene replacement. Cells expressing only Cdk7as are sensitive to growth inhibition by bulky, non-hydrolyzable ATP analogs. Inactivation of Cdk7as in synchronous cell populations rapidly impedes CDK activation and cell-cycle progression. Selective inhibition of Cdk7as during G1 curtails activating phosphorylation of Cdk2 and delays S phase entry, proving that Cdk7 is the Cdk2-activating kinase in vivo. Inhibition of Cdk7as during S/G2 progression prevents mitotic entry and abolishes Cdk1 activation by an unexpected mechanism: disrupted binding of Cdk1 to cyclin B. The block to complex assembly due to Cdk7-inhibition is reproduced in extracts of the mutant cells. Because Cdk1 requires a cyclin partner to be phosphorylated efficiently by Cdk7 (Fisher and Morgan, 1994), our results indicate that the two obligate steps in Cdk1 activation—cyclin-binding and phosphorylation by CAK—are mutually dependent and therefore likely to occur in concert.
Results
Engineered sensitivity to single-kinase inhibition in a human colon cancer cell
To replace wild-type Cdk7 with Cdk7as, we performed homology-directed gene replacement in HCT116 human colon carcinoma cells, with recombinant adeno-associated virus (rAAV) vectors (Kohli et al., 2004) (Figure 1A). In the first round of infection and selection, we obtained correctly targeted, Cdk7as/+ heterozygous clones (Figure 1B and data not shown). After excision of the neor drug-resistance marker with Cre recombinase, the second wild-type allele was targeted by repeating the cycle of rAAV infection, drug selection and screening by PCR. We confirmed the isolation of homozygous, Cdk7as/as cell clones by Southern blot hybridization (Figure 1B). Because the F91G mutation decreased electrophoretic mobility (Larochelle et al., 2006), we could also detect expression of Cdk7as (and absence of wild-type Cdk7 in Cdk7as/as cells) in immunoblots (Figure 1C, bottom).
Figure 1. Generation of Cdk7as/as HCT116 cells.
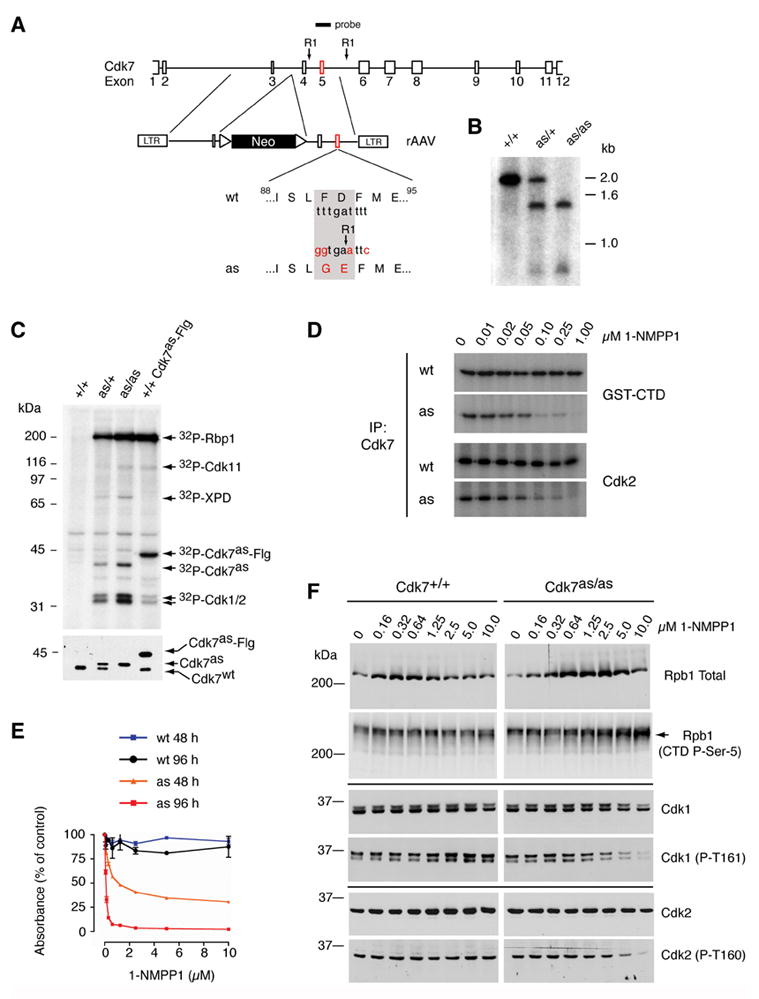
(A) The rAAV targeting vector, containing sequences from the Cdk7 locus flanking a neomycin resistance (neor) marker bounded by loxP sites. Exon 5 contained the F91G/D92E mutation and a novel EcoRI site for purposes of genotyping. The diagram indicates positions of Cdk7-derived sequences in the targeting vector and of the labeled hybridization probe, relative to EcoRI sites (RI) in the fourth and fifth introns. (B) Southern blot hybridization to EcoRI-digested genomic DNA detects single bands of ~2.0 and ~1.4 kb in the wild-type (+/+) and homozygous mutant (as/as) DNA, respectively, and an equal mixture of the two in heterozygotes (as/+). (C) Labeling with [γ-32P]N6-(benzyl)-ATP in whole-cell extracts of indicated genotypes (top). Identities of proteins labeled by both endogenous Cdk7as and recombinant, Flag-tagged Cdk7as (Cdk7as-Flg) in wild-type extract (last lane)—Rpb1 (the largest subunit of Pol II), Cdk1, -2 and -11—were inferred from similarity to the published pattern (Larochelle et al., 2006). The ~80 kDa polypeptide labeled by endogenous Cdk7as was identified by immunoprecipitation with anti-XPD antibodies. Immunoblot of kinase reaction mixtures (bottom) detects Cdk7wt and slower-migrating Cdk7as in the expected ratios; the two were expressed equally in heterozygous cells prior to excision of the neor gene (not shown), obviating the need for marker excision in homozygous Cdk7as/as cells. (D) Inhibition of Cdk7 immunoprecipitated from Cdk7as/as (AS) but not Cdk7+/+ (WT) cells by 1-NMPP1. The inhibitor was added at the indicated concentrations to GST-CTD or Cdk2 kinase assays, which contained 200 μM unlabeled ATP. (E) The dose-response of wild-type (wt) and Cdk7as/as (as) cells to 1-NMPP1 measured at 48 and 96 h of treatment by cell viability (MTT) assay. Error bars denote standard error of the mean in triplicate samples from a single representative experiment. (F) Dose-dependent effects of Cdk7 inhibition on phosphorylation of Cdk1, Cdk2 and Pol II in vivo. Asynchronous populations of Cdk7+/+ and Cdk7as/as HCT116 cells were incubated 14 h with the indicated concentrations of 1-NMPP1. Extracts were prepared and analyzed by immunoblotting with antibodies to: the amino-terminus of the Pol II subunit Rpb1 (Rpb1 Total); the Rpb1 CTD phosphorylated at the Ser5 position (CTD P-Ser-5); total Cdk1; Cdk1 phosphorylated on the T-loop (Cdk1 (P-T161)); total Cdk2 ; and Cdk2 phosphorylated on the T-loop (Cdk2 (P-T160)). Arrow at right indicates position of phosphorylated Rpb1 isoform that accumulates in Cdk7as/as cells treated with 1-NMPP1, which has a faster mobility than the slowest-migrating form (see also Supplemental Figure 1).
To confirm that Cdk7as expressed in HCT116 cells was functional, we measured phosphorylation in whole-cell extracts supplemented with the radioactive substrate analog, N6-(benzyl)-ATP (Figure 1C, top). Specific labeling of Cdk7 substrates occurred in the heterozygous (as/+) and homozygous (as/as) mutant, but not wild-type (+/+), cell extracts. Labeling by endogenous Cdk7as was dependent on gene dosage (compare signals in heterozygous and homozygous extracts), and qualitatively and quantitatively similar to labeling by the purified recombinant enzyme in wild-type cell extracts (Figure 1C, last lane). By immunoprecipitation, we identified the one polypeptide labeled uniquely in Cdk7as cell extracts as the DNA helicase XPD/ERCC2, a component of both TFIIH and a quaternary Cdk7/cyclin H/Mat1/XPD complex (Drapkin et al., 1996; Reardon et al., 1996; Larochelle et al., 2001; Chen et al., 2003). Endogenous XPD was not phosphorylated by exogenous Cdk7as, possibly because it was physically associated with (and preferentially phosphorylated by) wild-type Cdk7 in the extract.
To test whether Cdk7as expressed endogenously in human cells was sensitive to inhibition by the non-hydrolyzable analog 1-NMPP1, as was the recombinant enzyme expressed with baculoviruses and purified from insect cells (Larochelle et al., 2006), we immunoprecipitated Cdk7 from Cdk7as/as or Cdk7+/+ cells and tested its kinase activity towards both a Pol II CTD-containing fusion protein (GST-CTD) and human Cdk2. Cdk7 recovered from the mutant, but not the wild-type, cells was inhibited by 1-NMPP1, with an IC50 of ~50 nM with either substrate (Figure 1D). Replacement of wild-type Cdk7 with Cdk7as also rendered growth of HCT116 cells sensitive to 1-NMPP1. In the absence of the drug, the wild-type and Cdk7as/as cells had population doubling times of ~17.9 and ~20.2 h, respectively, with similar cell-cycle distributions in asynchronous culture (data not shown), indicating minimal impairment of Cdk7 function by the F91G mutation per se. The homozygous Cdk7as/as cells were sensitive to 1-NMPP1, however, with an IC50 ~100 nM measured by cell viability (MTT) assays performed after 96 h of drug exposure (Figure 1E). In contrast, wild-type HCT116 cells were resistant to 10 μM 1-NMPP1. From this we conclude that catalytic activity of Cdk7 is required for viability, and can be shut off selectively in vivo by a bulky ATP analog in the Cdk7as/as cell line.
We observed dose-dependent decreases in phosphorylation of Cdk1 and -2, detected by phospho-T-loop-specific antibodies, with addition of 1-NMPP1 to Cdk7as/as cells (Figure 1F). In contrast, phosphorylation of the Cdk7 target site within the heptad repeat of the Pol II CTD, Ser5, was not diminished by Cdk7 inhibition. Instead, reactivity with an antibody specific for that modification increased after 1-NMPP1 treatment. The increased signal appeared, however, in a distinct electrophoretic isoform, which accumulated to high levels only in the 1-NMPP1-treated, Cdk7as/as cells. The same isoform accumulated in Cdk7as/as cells treated with 1-NMPP1 after release from serum starvation (Supplemental Figure 1). Inhibition of Cdk7 in vivo therefore caused changes in phosphorylation state of three of its suspected substrates.
Cdk7 inhibition during G1 delays S phase entry
To test whether Cdk7 activity is required during G1, we synchronized wild-type or Cdk7as/as HCT116 cells by serum withdrawal for 48 h. Cells of either genotype released from serum starvation in the absence of drugs proceeded synchronously through G1, entered S phase in ~8–10 h and began to accumulate with a G2 DNA content by 15 h. Addition of 10 μM 1-NMPP1 retarded G1/S progression by the mutant but not the wild-type cells (Figure 2A, C). When added simultaneously with serum to the Cdk7as/as cells, 1-NMPP1 prevented any progression into S phase in the next 15 h. After 24 h, there was evidence of progression into S-phase by a fraction of Cdk7as/as cells released from serum starvation directly into medium containing 1-NMPP1, while a fraction remained in G1 (Supplemental Figure 2). The addition of 1-NMPP1 3 h or 6 h after serum addition delayed S-phase entry by ~7 h or by ~3 h, respectively (Figure 2A).
Figure 2. Inhibition of Cdk7 prevents Cdk2 activation and delays G1/S progression.
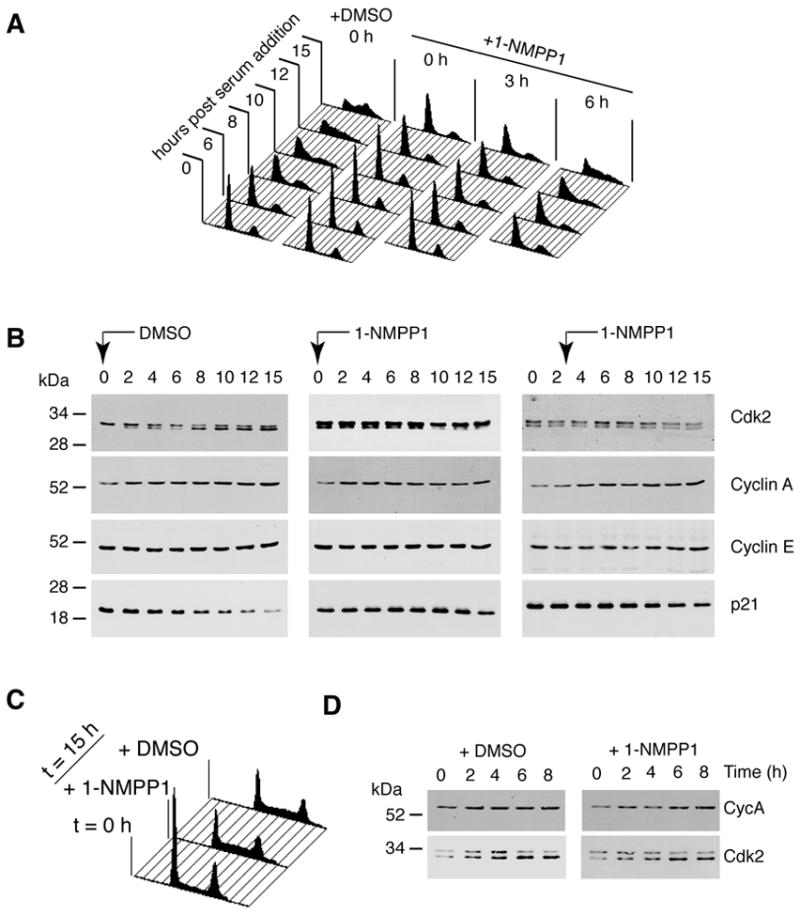
(A) Cells were arrested by serum starvation, then released into serum-containing medium with DMSO or 10 μM 1-NMPP1 added at the indicated times. We monitored DNA content at indicated times by flow cytometry. (B) Levels of Cdk2, cyclin A (Cyc A), cyclin E (Cyc E) and p21 were measured in immunoblots of whole-cell extracts at indicated times after release in DMSO-treated cells, or cells treated with 1-NMPP1 0 and 3 h after release. The higher-mobility isoform of Cdk2 is phosphorylated on Thr160 of the T-loop. Its apparent absence at time 0 in the DMSO-treated cells is anomalous; the other two samples taken at time 0 are more representative (see also Figure 3). (C) Wild-type HCT116 cells were synchronized by serum starvation for 48 h, released into fresh medium containing DMSO or 1-NMPP1 as indicated and collected for flow cytometry to measure DNA content at 15 h. (D) Extracts from DMSO- or 1-NMPP1-treated, wild-type HCT116 cells, collected at indicated times after release from serum starvation, were immunoblotted with antibodies to cyclin A and Cdk2, as indicated.
Cdk7 is a Cdk2-activating kinase in vivo
To determine whether the S-phase delay was due to a CAK defect, we monitored Cdk2 T-loop phosphorylation over time after serum stimulation in mock- (DMSO-) or 1-NMPP1-treated, wild-type and Cdk7as/as cells (Figure 2B, D). Cdk2 T-loop (Thr160) phosphorylation was usually detectable even after 48 h of serum starvation, and increased in control cell populations as they traversed G1 and S phase, until the majority of Cdk2 was in the faster-migrating electrophoretic isoform diagnostic of Thr160 phosphorylation (Gu et al., 1992). Addition of 1-NMPP1 to Cdk7as/as cells at 0 or 3 h prevented any increase in phosphorylated Cdk2 (Figure 2B). In contrast, 1-NMPP1 added at the same time as serum had no effect on Cdk2 T-loop phosphorylation in wild-type HCT116 cells (Figure 2D).
In the mock-treated mutant cells, Cdk2 T-loop phosphorylation and associated kinase activity continued to rise for at least 14 h after release (Figure 2B, 3A). Addition of 1-NMPP1 at 6 h caused prompt cessation of Cdk2 activation and partial loss of pre-existing T-loop phosphorylation (Figure 3A). In general, inhibition of Cdk7 implemented after addition of serum did not cause complete disappearance of the faster-migrating Cdk2 (Figures 2B, 3A). The phosphorylated form could be abolished, however, by a 9-h treatment with 1-NMPP1 during serum starvation (Figure 3B), indicating that Cdk7 phosphorylates Cdk2 even in cells deprived of serum. The fraction of Cdk2 resistant to dephosphorylation, which was low or absent in serum-starved cells (Figure 3B), increased with time after re-addition of serum (Figures 2B, 3A). This probably reflects the stabilization of T-loop modification by cyclins, which accumulate during G1 progression (see Discussion). The kinase activity of this apparently phosphatase-resistant Cdk2 population is stable for at least 8 h in the absence of Cdk7 activity (Figure 3A), and might be sufficient to support slow G1/S progression in the absence of ongoing T-loop phosphorylation (Figure 2A and Supplemental Figure 2).
Figure 3. Cdk7 is the Cdk2-activating kinase in vivo.
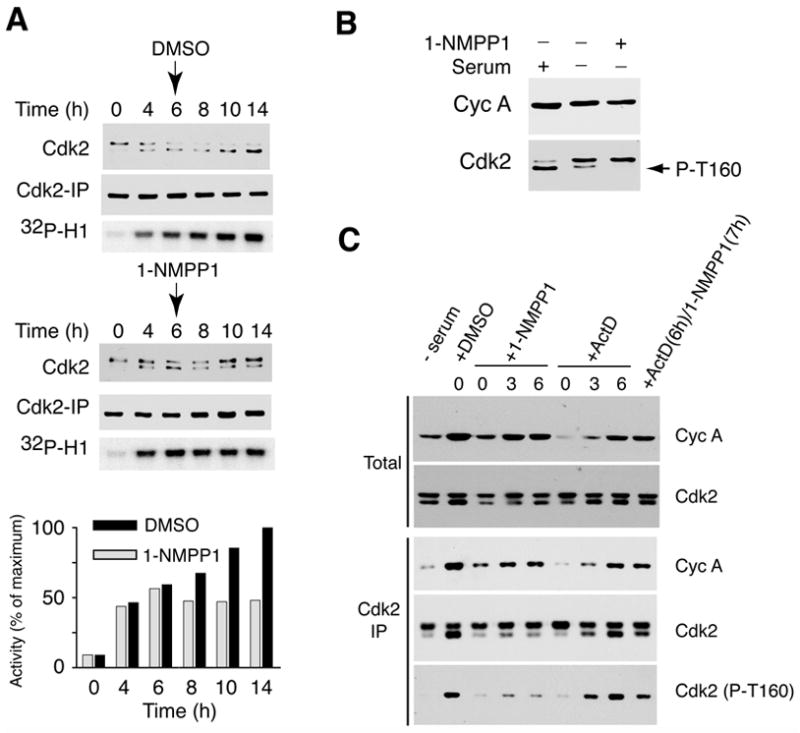
(A) Addition of 1-NMPP1 6 h after serum stimulation halts Cdk2 activation. Cdk7as/as cells were released from serum starvation and harvested at indicated times for extract preparation and measurement of Cdk2 isoforms in total extract (top), and of recovered Cdk2 (middle) and histone H1 kinase activity (bottom) in anti-Cdk2 immunoprecipitates. (Cdk2 isoforms were not resolved in the immunoprecipitated samples.) DMSO or 1-NMPP1 was added as indicated at 6 h. Incorporation of 32P into histone H1 was quantified by Phosphorimager. (B) Inhibition of Cdk7 decreases Cdk2 T-loop phosphorylation in serum-starved cells. In first lane, serum-starved cells were incubated with serum for 15 h. In next two lanes, cells were treated with DMSO or 10 μM 1-NMPP1 in serum-free medium 9 h before extract preparation. (C) CAK inhibition is responsible for defective T-loop phosphorylation in 1-NMPP1-treated cells. Cells were treated with DMSO, 1-NMPP1 and/or actinomycin D (Act D), at times indicated above each lane, and harvested 15 h after release (except for first lane, time 0). Cyclin A accumulation (top) and Cdk2 levels (second from top) were measured in total lysates. Cdk2 immune complexes were isolated and probed with anti-cyclin A (third from top), anti-Cdk2 (fourth from top) or phospho-T-loop-specific (Cdk2 (T160-P), bottom) antibodies.
We also measured levels of cyclins and CDK inhibitors (CKIs) after serum stimulation. During G1, levels of cyclin E, which are known to be elevated in HCT116 cells (Lu et al., 2000), remained more-or-less constant in both mock- and 1-NMPP1-treated cells of either genotype. Cyclin A gradually accumulated in G1 and S phase in the mock-treated mutant cells; the increase was attenuated by addition of 1-NMPP1 (Figures 2B, 3C). Similarly, during the first 6–8 h, levels of the CKIs p21 (Figure 2B) and p27 (data not shown) were not affected by Cdk7 inhibition. At later times in the control cells, p21 decreased, possibly due to its destabilization in S phase (Bornstein et al., 2003); this response was delayed by treatment of Cdk7as/as cells with 1-NMPP1 at 0 or 3 h (Figure 2B). In contrast to the rapid loss of Cdk2 T-loop phosphoryation, the changes in cyclin A and p21 levels occurred several hours after addition of the analog, and are likely to be secondary effects of the cell-cycle delays caused by Cdk7 inhibition.
The rapid arrest and reversal of Cdk2 T-loop phosphorylation (and the modest, delayed effects on cyclin and CKI levels) suggested direct inhibition of CAK by the analog. To rule out an indirect, transcriptional mechanism, we compared effects of 1-NMPP1—on cell cycle progression, cyclin A accumulation, Cdk2/cyclin A complex formation and Cdk2 phosphorylation—with those of the DNA intercalator actinomycin D, which represses transcription globally in mammalian cells (Lam et al., 2001). Actinomycin D arrested the cell cycle when added with, or 3 or 6 h after, serum (Supplemental Figure 3), and was at least as potent as 1-NMPP1 in limiting cyclin A accumulation by the end of the 15-h time course (Figure 3C). With either drug treatment, Cdk2/cyclin A complex formation, measured by immunoprecipitation with anti-Cdk2 followed by immunoblotting with anti-cyclin A antibodies, was proportional to total cyclin A levels (Figure 3C). In contrast, actinomycin D was less effective than 1-NMPP1, added 3 or 6 h after serum, at inhibiting Cdk2 T-loop phosphorylation, detected by probing anti-Cdk2 immunoprecipitates with isoform-specific antibodies (Figure 3C). Moreover, inhibition of Cdk7 blocked Cdk2 phosphorylation independent of ongoing transcription; 1-NMPP1 added at 7 h diminished phospho-Thr160 in cells previously exposed to actinomycin D for 1 h. Taken together, the data indicate that Cdk7 is the physiologic Cdk2-activating kinase.
Inhibiting Cdk7 during S/G2 prevents Cdk1 activation and mitotic entry
To test the requirement for Cdk7 in mitosis, we pre-synchronized cells at the G1/S boundary with two sequential thymidine blocks. They were then released into medium lacking thymidine and monitored for DNA content by flow cytometry (Figure 4A). In the culture treated with DMSO, S phase was completed by ~7 h and, by 10–13 h, the majority of cells had passed mitosis, as indicated by accumulation with a G1 DNA content. Addition of 10 μM 1-NMPP1 immediately upon release (0 h) delayed, but did not prevent, completion of S phase. By ~10 h, cells had apparently reached G2, but there was no evidence of passage through mitosis by 13 h (i.e. no re-accumulation in G1). When the inhibitor was added at 2 or 4 h, S-phase kinetics appeared nearly normal, but cells accumulated with a G2 DNA content, indicating they were unable to pass mitosis.
Figure 4. Cdk7 function is required for mitotic entry.
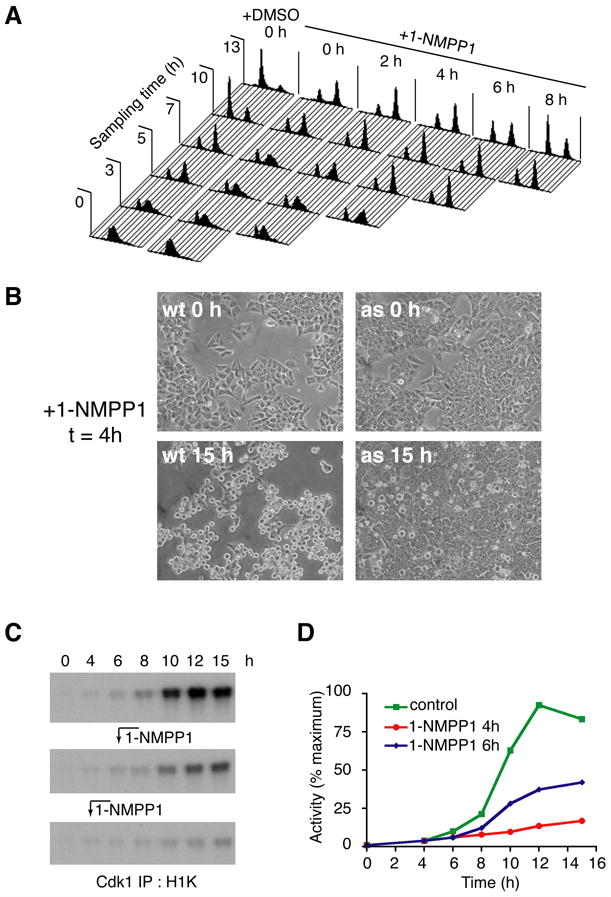
(A) Execution point determination of a Cdk7-dependent function in S/G2. Cdk7as/as cells were arrested at the G1/S boundary by double-thymidine block, then released into fresh medium without or with 10 μM 1-NMPP1 added at the indicated times. To monitor cell cycle progression, we measured DNA content at indicated times by flow cytometry. Note: after release from the thymidine block, we consistently observed a minor population of cells that remained arrested with a G1 DNA content (evident, for example, in the profiles of the DMSO control culture at 3, 5 and 7 h). (B) Inhibition of Cdk7 at 4 h after release from double-thymidine block prevents mitotic entry. Wild-type (wt) and mutant (as) cells were released into nocodazole-containing medium; 1-NMPP1 was added at 4 h and cells were examined by phase-contrast microscopy at 15 h. (C) Cdk1-associated histone H1 kinase activity in cells released from G1/S (double-thymidine) block into nocodazole-containing medium. Cells were mock-treated (top), or treated with 1-NMPP1 at 6 h (middle) or 4 h (bottom) after release. (D) Incorporation of 32P into histone H1 in (C) was quantified by Phosphorimager.
When 1-NMPP1 was added 6 h after release from a double-thymidine block, roughly equal populations of cells accumulated in G1 and G2 by the end of the experiment (Figure 4A). This suggests that, by 6 h post-release, roughly half the cells had fulfilled the requirement for Cdk7 to promote mitosis. When 1-NMPP1 was added 2 h later, the majority of cells passed through mitosis into the next G1 phase. In parallel experiments, cells were released into medium containing the microtubule-depolymerizing agent nocodazole to arrest them in mitosis. Adding 1-NMPP1 0, 2 or 4 h after release blocked entry to mitosis, as revealed by a paucity of rounded cells detached from the dish at the end of the experiment (Figure 4B and data not shown). Addition of 1-NMPP1 at 6 h yielded a mixture of rounded and adherent cells, whereas later addition did not prevent mitosis in the majority of cells (data not shown). We therefore infer an execution point, near the end of S phase, for a Cdk7-dependent function required to enter mitosis.
We investigated the biochemical basis of the Cdk7 requirement, by measuring the kinetics of Cdk1 activation in cells released from a double-thymidine block into nocodazole-containing medium (Figures 4C, D). In the mock-treated culture, a sharp increase in Cdk1-associated kinase activity occurred ~10 h after release from the G1/S block. Addition of 1-NMPP1 at 4 h or 6 h inhibited Cdk1 activation by ~80% or ~50%, respectively. In both populations, what Cdk1 activation there was occurred on schedule, at ~10 h post-release. Because ~50% of cells exposed to 1-NMPP1 at 6 h eventually entered mitosis (Figure 4A), the likely explanation is that Cdk1 activation occurred normally in those cells, and not at all in those arrested in G2.
Cdk7 activity is required for assembly of Cdk1/cyclin B complexes in vivo
To determine the mechanism by which inhibition of Cdk7 prevented activation of Cdk1, we measured levels of total cyclin B and Cdk1/cyclin B complexes in cells treated with 1-NMPP1 at various times after release from a double-thymidine block. In mock-treated cells, cyclin B accumulated as cells progressed from G1/S into mitosis (Figure 5A). Cdk1/cyclin B complexes, measured by anti-cyclin B immunoblot after anti-Cdk1 immunoprecipitation (Figure 5B), formed throughout S and G2 in mock-treated cells, with the bulk of assembly occurring 6 h or more after removal of thymidine. Cdk1 and cyclin B failed to bind efficiently, however, in cells treated with 1-NMPP1 4 or 6 h after release from the G1/S block. In fact, after addition of the analog, the levels of cyclin B co-precipitated with Cdk1 decreased at the 8-h point and remained low throughout the time course, suggesting the disassembly of pre-formed Cdk1/cyclin B complexes. The reciprocal experiment—measurement of Cdk1 in anti-cyclin B immunoprecipitates—produced essentially the same result (Figure 5C). In mock-treated cells, Cdk1/cyclin B complexes increased throughout G2/M; the abrupt increase in kinase activity at ~10 h (Figure 4C) coincided with the dephosphorylation of the inhibitory Tyr15 residue, resulting in increased electrophoretic mobility of Cdk1 (Gu et al., 1992). The inhibitor decreased Cdk1/cyclin B complex formation when added 4 or 6 h after release from thymidine. Some complexes that did form underwent timely dephosphorylation of Tyr15, however, perhaps suggesting that activation of Cdk1 occurred normally in a fraction of cells in each population that had passed the Cdk7 execution point prior to inhibitor addition.
Figure 5. Cdk7 activity is required for Cdk1/cyclin B assembly in vivo.
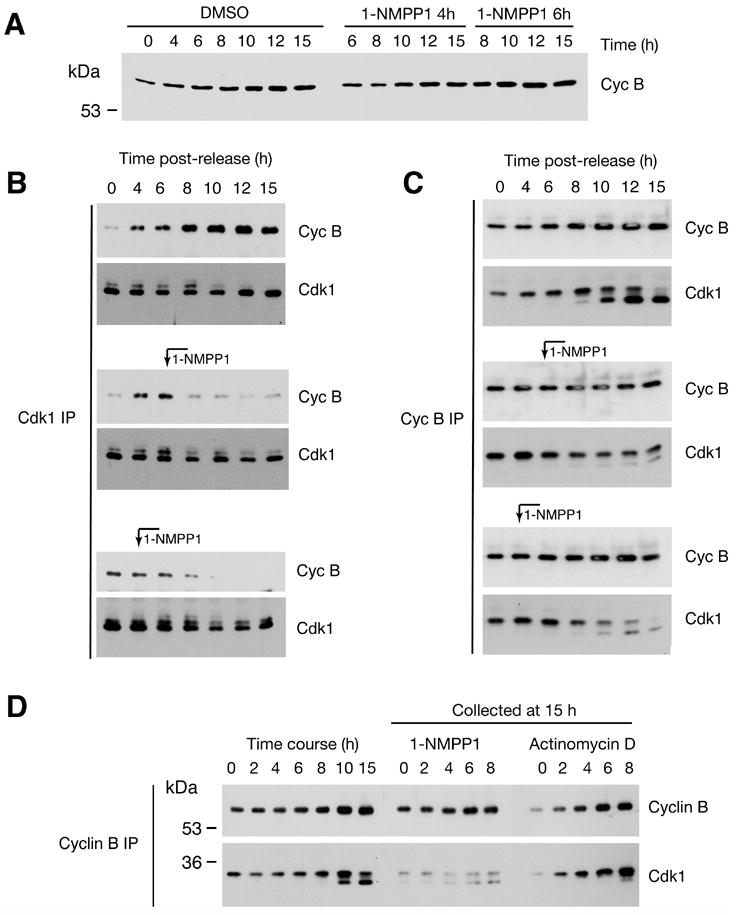
(A) Total cyclin B levels in Cdk7as/as cells released from double-thymidine block into nocodazole-containing medium and mock-treated, or treated with 1-NMPP1 at indicated times. (B) Cdk1/cyclin B assembly was measured by immunoprecipitation with anti-Cdk1 antibodies, followed by immunoblotting with anti-cyclin B (Cyc B) and anti-Cdk1 antibodies. Where indicated, 1-NMPP1 was added at 4 or 6 h. (C) Cdk1/cyclin B assembly was measured by immunoprecipitation with anti-cyclin B (Cyc B) antibodies, followed by immunoblotting with anti-cyclin B and anti-Cdk1 antibodies. 1-NMPP1 was added at indicated time points. Cdk1 exists in two isoforms; the slower-migrating one is phosphorylated on Tyr15, and is enriched in complexes with cyclin B until cells enter mitosis. (D) Comparison of Cdk1/cyclin B binding in cells treated with 1-NMPP1 or actinomycin D. In the first seven lanes, Cdk1/cyclin B assembly was monitored at indicated times as Cdk7as/as cells progressed from a G1/S (double-thymidine) to a mitotic (nocodazole) arrest in the absence of 1-NMPP1. In the last ten lanes, 10 μM 1-NMPP1 or 10 μg/ml actinomycin D was added at indicated time after release from thymidine- into nocodazole-containing medium, and cells were harvested at 15 h.
Because Cdk7-inhibition starting at 4 h diminished cyclin B levels measured 15 h after release from G1/S (Figure 5A), we asked whether limiting cyclin B could account for the impaired complex formation. Actinomycin D added any time after release caused decreases in cyclin B accumulation, measured at 15 h, equal to or greater than those caused by 1-NMPP1, added at the same time to parallel cultures. In contrast, Cdk1/cyclin B complex assembly appeared normal, i.e. proportional to cyclin B levels, in the actinomycin D-treated cells (Figure 5D). The cells did not enter mitosis, however, even when actinomycin D was added 8 h after thymidine removal (data not shown). This is likely due to activation of the G2/M checkpoint; cyclin B-bound Cdk1 remained in the slower-migrating isoform, indicating persistent inhibitory phosphorylation of Tyr15.
These results suggested that CAK activity is required to assemble Cdk1/cyclin B complexes in vivo, even though they can form independent of T-loop phosphorylation in vitro (Desai et al., 1995). To test this idea, we attempted to drive Cdk1/cyclin B complex assembly in extracts from wild-type and Cdk7as/as HCT116 cells with an excess of purified, Myc epitope-tagged cyclin B. We measured assembly of the added cyclin B into active complexes with endogenous Cdk1 that could be immunoprecipitated with anti-Myc antibodies (Figure 6A). Assembly and activation were inhibited by 2 μM 1-NMPP1 in the mutant, but not the wild-type, extract and could be rescued by addition of either purified, wild-type Cdk7/cyclin H/Mat1, or fission yeast Csk1, which is a dedicated CAK with no known, non-CDK targets. These experiments were performed in extracts from cells arrested in mitosis with nocodazole, to avoid inhibition due to phosphorylation of Tyr15 and thereby allow accurate quantification of Cdk1-associated kinase activity. We observed identical effects on Cdk1/cyclin B assembly in G2 extracts: inhibition by 1-NMPP1 in the mutant but not the wild-type extracts, and rescue by purified, wild-type Cdk7 complexes (Figure 6B and data not shown). Purified Cdk7as complexes, however, failed to restore efficient Cdk1/cyclin B assembly in the 1-NMPP1-treated, G2 extract from Cdk7as/as cells (Figure 6B). We conclude that CAK activity is required for Cdk1/cyclin B complex formation in vivo and in whole-cell extracts in vitro.
Figure 6. A CAK requirement for Cdk1/cyclin B assembly recapitulated in vitro.
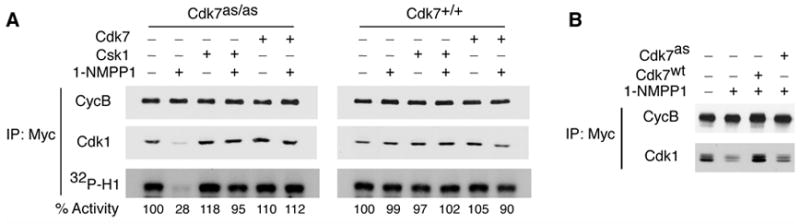
(A) CAK is required for Cdk1/cyclin B assembly in whole-cell extracts. Mitotic extracts from Cdk7as/as or Cdk7+/+ cells were pre-incubated with or without 1-NMPP1 as indicated and supplemented with purified, recombinant Myc-tagged cyclin B. An ATP-regenerating system and, where indicated, either Cdk7/cyclin H/Mat1 complex or Csk1 was added, and extracts were incubated 90 min at room temperature. Samples were immunoprecipitated with anti-Myc antibody and immunoblotted with anti-cyclin B (CycB) or anti-Cdk1 antibodies, and tested for histone H1 kinase activity (32P-H1). Incorporation was quantified by Phosphorimager and indicated below each lane, relative to control (-1-NMPP1, -CAK) defined as 100%. (B) As in (A), except that all reactions contained Myc-tagged cyclin B added to extract from Cdk7as/as cells synchronized in G2. Where indicated, extracts were treated with 2 μM 1-NMPP1 (+) or DMSO (−) and supplemented with wild-type (Cdk7wt) or analog-sensitive (Cdk7as) CAK.
Discussion
Cdk7 is the CDK-activating kinase in vivo
By chemical genetics, we have demonstrated a requirement for Cdk7 to activate both Cdk1 and -2 in vivo. This validates the assignment of general CAK function to Cdk7, originally made on the basis of biochemical analyses but since questioned for lack of conclusive genetic proof (reviewed by Harper and Elledge, 1998; Abbas and Dutta, 2006). At least two other CDKs implicated in mammalian cell-cycle control—Cdk4 and -11—were among the proteins selectively phosphorylated by Cdk7as in crude extracts (Larochelle et al., 2006). Therefore, the metazoan cell-cycle machinery probably relies on a unitary T-loop phosphorylation system.
That system appears to operate on two of its targets—Cdk1 and -2—with different kinetics in vivo, possibly explaining the different effects, on G1/S and G2/M transitions, of Cdk7-inhibition. During G2 progression, we could precisely define an execution point for Cdk7—a time after which CAK-inhibition was ineffectual in preventing mitotic entry. Prior to that point, the effect of inhibiting Cdk7, on Cdk1/cyclin B assembly and activation, was near-total. This is consistent with the observation that Cdk1 is dephosphorylated at each mitosis (Lorca et al., 1992), and the consequent requirement that it be phosphorylated de novo in G2, concomitant with cyclin-binding. In contrast, the G1/S transition was delayed but not completely blocked by Cdk7-inhibition imposed at any time within ~6 h after release from serum starvation; with increasing time after release, the delay grew shorter. Cdk2 phosphorylation is relatively stable; it persisted even during serum starvation and was slow to disappear in the absence of CAK activity (Figures 2, 3). The amount of Cdk2 resistant to Thr160 dephosphorylation roughly correlated with the level of cyclin A in the cell at the time Cdk7 was inhibited, and might therefore be explained by the strong preferences for monomeric Cdk2 of the known T-loop phosphatases (Poon and Hunter, 1995; Cheng et al., 1999). Differences between Cdk1 and -2 in the rate of T-loop phosphate turnover might also explain why Drosophila temperature-sensitive mutants could uncover the role of Cdk7 in activating Cdk1 at mitosis, but not in activating Cdk2 and promoting S-phase entry (Larochelle et al., 1998).
A CAK requirement in Cdk1/cyclin B assembly: implications for mitotic regulation
We have gained an insight into mammalian cell-cycle control: efficient assembly of the mitotic CDK is not a simple function of cyclin B concentration, but depends also on the enzymatic activity of Cdk7. There is precedent in the case of Cdk1 and cyclin A, which require T-loop phosphorylation for their stable association in vitro (Desai et al., 1995) and in vivo (Larochelle et al., 1998). Similarly, a Cdk7/cyclin H/Mat1 complex can be formed, independent of T-loop phosphorylation, from recombinant subunits in vitro or by overexpression in vivo, but the unphosphorylated complex is thermolabile in vivo and in vitro (Larochelle et al., 2001). There is also crystallographic evidence that Cdk2/cyclin A complexes are stabilized by interaction between the T-loop phosphothreonine and conserved, basic residues of the cyclin (Russo et al., 1996). The binding energy provided by this interaction is not necessary for Cdk2 to form complexes with cyclins in vitro or, apparently, with cyclins A and E in vivo (Figure 3C, Supplemental Figure 4). Cdk1-cyclin B binding appears to depend, however, on T-loop phosphorylation under physiologic conditions. Interestingly, Cdk1 variants with mutations of the T-loop threonine were defective in binding cyclin B in Xenopus extracts (Ducommun et al., 1991; Lee et al., 1994), but both a T161A mutant and unphosphorylated, wild-type Cdk1 bound cyclin B efficiently in a defined system containing only purified proteins (Desai et al., 1995). This suggests the inability of Cdk1 and cyclin B to assemble in the absence of CAK activity might be actively imposed, in vivo and in extracts, by destabilizing factors that enforce coupling between T-loop phosphorylation and cyclin-binding.
It was suspected that the activity of Cdk7—the level of which remains constant throughout the cell cycle—is governed passively in vivo by availability of its protein substrates and/or stability of its phosphoprotein products, both determined primarily by cyclin-binding (reviewed by Morgan, 1997). Here we uncover a more complex relationship—mutual dependence—between the two, obligate steps of Cdk1 activation in vivo: cyclin-binding and T-loop phosphorylation. Cdk1/cyclin B assembly is also sensitive to inhibition of Cdk7 in whole-cell extracts of synchronized Cdk7as/as cells (Figure 6). We could rescue the assembly defect with wild-type Cdk7 or fission yeast Csk1: structurally dissimilar kinases with only a substrate—the Cdk1 T-loop—in common. The interdependence of the two CDK-activating steps implies a concerted reaction with no inactive, unphosphorylated complex as an intermediate (Figure 7). That would necessitate tight coupling with inhibitory mechanisms, such as Tyr15 phosphorylation or nuclear exclusion of cyclin B (Jin et al., 1998), in order to prevent precocious activation of Cdk1/cyclin B leading to premature mitosis.
Figure 7. A concerted pathway of Cdk1/cyclin B assembly and activation.
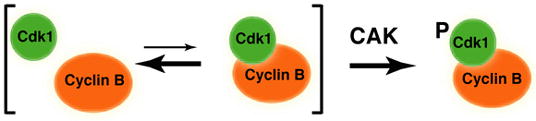
Cdk1 activation is a concerted reaction in vivo, with formation of a stable Cdk1/cyclin B complex occurring simultaneously with T-loop phosphorylation by Cdk7, as suggested by the interdependence of the two steps in vitro and in vivo. This pathway, if uncoupled from inhibitory mechanisms, would not generate an unphosphorylated Cdk1/cyclin B complex as an inactive intermediate.
Chemical genetics in human somatic cells
In theory, an analog-sensitive version of any protein kinase can be engineered (Bishop et al., 2000) and expressed from its endogenous promoter by gene replacement with rAAV vectors (Papi et al., 2005). We combined the two strategies to investigate Cdk7—an attractive anti-cancer drug target because of its dual functions in gene expression and cell proliferation (Fisher, 2005; Lolli and Johnson, 2005). Phosphorylation of both Cdk1-Thr161 and Cdk2-Thr160 decreased after Cdk7 inhibition, in both time- and dose-dependent fashion, consistent with Cdk7 being the sole kinase responsible for CDK activation. In contrast, Ser5 of the Pol II CTD is targeted by multiple kinases in vitro (Ramanathan et al., 2001) and, apparently, in vivo. Inactivating Cdk7 led to an apparent increase in net phosphorylation by other Ser5 kinases, but produced a Pol II isoform of altered electrophoretic mobility (Figure 1F and Supplemental Figure 1). This suggests a derangement of normal CTD phosphorylation patterns, and probably severe transcriptional dysfunction, which could explain why Cdk7as/as cells were sensitive to killing (Figure 1E) by prolonged exposure to 1-NMPP1 at doses insufficient to arrest the cell cycle (data not shown).
The chemical-genetic tools developed here can now be applied to investigate the connection between cell-division and transcription machineries maintained by Cdk7, in its dual roles as the CDK-activating and TFIIH-associated kinase (reviewed by Nigg, 1996; Fisher, 2005). The chemical-genetic approach ensures that cytotoxicity is Cdk7-specific, i.e. not due to off-target effects of the analog, and should therefore allow us to evaluate CAK/TFIIH as a possible therapeutic target in human cancer cells. The Cdk7as/as phenotype, moreover, will provide metrics—signatures of cell-cycle and transcriptional disruption—by which to evaluate potentially selective inhibitors of wild-type Cdk7 isolated through screens of chemical libraries.
Experimental Procedures
Gene targeting
To introduce the F91G mutation (Larochelle et al., 2006), together with an EcoRI restriction site, into the cdk7 locus, 5’ and 3’ arms were PCR-amplified from a human BAC clone (RP11-124A2; Children’s Hospital of Oakland Research Institute) and cloned on either side of a central loxP-neoR-loxP cassette in pBluescript derivative pNX. The mutations were introduced into the right homology arm (exon 5) by PCR amplification; the entire insert was then excised by NotI digestion and ligated to a pAAV vector backbone, yielding pAAV-cdk7as. Transfection of HEK293 cells, isolation of rAAV particles, and infection of HCT116 cells were performed as described (Kohli et al., 2004). Additional information, including sequences of primers used in this study, is available upon request.
Biochemical and immunological methods
Immunoblotting and immunoprecipitation, and kinase assays of immune complexes, were carried out as previously described (Wohlbold et al., 2006). To measure Cdk1/cyclin B assembly, we pre-incubated extracts (200 μg total protein) from cells in mitosis or G2 with 2 μM 1-NMPP1 or DMSO, then added 500 ng purified cyclin B1, amino-terminally tagged with hexahistidine and the Myc epitope (gift of D. Morgan, University of California, San Francisco), and an ATP-regenerating system (Larochelle et al., 2006). Where indicated, incubations were supplemented with 400 ng purified Csk1 (Larochelle et al., 2006) or 600 ng wild-type or analog-sensitive, T-loop-phosphorylated Cdk7/cyclin H/Mat1 complex (Larochelle et al., 2001; Larochelle et al., 2006). After 90 min at room temperature, Myc-cyclin B and associated proteins were immunoprecipitated with anti-Myc antibodies and immune complexes were subjected to immunoblotting, with anti-Myc and anti-Cdk1 antibodies, and tested for histone H1 kinase activity. Antibodies used in this study were: anti-Cdk7 (MO1.1), from Sigma; anti-Cdk1 (C19), -Cdk2 (D-12 and M2), -cyclin B1 (GN51 and H-433), -cyclin A (H-432) -cyclin E (HE111 or M20), and -Rpb1 amino-terminus (N20), from Santa Cruz Biotechnology; anti-Rpb1 CTD phospho-Ser5 (polyclonal), from Abcam (used in Figure 1F); anti-Myc (9E10) and -Rpb1 CTD phospho-Ser5 (monoclonal H14, used in Supplemental Figure 1), from Covance; anti-Cdk1-Thr161 and -Cdk2-Thr160, from Cell Signaling Technologies; and anti-p21 (Ab-11), from Neo Markers.
Chemical genetic methods
The labeled analog, [γ-32P]N6-(benzyl)-ATP was generated enzymatically with nucleoside diphosphate kinase (NDPK) as previously described (Kraybill et al., 2002), and labeling by endogenous Cdk7as was performed exactly as described previously for labeling by the recombinant enzyme (Larochelle et al., 2006). The inhibitory analog 1-NMPP1 (Bishop et al., 2000) was dissolved and stored in DMSO and used at final concentrations indicated.
Cell synchronization, cell-cycle analysis and viability assays
Wild-type or Cdk7as/as HCT116 cells were synchronized by incubation in serum-free medium for 48 h and released into medium containing 10% fetal calf serum. Synchronization with thymidine or nocodazole, and analysis of cell-cycle distribution by flow cytometry, were performed as previously described (Papi et al., 2005). Cell viability was measured by 3,[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay as described (van der Kuip et al., 2001).
Supplementary Material
Acknowledgments
We thank D. Morgan (University of California, San Francisco) for cyclin B and NDPK expression vectors, S. Keeney for critical review of the manuscript, and H. Hochegger and S. Takeda (Kyoto University), for communicating their pre-publication results obtained with analog-sensitive Cdk1 in vertebrate cells. This work was supported by a post-doctoral fellowship of the Deutsche Forschungsgemeinschaft to L.W., and by U.S. National Institutes of Health grants GM56985 to R.P.F., CA107342 to P.V.J. and EB001987 to K.M.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas T, Dutta A. CDK2-Activating Kinase (CAK): More Questions than Answers. Cell Cycle. 2006;5:1123–1124. doi: 10.4161/cc.5.10.2819. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Chen J, Larochelle S, Li X, Suter B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature. 2003;424:228–232. doi: 10.1038/nature01746. [DOI] [PubMed] [Google Scholar]
- Cheng A, Ross KE, Kaldis P, Solomon MJ. Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes Dev. 1999;13:2946–2957. doi: 10.1101/gad.13.22.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismowski MJ, Laff GM, Solomon MJ, Reed SI. KIN28 encodes a C-terminal domain kinase that controls mRNA transcription in Saccharomyces cerevisiae but lacks cyclin-dependent kinase-activating kinase (CAK) activity. Mol Cell Biol. 1995;15:2983–2992. doi: 10.1128/mcb.15.6.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai D, Wessling HC, Fisher RP, Morgan DO. The effect of phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol Cell Biol. 1995;15:345–350. doi: 10.1128/mcb.15.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapkin R, Le Roy G, Cho H, Akoulitchev S, Reinberg D. Human cyclin-dependent kinase-activating kinase exists in three distinct complexes. Proc Natl Acad Sci USA. 1996;93:6488–6493. doi: 10.1073/pnas.93.13.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducommun B, Brambilla P, Felix MA, Franza BR, Karsenti E, Draetta G. cdc2 phosphorylation is required for its interaction with cyclin. EMBO J. 1991;10:3311–3319. doi: 10.1002/j.1460-2075.1991.tb04895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza FH, Farrell A, Erdjument-Bromage H, Tempst P, Morgan DO. A cyclin-dependent kinase-activating kinase (CAK) in budding yeast unrelated to vertebrate CAK. Science. 1996;273:1714–1717. doi: 10.1126/science.273.5282.1714. [DOI] [PubMed] [Google Scholar]
- Feaver WJ, Svejstrup JQ, Henry NL, Kornberg RD. Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell. 1994;79:1103–1109. doi: 10.1016/0092-8674(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell. 1994;78:713–724. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992;11:3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The role of Cdk7 in CAK function, a retro-retrospective. Genes Dev. 1998;12:285–289. doi: 10.1101/gad.12.3.285. [DOI] [PubMed] [Google Scholar]
- Hermand D, Pihlak A, Westerling T, Damagnez V, Vandenhaute J, Cottarel G, Mäkelä TP. Fission yeast Csk1 is a CAK-activating kinase (CAKAK) EMBO J. 1998;17:7230–7238. doi: 10.1093/emboj/17.24.7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol. 1998;141:875–885. doi: 10.1083/jcb.141.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldis P, Solomon MJ. Analysis of CAK activities from human cells. Eur J Biochem. 2000;267:4213–4221. doi: 10.1046/j.1432-1327.2000.01455.x. [DOI] [PubMed] [Google Scholar]
- Kaldis P, Sutton A, Solomon MJ. The Cdk-activating kinase (CAK) from budding yeast. Cell. 1996;86:553–564. doi: 10.1016/s0092-8674(00)80129-4. [DOI] [PubMed] [Google Scholar]
- Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraybill BC, Elkin LL, Blethrow JD, Morgan DO, Shokat KM. Inhibitor scaffolds as new allele specific kinase substrates. J Am Chem Soc. 2002;124:12118–12128. doi: 10.1021/ja0264798. [DOI] [PubMed] [Google Scholar]
- Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:RESEARCH0041. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13:55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Chen J, Knights R, Pandur J, Morcillo P, Erdjument-Bromage H, Tempst P, Suter B, Fisher RP. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 2001;20:3749–3759. doi: 10.1093/emboj/20.14.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Pandur J, Fisher RP, Salz HK, Suter B. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev. 1998;12:370–381. doi: 10.1101/gad.12.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Saiz JE, Barton WA, Fisher RP. Cdc2 activation in fission yeast depends on Mcs6 and Csk1, two partially redundant Cdk-activating kinases CAKs) Curr Biol. 1999;9:441–444. doi: 10.1016/s0960-9822(99)80194-8. [DOI] [PubMed] [Google Scholar]
- Lee TH, Turck C, Kirschner MW. Inhibition of cdc2 activation by INH/PP2A. Mol Biol Cell. 1994;5:323–338. doi: 10.1091/mbc.5.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu C, Galaktionov K. p42, a novel cyclin-dependent kinase-activating kinase in mammalian cells. J Biol Chem. 2004;279:4507–4514. doi: 10.1074/jbc.M309995200. [DOI] [PubMed] [Google Scholar]
- Lolli G, Johnson LN. CAK-Cyclin-dependent Activating Kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle. 2005;4:572–577. [PubMed] [Google Scholar]
- Lorca T, Labbé JC, Devault A, Fesquet D, Capony JP, Cavadore JC, Le Bouffant F, Dorée M. Dephosphorylation of cdc2 on threonine 161 is required for cdc2 kinase inactivation and normal anaphase. EMBO J. 1992;11:2381–2390. doi: 10.1002/j.1460-2075.1992.tb05302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Shih C, Teicher BA. Expression of pRB, cyclin/cyclin-dependent kinases and E2F1/DP-1 in human tumor lines in cell culture and in xenograft tissues and response to cell cycle agents. Cancer Chemother Pharmacol. 2000;46:293–304. doi: 10.1007/s002800000136. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Cyclin-dependent kinases: engines, clocks and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Cyclin-dependent kinase 7: at the cross-roads of transcription, DNA repair and cell cycle control? Curr Opin Cell Biol. 1996;8:312–317. doi: 10.1016/s0955-0674(96)80003-2. [DOI] [PubMed] [Google Scholar]
- Papi M, Berdougo E, Randall CL, Ganguly S, Jallepalli PV. Multiple roles for separase auto-cleavage during the G2/M transition. Nat Cell Biol. 2005;7:1029–1035. doi: 10.1038/ncb1303. [DOI] [PubMed] [Google Scholar]
- Poon RYC, Hunter T. Dephosphorylation of Cdk2 Thr160 by the cyclin-dependent kinase-interacting phosphatase KAP in the absence of cyclin. Science. 1995;270:90–93. doi: 10.1126/science.270.5233.90. [DOI] [PubMed] [Google Scholar]
- Ramanathan Y, Rajpara SM, Reza SM, Lees E, Shuman S, Mathews MB, Pe’ery T. Three RNA polymerase II carboxyl-terminal domain kinases display distinct substrate preferences. J Biol Chem. 2001;276:10913–10920. doi: 10.1074/jbc.M010975200. [DOI] [PubMed] [Google Scholar]
- Reardon JT, Ge H, Gibbs E, Sancar A, Hurwitz J, Pan ZQ. Isolation and characterization of two human transcription factor IIH (TFIIH)-related complexes: ERCC2/CAK and TFIIH*. Proc Natl Acad Sci USA. 1996;93:6482–6487. doi: 10.1073/pnas.93.13.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Londesborough A, Korsisaari N, Pihlak A, Lehtonen E, Henkemeyer M, Mäkelä TP. Inability to enter S phase and defective RNA polymerase II CTD phosphorylation in mice lacking Mat1. EMBO J. 2001;20:2844–2856. doi: 10.1093/emboj/20.11.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Pavletich NP. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nature Struct Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- Saiz JE, Fisher RP. A CDK-activating kinase network is required in cell cycle control and transcription in fission yeast. Curr Biol. 2002;12:1100–1105. doi: 10.1016/s0960-9822(02)00903-x. [DOI] [PubMed] [Google Scholar]
- Sutton A, Freiman R. The Cak1p protein kinase is required at G1/S and G2/M in the budding yeast cell cycle. Genetics. 1997;147:57–71. doi: 10.1093/genetics/147.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuret JY, Valay JG, Faye G, Mann C. Civ1 (CAK in vivo), a novel Cdk-activating kinase. Cell. 1996;86:565–576. doi: 10.1016/s0092-8674(00)80130-0. [DOI] [PubMed] [Google Scholar]
- van der Kuip H, Goetz AW, Miething C, Duyster J, Aulitzky WE. Adhesion to fibronectin selectively protects Bcr-Abl+ cells from DNA damage-induced apoptosis. Blood. 2001;98:1532–1541. doi: 10.1182/blood.v98.5.1532. [DOI] [PubMed] [Google Scholar]
- Wallenfang MR, Seydoux G. cdk-7 is required for mRNA transcription and cell cycle progression in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA. 2002;99:5527–5532. doi: 10.1073/pnas.082618399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold L, Larochelle S, Liao JC, Livshits G, Singer J, Shokat KM, Fisher RP. The cyclin-dependent kinase (CDK) family member PNQALRE/CCRK supports cell proliferation but has no intrinsic CDK-activating kinase (CAK) activity. Cell Cycle. 2006;5:546–554. doi: 10.4161/cc.5.5.2541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.