

Abstract
VEGF and TGF-β1 are potent angiogenesis inducers with opposing effects on endothelial cells. TGF-β1 induces apoptosis; VEGF protects endothelial cells from apoptosis. We found that TGF-β1 promotes endothelial cell expression of FGF-2, which up-regulates VEGF synthesis. Inhibition of VEGF signaling through VEGF receptor 2 (flk-1) abrogates TGF-β1-induced apoptosis and p38MAPK activation. Inhibition of p38MAPK blocks TGF-β1-induced apoptosis, showing that VEGF/flk-1-mediated activation of p38MAPK is required for TGF-β1 induction of apoptosis. In the absence of TGF-β1, VEGF activates p38MAPK and promotes endothelial cell survival. However, in context with TGF-β1, VEGF/flk-1-mediated activation of p38MAPK results in apoptosis. Thus, cross-talk between TGF-β1 and VEGF signaling converts VEGF/flk-1-activated p38MAPK into a proapoptotic signal. This finding illustrates an unexpected role of VEGF and indicates that VEGF can be pharmacologically converted into an apoptotic factor, a novel approach to antiangiogenesis therapy.
Keywords: angiogenesis, MAPK kinase, p38, VEGF receptor, cancer
VEGF, FGF-2, and TGF-β1 play prominent roles in angiogenesis, the formation of capillaries from preexisting blood vessels (1–3). VEGF controls a variety of endothelial cell functions involved in angiogenesis and protects endothelial cells from apoptosis (2). Multiple stimuli, including hypoxia, cytokines, and growth factors, control VEGF expression (2). FGF-2 and TGF-β1 induce endothelial cell expression of VEGF, which mediates the angiogenic activity of FGF-2 in vitro and in vivo (4, 5).
Two tyrosine kinase receptors, flt-1 [VEGF receptor 1 (VEGFR1)] and flk-1 (VEGFR2) transduce the activity of VEGF. flk-1 has been implicated in endothelial cell proliferation and survival; flt-1 has been implicated in chemotaxis and vascular permeability (2). Protein kinase B (or Akt) and MAPK are components of the signaling mechanism activated by flk-1 (6, 7). VEGF, flk-1, and flt-1 are indispensable for angiogenesis, and their genetic deficiency causes embryonic lethality with blood vessel disorganization, endothelial cell overgrowth, or impaired endothelial cell development (8, 9).
In tumors VEGF expression correlates with tumor vascularity and progression. Inhibition of VEGF results in decreased tumor vascularity and growth, implicating VEGF as the major tumor angiogenesis factor (2). Pharmacological treatments targeting VEGF are currently used or in clinical trials for the therapy of malignancies and other conditions involving increased angiogenesis (10).
FGF-2 controls a variety of endothelial cell functions through paracrine or autocrine mechanisms (1, 11, 12) and up-regulates endothelial cell expression of VEGF (4). Mice genetically deficient in FGF-2 have no apparent defects related to impaired angiogenesis (13, 14). However, their endothelial cells show impaired migration in vitro, a defect resulting from lack of ERK1/2 activation (15).
TGF-β1 is an influential regulator of tissue morphogenesis and a potent inhibitor of proliferation for most cell types (3). Half of mice genetically deficient in TGF-β1 die in utero and show defective vasculogenesis, a phenotype consistent with abundant TGF-β1 expression in endothelial precursors (16). TGF-β1 has multiple effects on endothelial cells. In vivo TGF-β1 induces vessel formation (17–19). However, in vitro it inhibits endothelial cell proliferation (20), migration and proteolytic activity, opposes the stimulatory effect of FGF-2 on these cell functions (21), and down-regulates VEGFR-2 expression (22). Intriguingly, TGF-β1 promotes endothelial cell apoptosis in vitro and in vivo (20, 23) but up-regulates endothelial cell expression of VEGF (5), an apparent discrepancy because VEGF protects endothelial cells from apoptosis (2).
During angiogenesis apoptosis is required for pruning the forming vascular network, and inhibition of apoptosis results in formation of abnormal vessels (23). In addition, apoptosis controls cell functions required for angiogenesis in vitro and in vivo (24–26). However, uncontrolled apoptosis blocks angiogenesis, and a number of antiangiogenesis treatments are aimed to induce endothelial cell apoptosis (27).
TGF-β signaling is specifically mediated by the Smad proteins (3, 28). In addition, increasing evidence implicates the MAPKs in the regulation of TGF-β signaling (29), indicating interactions between the Smads and signaling pathways activated by other growth factors.
VEGF, TGF-β1 and FGF-2 are often co-expressed in tissues in which angiogenesis occurs. However, their interactions are poorly understood. We have previously shown that FGF-2 up-regulates endothelial cell expression of VEGF, which mediates the angiogenic activity of FGF-2 in vitro and in vivo (4). Because TGF-β1 opposes the effects of FGF-2 and VEGF on angiogenesis (21) we investigated the potential role of FGF-2 and VEGF in the control of TGF-β1 activity on endothelial cells. Unexpectedly, we found that VEGF, a survival factor, acts in concert with TGF-β1 to induce endothelial cell apoptosis.
Results
TGF-β1 Induces Endothelial Cell Expression of FGF-2 and VEGF.
We have previously shown that FGF-2 induces endothelial cell expression of VEGF (4). Because the effects of FGF-2 and VEGF on endothelial cells are opposed by TGF-β1 (21), we hypothesized that TGF-β1 controls FGF-2 and/or VEGF activity. Therefore, we characterized FGF-2 and VEGF levels in endothelial cells treated with TGF-β1. The results showed that TGF-β1 induced dose-dependent up-regulation of both FGF-2 and VEGF expression (Fig. 1A).
Fig. 1.

TGF-β1 induces endothelial cell expression of FGF-2 and VEGF. (A) Western blotting analysis of FGF-2 in extracts (Top) and VEGF in conditioned medium (Middle) of BCE cells treated with the indicated concentrations of TGF-β1 for 6 h. (Bottom) ERK2 as loading control. The bands shown in Top represent 23-kDa, 21-kDa, and 18-kDa FGF-2. Similar results were obtained with different clones of BCE cells, bovine aortic endothelial cells, HUVEC, and human microvascular endothelial cells. (B) Northern blotting analysis of VEGF mRNA in BCE cells incubated with control medium (Control), 10 ng/ml FGF-2, or 1 ng/ml TGF-β1 in the presence of FGF-2 mAb or n.i. IgG (10 μg/ml). G3PDH: loading control. (C) Western blotting analysis of VEGF in medium conditioned by FGF-2+/+ or FGF-2−/− endothelial cells incubated with control medium (C), FGF-2 (10 ng/ml), or TGF-β1 (1 ng/ml). Forty microliters of concentrated conditioned medium from confluent cultures was loaded for all of the samples. These experiments were repeated three times with comparable results.
TGF-β1 induction of VEGF expression was rapid (3 h), persisted for at least 24 h (Fig. 7, which is published as supporting information on the PNAS web site), and involved up-regulation of the VEGF mRNA level (Fig. 1B). Anti-FGF-2 antibody abolished this effect (Fig. 1B), indicating that endogenous FGF-2 mediates TGF-β1 up-regulation of VEGF expression (4). Consistent with this finding, TGF-β1 induced VEGF expression in endothelial cells from WT but not from FGF-2 knockout (FGF-2−/−) mice (Fig. 1C), showing that TGF-β1 induces endothelial cell expression of FGF-2, which in turn up-regulates VEGF synthesis through an autocrine mechanism.
Inhibition of VEGF Blocks TGFβ-1 Induction of Endothelial Cell Apoptosis.
Because TGF-β1 inhibits endothelial cell functions controlled by FGF-2 and VEGF, we hypothesized that the up-regulation of FGF-2 and VEGF expression by TGF-β1 represents a feedback mechanism to control TGF-β1 activity. Therefore, we tested the effect of anti-FGF-2 and anti-VEGF antibodies on cell proliferation, migration, type 1 plasminogen activator inhibitor, and urokinase plasminogen activator expression in endothelial cells treated with TGF-β1. In all experiments the antibodies neither inhibited nor increased the effect of TGF-β1 (data not shown). However, VEGF antibody strongly reduced the number of floating cells in TGF-β1-treated cultures, suggesting that inhibition of endogenous VEGF blocked TGF-β1-induced apoptosis. Therefore, we used the annexin V binding assay to characterize apoptosis in endothelial cells treated with TGF-β1 in the presence or absence of anti-FGF-2 or anti-VEGF antibodies (Fig. 2A). TGF-β1 increased the number of apoptotic cells from 2% in control cultures to 12–15%. In cells treated with TGF-β1 and anti-FGF-2 antibody apoptosis was reduced nonsignificantly. Conversely, cells treated with TGF-β1 in the presence of anti-VEGF antibody had an apoptotic index comparable to that of untreated controls, indicating that inhibition of VEGF completely abolished the apoptotic effect of TGF-β1. In the absence of TGF-β1, anti-VEGF antibody had no effect on apoptosis. In contrast, anti-FGF-2 antibody strongly increased the number of apoptotic cells (Fig. 2A). Thus, anti-FGF-2 antibody did not block TGF-β1 up-regulation of apoptosis because it induces programmed cell death even in the absence of TGF-β1. This conclusion is in agreement with previous findings that antibodies to FGF-2 deprive endothelial cells of endogenous antiapoptotic signaling (30, 31).
Fig. 2.

VEGF mediates the apoptotic activity of TGF-β1. (A) Annexin V binding assay. BCE cells were serum-starved (0.5% CS) overnight and incubated for 6 h with control medium (Control) or TGF-β1 (1 ng/ml) with or without n.i. IgG, VEGF mAb, or FGF-2 mAb (10 μg/ml). Control cells were incubated with FGF-2 (10 ng/ml) or VEGF (30 ng/ml) or with the respective antibodies in the absence of TGF-β1. Histograms show mean ± SD of percentage of apoptotic cells in three separate experiments. ∗, P < 0.05 (sample vs. Control). (B and C) Western blotting analysis of PARP degradation. (B) HUVEC incubated with either control medium (Control) or 1 ng/ml TGF-β1 with or without n.i. IgG or VEGF mAb (10 μg/ml) for 6 h. (C) HUVEC incubated for 30 h in medium containing 0.5% FCS in the absence (Control) or presence of VEGF (30 ng/ml). ERK2: loading control. (D) Western blotting analysis of caspase 3 cleavage in HUVEC incubated in the absence (C) or presence of the indicated concentrations of TGF-β1. ERK2: loading control. (E) Western blotting analysis of cleaved caspase 3 in FGF-2+/+ or FGF-2−/− endothelial cells treated for 6 h with control medium (Control) or the indicated reagents: FGF-2 (10 ng/ml), TGF-β1 (1 ng/ml), or VEGF (30 ng/ml).
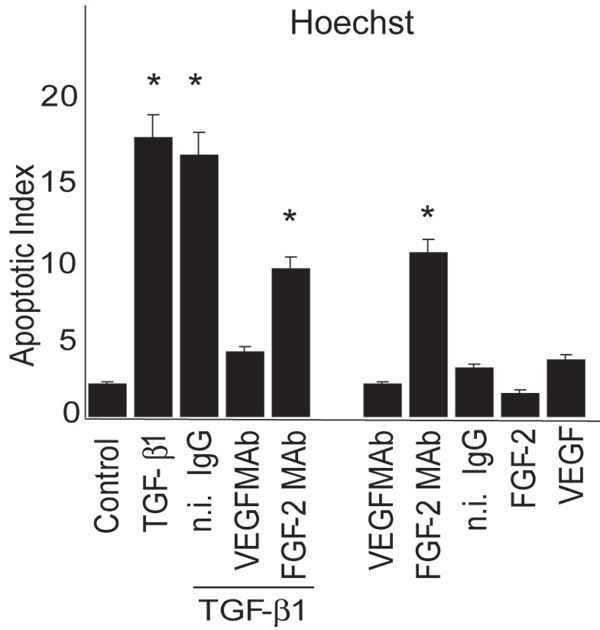
Consistent with the annexin V binding results, analysis of nuclear chromatin condensation by Hoechst staining (Fig. 8, which is published as supporting information on the PNAS web site) or poly (ADP-ribose) polymerase (PARP) degradation (Fig. 2B) also showed that anti-VEGF antibody abolished the apoptotic effect of TGF-β1. As a positive control for apoptosis we serum-starved the cells for 30 h, a treatment known to induce endothelial cell apoptosis (32). As expected, addition of VEGF to the culture medium abolished apoptosis induced by serum starvation (Fig. 2C) (32), showing that VEGF can have either proapoptotic or antiapoptotic activity depending on the apoptotic stimulus and/or the presence of other growth factors. To gain insight into the mechanism of TGF-β1 induction of endothelial cell apoptosis, we characterized the effect of TGF-β1 on activation of caspase 3, an effector of programmed cell death. As shown in Fig. 2D, TGF-β1 induced caspase 3 activation with a dose-dependent effect that paralleled the up-regulation of FGF-2 and VEGF expression (Fig. 1A). Thus, these data indicated that endogenous VEGF is necessary for the apoptotic effect of TGF-β1 on endothelial cells.
To validate this hypothesis we tested the effect of TGF-β1 on apoptosis of FGF-2−/− endothelial cells, which do not express VEGF in response to TGF-β1 (Fig. 1C). FGF-2−/− and WT endothelial cells have comparable growth rates, and exogenous FGF-2 has a similar proliferative effect on the two cell types (data not shown). In absence of TGF-β1, the level of apoptosis was considerably higher in FGF-2−/− than in WT cells, and was strongly down-regulated by exogenous FGF-2, consistent with our observation that FGF-2 antibody up-regulates endothelial cell apoptosis (Fig. 2B). TGF-β1 induced apoptosis in WT but not in FGF-2−/− cells. However, treatment of FGF-2−/− cells with both TGF-β1 and VEGF resulted in increased apoptosis, showing that TGF-β1 and VEGF have a synergistic effect on apoptosis in these cells (Fig. 2E). Thus, these results showed that VEGF signaling is required for TGF-β1 induction of endothelial cell apoptosis.
Inhibition of VEGF Specifically Blocks TGF-β1 Induction of Apoptosis with a Mechanism Independent of Cell Proliferation.
Induction of endothelial cell apoptosis by TGF-β1 may be cell cycle-dependent, and endothelial cells may require endogenous VEGF to progress in the cell cycle. Therefore, the inhibitory effect of VEGF antibody on TGF-β1-induced apoptosis might result indirectly from the potential antiproliferative action of the antibody. To test this hypothesis we characterized the effect of TGF-β1 and VEGF antibody on BrdU uptake, a measurement of the percentage of cells in S phase. Under our experimental conditions (confluent cells in 0.5% serum) ≈5% of the endothelial cells were in S phase (vs. 15% in 10% serum). Neither TGF-β1 nor VEGF antibody, nor a combination of these reagents altered the percentage of proliferating cells relative to untreated cells. In contrast, TGF-β1 increased the apoptotic rate to 15–20%, and VEGF antibody abolished this effect (Fig. 3A and B). Therefore, these results showed that VEGF antibody blocks TGF-β1 induction of apoptosis by a mechanism independent of the effects of these cytokines on cell proliferation.
Fig. 3.

VEGF specifically mediates TGF-β1 induction of apoptosis with a mechanism independent of its proliferative activity. (A) BrdU uptake (% positive cells) by BCE cells in medium containing 10% FCS (Control 10% serum) or grown overnight in 0.5% FCS and incubated for 6 h without (Control) or with TGF-β1 (1 ng/ml) with or without VEGF antibody (VEGF mAb) or n.i. IgG (10 μg/ml) or with anti-VEGF antibody alone. Means ± SE of a representative experiment are shown. (B) Apoptotic index (%). The same cells were immunostained with antibody to cleaved caspase 3 and counterstained with DAPI. The bars show the percentage of cleaved caspase 3-positive cells per total number of nuclei counted (1,000). Means ± SE of a representative experiment are shown. (C) Western blotting analysis of cleaved caspase 3 in BCE cells incubated without (Control) or with TGF-β1 (1 ng/ml) or TNF-α (10 ng/ml plus 1 μg/ml cycloheximide) with or without anti-VEGF antibody (VEGF mAb) or n.i. IgG (10 μg/ml). (Lower) BCE cells irradiated with UVB (20 mJ/cm2) or treated with H2O2 (400 μM) and incubated for 6 h with the indicated reagents. In B and C, ERK2 was used as a loading control. (D) Immunocytochemical analysis of caspase 3 activation in cells treated as described above with TGF-β1 or TNF-α and the indicated reagents. The apoptotic index was measured as described in B. These experiments were repeated twice with comparable results.
To rule out other nonspecific effects of VEGF inhibition we tested whether VEGF antibody blocks endothelial cell apoptosis induced by treatments other than TGF-β1. TNF-α, oxidative stress and UV light (UVB), known inducers of programmed cell death, up-regulated endothelial cell apoptosis with an effect comparable to that of TGF-β1. However, VEGF antibody did not reduce apoptosis induced by these treatments (Fig. 3 C and D). Thus, VEGF antibody blocks TGF-β1 induction of apoptosis but has no effect on other inducers of endothelial cell apoptosis.
Down-Regulation of VEGFR-2 (flk-1) Abrogates the Apoptotic Activity of TGF-β1 on Endothelial Cells.
To investigate which VEGFR mediates the apoptotic activity of TGF-β1 we first characterized the effect of TGF-β1 on flk-1 and flt-1 expression. Treatment of endothelial cells with TGF-β1 for 6 h did not alter the levels of either VEGFR (Fig. 4A and B). However, consistent with previous reports, TGF-β1 down-regulated flk-1 expression in cells treated for 24–30 h (data not shown). Because apoptosis induction by TGF-β1 occurs within 6 h of incubation (Fig. 1), these results showed that TGF-β1 induces apoptosis in endothelial cells that express both VEGFRs.
Fig. 4.

flk-1 mediates the apoptotic activity of TGF-β1. (A and B) Western blotting analysis of flt-1 and flk-1 expression in HUVEC incubated without (−) or with (+) TGF-β1 (1 ng/ml) for 6 h. ERK2: loading control. (C and D) Western blotting. (E) RT-PCR. Shown is flt-1 or flk-1 expression in HUVEC transfected with flt-1 or flk-1 siRNAs (+) or mock-transfected (−). ERK2 and β-actin: loading controls. Control, nontransfected cells; siRNA −, mock-transfected cells. (F) Western blotting analysis of PARP degradation in HUVEC transfected with flt-1 or flk-1 siRNAs or mock-transfected (Control). These experiments were repeated three times with similar results.
To identify which VEGFR mediates the apoptotic effect of TGF-β1 we used siRNAs to inhibit either flt-1 or flk-1 expression. Transfection of endothelial cells with flt-1 siRNAs blocked flt-1 expression without affecting flk-1 expression. Conversely, flk-1 siRNAs abolished flk-1 expression without affecting flt-1 levels (Fig. 4 C–E), indicating that the inhibitory action of the flk-1 and flt-1 siRNAs did not reflect nonspecific effects of the siRNAs transfection. Inhibition of flk-1 or flt-1 expression did not alter the basal level of apoptosis in the absence of TGF-β1 (Fig. 4F). However, flk-1 siRNAs completely blocked TGF-β1 induction of apoptosis, whereas inhibition of flt-1 expression was ineffective (Fig. 4F). Consistent with these results, TGF-β1 did not induce apoptosis in human smooth muscle cells nor in porcine aortic endothelial cells devoid of flk-1 (Fig. 9, which is published as supporting information on the PNAS web site). These results showed that TGF-β1 induction of endothelial cell apoptosis requires signaling through VEGFR-2 (flk-1).
TGF-β1 Induction of Endothelial Cell Apoptosis Requires VEGF/flk-1-Mediated Activation of p38MAPK.
Induction of endothelial cell apoptosis by TGF-β1 involves p38MAPK activation (33), and in a variety of cell types TGF-β1 activates p38MAPK through a Smad-independent mechanism (34, 35). Therefore, we hypothesized that TGF-β1 activates MAPKs in endothelial cells through the action of endogenous VEGF. Because p38MAPK and ERK1/2 are activated by VEGF, we characterized the activation of these MAPKs in endothelial cells treated with TGF-β1 for 6 h, a time at which apoptosis occurs. TGF-β1 induced strong activation of ERK1/2 and p38MAPK, which was blocked by neutralizing anti-VEGF antibody (Fig. 5A). In both TGF-β1- and VEGF-treated cells down-regulation of flt-1 did not affect ERK1/2 activation but slightly up-regulated p38MAPK activation. In contrast, inhibition of flk-1 expression abolished both ERK1/2 and p38MAPK activation (Fig. 5B), showing that TGF-β1 activates these signaling pathways through the action of VEGF.
Fig. 5.

VEGFR-2 mediates MAPK activation by TGF-β1. (A and B) Western blotting analysis of p38MAPK [(p)p38] and ERK1/2 [(p)ERK1/2] activation. (A) HUVEC incubated with either TGF-β1 (1 ng/ml) or control medium (Control) with or without n.i. IgG or VEGF mAb (10 μg/ml). (B) HUVEC transfected with flt-1 or flk-1 siRNAs or with transfection reagent alone (Control) and incubated without (−) or with TGF-β1 (1 ng/ml) for 6 h, or with VEGF (30 ng/ml) for 15 min. ERK2: loading control. (C and D) Immunocytochemical analysis of p38MAPK activation. (C) HUVEC incubated with control medium (Upper) or TGF-β1 (1 ng/ml; Lower). (D) HUVEC transfected with flt-1 or flk-1 siRNAs (Lower) or mock-transfected (Upper) and treated with TGF-β1 or control medium (Control). The cells were double-stained with phospho-p38MAPK antibody [(p)p38] and with DAPI to detect apoptotic nuclei. FITC-labeled anti-rabbit IgG antibodies show active p38MAPK. (Magnification: ×200.) (E) Western blotting analysis of PARP degradation in HUVEC treated with TGF-β1 (1 ng/ml) in the presence or absence of the p38MAPK inhibitor SB202190 (10 μM) or the MEK1/2 inhibitor UO126 (10 μM). UO126 abolished ERK1/2 activation and SB202190 blocked p38MAPK activity as assessed by Western blotting with antibodies to active ERK1/2 and in vitro kinase assay with ATF-2, a p38MAPK substrate (data not shown). ERK2: loading control. (F) Western blotting analysis of p38MAPK expression (p38) and activation [(p)p38] and caspase 3 activation in HUVEC transfected with siRNA to p38MAPK or control siRNA to lamin A/C, and incubated for 6 h in the presence or absence of TGF-β1 (1 ng/ml). ERK2: loading control. These experiments were repeated three times with comparable results.
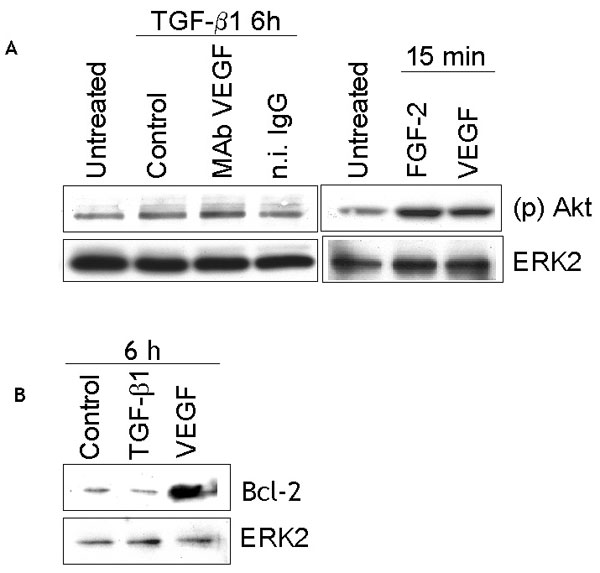
VEGF protects endothelial cells from p38MAPK-mediated apoptosis by up-regulating activation of the PI3K-Akt-Bcl-2 pathway (36). Therefore, we tested the hypothesis that TGF-β1 induces endothelial cell apoptosis by down-regulating activation of the Akt survival pathway. The results (Fig. 10, which is published as supporting information on the PNAS web site) showed that whereas VEGF rapidly up-regulated Akt activation and Bcl-2 levels, TGF-β1 did not alter the levels of Bcl-2 or active Akt relative to control, untreated cells.
p38MAPK transduces the apoptotic signal of TGF-β1, whereas ERK1/2 relays prosurvival signaling in a variety of cells including endothelial cells (37–39). In TGF-β1-treated endothelial cells p38MAPK activation occurred only in cells that showed the nuclear chromatin condensation typical of apoptosis, whereas cells devoid of apoptotic nuclei showed no active p38MAPK (Fig. 5C). Inhibition of flk-1 expression down-regulated the number of active p38MAPK-positive/apoptotic cells, whereas down-regulation of flt-1 expression had no such effect (Fig. 5D). Pharmacological inhibition of p38MAPK blocked TGF-β1 induction of endothelial cell apoptosis, whereas inhibition of ERK1/2 activation increased TGF-β1-induced apoptosis (Fig. 5E), indicating that p38MAPK mediates the apoptotic signaling of TGF-β1 in endothelial cells. To test this hypothesis we down-regulated p38MAPK expression by specific siRNAs. In control siRNA-transfected cells TGF-β1 induced both apoptosis and p38MAPK activation. In contrast, transfection with p38MAPK siRNA abrogated both TGF-β1 activation of p38MAPK and induction of apoptosis (Fig. 5F). Thus, TGF-β1 induction of endothelial cell apoptosis requires VEGF/flk-1-mediated activation of p38MAPK.
Discussion
The ability to promote endothelial cell apoptosis and expression of FGF-2 and VEGF at the same time is an intriguing feature of TGF-β1, a potent angiogenesis inducer. Both FGF-2 and VEGF are endothelial cell survival factors. How can TGF-β1 induce endothelial cell apoptosis while up-regulating expression of survival factors? How can TGF-β1 induce angiogenesis while promoting endothelial cell apoptosis? The prosurvival or proapoptotic activity of growth factors can be modified depending on the context of other signaling molecules (40). The data reported here show several features of the interactions among TGF-β1, FGF-2, and VEGF, growth factors often coexpressed in tissues in which angiogenesis occurs. TGF-β1, FGF-2, and VEGF act in series on endothelial cells through a cascade of autocrine or paracrine reactions (Fig. 6). An important consequence of these growth factor interactions is TGF-β1-induced conversion of endothelial cell VEGF signaling. VEGF protects endothelial cells from apoptosis. However, in context with TGF-β1, VEGF provides endothelial cells with permissive signaling for apoptosis induction by TGF-β1.
Fig. 6.

Control of endothelial cell apoptosis by TGF-β1–VEGF interaction. In the absence of TGF-β1, VEGF induces endothelial cell activation of p38MAPK through flk-1 and promotes endothelial cell survival (Left). TGF-β1 induces endothelial cell production of FGF-2, which up-regulates VEGF expression (Right). Cross-talk between VEGF- and TGF-β1-specific signaling pathways converts VEGF/flk-1-activated p38MAPK into a proapoptotic signal.
Our data show that the apoptotic activity of TGF-β1 on endothelial cells is controlled by the opposing effects of FGF-2 and VEGF. Abrogation of FGF-2 signaling up-regulates endothelial cell apoptosis (Fig. 2 A and E). In contrast, inhibition of VEGF signaling abolishes the apoptotic activity of TGF-β1 (Figs. 2 A and B, 3 B–D, and 4F). Although FGF-2 mediates the expression of VEGF required for TGF-β1 induction of apoptosis, neutralizing antibody to FGF-2 does not inhibit the apoptotic effect of TGF-β1. This seeming discrepancy is explained by our findings that antibody-mediated inhibition or genetic deficiency of FGF-2 increases endothelial cell apoptosis in the absence of TGF-β1. Thus, FGF-2 has a dual role in the control of endothelial cell apoptosis. On one hand, FGF-2 up-regulates VEGF expression, which is required for TGF-β1 induction of apoptosis; on the other hand, it protects endothelial cells from apoptosis through a VEGF-independent mechanism. TGF-β1 down-regulation of flk-1 expression may represent an additional mechanism for the control of endothelial cell apoptosis. In our experiments 6-h treatment of endothelial cells with TGF-β1 did not alter endothelial cell levels of VEGFRs. However, consistent with previous reports (22), TGF-β1 treatment for 15–24 h induced a 40–50% decrease in flk-1 expression. Thus, TGF-β1 rapidly induces apoptosis in endothelial cells in which flk-1 is expressed. Subsequently, down-regulation of flk-1 levels can down-regulate apoptosis.
TGF-β1 exerts its activity on a variety of cells through activation of the Smad proteins and other signaling pathways including the MAPKs. Our results show that in endothelial cells TGF-β1 activates p38MAPK through VEGF-mediated activation of flk-1 (Fig. 6). Inhibition of p38MAPK signaling blocks TGF-β1 induction of apoptosis. Therefore, TGF-β1 induction of endothelial cell apoptosis requires VEGF/flk-1-mediated activation of p38MAPK. In the absence of TGF-β1, VEGF also induces endothelial cell activation of p38MAPK but VEGF signaling results in cell survival and proliferation; in contrast, in the context of TGF-β1 signaling VEGF-induced activation of p38MAPK results in apoptosis. Thus, TGF-β1 renders VEGF signaling permissive for TGF-β1 induction of apoptosis. This effect likely involves cross-talk between TGF-β1- and VEGF-specific signaling pathways that can occur at multiple levels upstream of p38MAPK. Alternatively, parallel but independent signals from VEGF and from TGF-β1 may both be required for induction of endothelial cell apoptosis (Fig. 6).
Although TGF-β1 induces endothelial cells apoptosis, it promotes angiogenesis in vitro and in vivo (17–19, 26). Several mechanisms can explain this seeming contradiction. We found that the apoptotic effect of TGF-β1 on endothelial cells in vitro is transient (3–9 h) (33) and followed by refractoriness of the cells to TGF-β1 induction of apoptosis (data not shown). Previous reports have shown that the apoptotic activity of TGF-β1 is followed by endothelial cell proliferation and migration, with formation of capillary structures in vitro and in vivo (26). These effects can be controlled by multiple mechanisms. Our data show that TGF-β1 induction of apoptosis involves up-regulation of endothelial cell expression of FGF-2 and VEGF, which promote endothelial cell survival, proliferation and migration, functions required for angiogenesis. Whereas apoptosis prunes excess endothelial cells from the forming vascular network, adjacent cells proliferate and migrate (20, 23, 26). The up-regulation of FGF-2 and VEGF expression by TGF-β1 can thus provide endothelial cells with proliferative, migratory and survival signaling that can be relayed from apoptotic cells to adjacent endothelial cells. This conclusion is consistent with the hypothesis that apoptosis not only represents a mechanism for pruning the forming vascular network but is necessary to control endothelial cell functions required for angiogenesis (26).
Under normal conditions, vascular endothelium in vivo, with the exception of some tumor endothelial cells and aortic endothelial cells, does not produce VEGF (41). However, endothelial cells of forming capillaries express VEGF (4), and TGF-β1 induces endothelial cell apoptosis in vivo (24). Thus, it is possible that VEGF is also produced by vascular endothelium in tissues in which active TGF-β1 is present. In addition, in vivo VEGF derived from other cells types can activate signaling in endothelial cells. Whatever the source of VEGF in vivo, its prosurvival signaling can be converted by TGF-β1 into permissive signaling for TGF-β1 induction of endothelial cell apoptosis.
In conclusion, the cross-talk between TGF-β1, FGF-2 and VEGF has two important consequences. The coordination of both proapoptotic and prosurvival, proproliferative, and promigratory signals affords a fine modulation of opposing cell functions required for angiogenesis. More important, TGF-β1 converts VEGF activity from antiapoptotic to proapoptotic. This finding has a pharmacologically relevant implication. Uncontrolled endothelial cell apoptosis inhibits angiogenesis, and a number of antiangiogenesis treatments are aimed to induce endothelial cell apoptosis (27). Our finding that VEGF, the major tumor angiogenesis inducer, can be converted into a factor required for TGF-β1 induction of endothelial cell apoptosis indicates a novel approach to block tumor angiogenesis. Pharmacological treatments that mimic the effect of TGF-β1 on VEGF signaling could take advantage of high local levels of tumor-derived VEGF to induce uncontrolled endothelial cell apoptosis and efficiently block angiogenesis.
Materials and Methods
Materials.
Human purified or recombinant TGF-β1, human and mouse recombinant VEGF165, human recombinant TNF-α, neutralizing polyclonal antibody to human VEGF, goat anti-human flk-1 and flt-1 antibodies, and anti-human and anti-mouse VEGF antibodies were purchased from R & D Systems (Minneapolis, MN); human recombinant FGF-2 was from Gibco/BRL, Life Technologies (Rockville, MD); BrdU and anti-BrdU monoclonal antibody were from Roche Molecular Biochemicals (Indianapolis, IN); polyclonal antibodies to phosphorylated ERK1/2, PARP p85, and cleaved human caspase 3 were from Cell Signaling Technology (Beverly, MA); polyclonal antibodies to total ERK-2 and p38MAPK were from Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal antibody to phosphorylated p38MAPK and the MEK1/2 inhibitor UO126 were from Promega (Madison, WI); mouse and rabbit nonimmune (n.i.) IgG was from Sigma (St. Louis, MO); the p38 inhibitor SB202190 was from Calbiochem (La Jolla, CA); flt1, flk1, and p38 siRNAs were from Dharmacon RNA Technologies (Lafayette, CO); and lamin A/C siRNA was from TriLink Biotechnologies (San Diego, CA). Neutralizing monoclonal antibody to human FGF-2 (mAb 354F1) was a generous gift from Texas Biotechnology (Houston, TX).
Cells and Media.
Bovine aortic endothelial cells, bovine capillary endothelial cells (BCE cells), and FGF-2−/− endothelial cells were grown as described (4, 15). Human umbilical vein endothelial cells (HUVEC) (Cascade Biologics, Portland, OR) were grown in media recommended by the manufacturers and used between passages 3 and 5.
Induction of Apoptosis.
Subconfluent endothelial cells starved overnight in medium containing 0.5% serum were incubated for 6 h in starvation medium supplemented with the indicated concentrations of TGF-β1, TNF-α (10 ng/ml plus 1 μg/ml cycloheximide) (42), or H2O2 (400 μM) in the absence or presence of the indicated reagents.
Western and Northern Blotting.
Western blotting was performed as described in ref. 15, and Northern blotting was performed as described in ref. 4.
siRNA Transfection.
Subconfluent HUVEC were incubated with 200 pmol of human flk-1, flt-1, p38MAPK, or lamin A/C siRNA and 4 μl of Oligofectamine (Invitrogen, Carlsbad, CA) in serum-free medium for 4 h at 37°C, after which medium supplemented with 10% serum was added. The cells were used 48 h after transfection.
RT-PCR.
The following primers for human flk-1 and flt-1 were synthesized by IDT DNA Technologies (Coralville, IA) based on the published sequences (43): flt-1 (s, 5′-CGACCTTGGTTGTGGCTGACT; a, 5′-CGGTTCTGGTTGGTGGCTTTG); flk-1 (s, 5′-AACAAAGTCGGGAGAGGA; a, 5′-TGACAAGAAGTAGCCAGAAGA); and β-actin (s, 5′-ATCTGGGACCAACCTTCTAGAATGAG; a, 5′-CGTCATACTCCTGCTTGCTGATCCAC). cDNA was synthesized from 1 μg of total RNA by using SuperScript II RT (Invitrogen) and oligo(dT) 3′ primer. Two microliters of cDNA was amplified by PCR as described (43).
Annexin V Binding Assay.
Cells were detached with 2.2 mM EDTA in PBS, mixed with cells present in the supernatant, and stained with FITC-labeled annexin V and propidium iodide (PI; Roche Molecular Biochemicals) according to the manufacturer's instructions. Samples (104 events) were analyzed with a flow cytometer (Becton Dickinson, San Jose, CA), and cell distributions were determined with eCellQuest Software (Becton Dickinson). Annexin V+/PI− cells were considered apoptotic; annexin V+/PI+ cells were considered necrotic.
BrdU Uptake.
Cells grown on coverslips were incubated with BrdU (10 μM) for 1 h at 37°C. The cells were stained with BrdU antibody following the manufacturer's instructions, and with Hoechst 33342 (Sigma; 10 mg/ml) to evidence the nuclei. One thousand nuclei per coverslip were examined with a Axioskop 2 photomicroscope (Zeiss, Thornwood, NY) at ×400. The results are expressed as number of BrdU-positive nuclei/total number of nuclei × 100.
Statistical Analysis.
t tests on the equality of means were performed by using Stata 8.
Supplementary Material
Acknowledgments
We thank Drs. David Moscatelli, John S. Munger, Daniel B. Rifkin, and Edward Y. Skolnik for their critical reading of the manuscript, valuable comments, and suggestions. This work was supported by National Institutes of Health Grants R01 HL070203 and R01 HL070203-03S1 (to P.M.) and by funds from the Seymour Cohn Foundation for Cardiovascular Research.
Abbreviations
- BCE cell
bovine capillary endothelial cell
- HUVEC
human umbilical vein endothelial cell
- n.i.
nonimmune.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 3.Massague J, Blain SW, Lo RS. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 4.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. J Cell Biol. 1998;141:1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bostrom K, Zebboudj AF, Yao Y, Lin TS, Torres A. J Biol Chem. 2004;279:52904–52913. doi: 10.1074/jbc.M406868200. [DOI] [PubMed] [Google Scholar]
- 6.Cross MJ, Claesson-Welsh L. Trends Pharmacol Sci. 2001;22:201–207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 7.Lamalice L, Houle F, Jourdan G, Huot J. Oncogene. 2004;23:434–445. doi: 10.1038/sj.onc.1207034. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Nat Rev Drug Discovery. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 11.Mignatti P, Morimoto T, Rifkin DB. Proc Natl Acad Sci USA. 1991;88:11007–11011. doi: 10.1073/pnas.88.24.11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato Y, Rifkin DB. J Cell Biol. 1988;107:1199–1205. doi: 10.1083/jcb.107.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Proc Natl Acad Sci USA. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, et al. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pintucci G, Moscatelli D, Saponara F, Biernacki PR, Baumann FG, Bizekis C, Galloway AC, Basilico C, Mignatti P. FASEB J. 2002;16:598–600. doi: 10.1096/fj.01-0815fje. [DOI] [PubMed] [Google Scholar]
- 16.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Development (Cambridge, UK) 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 17.Madri JA, Pratt BM, Tucker AM. J Cell Biol. 1988;106:1375–1384. doi: 10.1083/jcb.106.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. Proc Natl Acad Sci USA. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang EY, Moses HL. J Cell Biol. 1990;111:731–741. doi: 10.1083/jcb.111.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollman MJ, Naumovski L, Gibbons GH. Circulation. 1999;99:2019–2026. doi: 10.1161/01.cir.99.15.2019. [DOI] [PubMed] [Google Scholar]
- 21.Saksela O, Moscatelli D, Rifkin DB. J Cell Biol. 1987;105:957–963. doi: 10.1083/jcb.105.2.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandriota SJ, Menoud PA, Pepper MS. J Biol Chem. 1996;271:11500–11505. doi: 10.1074/jbc.271.19.11500. [DOI] [PubMed] [Google Scholar]
- 23.Pollman MJ, Naumovski L, Gibbons GH. J Cell Physiol. 1999;178:359–370. doi: 10.1002/(SICI)1097-4652(199903)178:3<359::AID-JCP10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 24.Fierlbeck W, Liu A, Coyle R, Ballermann BJ. J Am Soc Nephrol. 2003;14:1349–1354. doi: 10.1097/01.asn.0000061779.70530.06. [DOI] [PubMed] [Google Scholar]
- 25.Segura I, Serrano A, De Buitrago GG, Gonzalez MA, Abad JL, Claveria C, Gomez L, Bernad A, Martinez AC, Riese HH. FASEB J. 2002;16:833–841. doi: 10.1096/fj.01-0819com. [DOI] [PubMed] [Google Scholar]
- 26.Choi ME, Ballermann BJ. J Biol Chem. 1995;270:21144–21150. doi: 10.1074/jbc.270.36.21144. [DOI] [PubMed] [Google Scholar]
- 27.Ferrara N, Kerbel RS. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Massague J. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez-Capelo A. Cytokine Growth Factor Rev. 2005;16:15–34. doi: 10.1016/j.cytogfr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Karsan A, Yee E, Poirier GG, Zhou P, Craig R, Harlan JM. Am J Pathol. 1997;151:1775–1784. [PMC free article] [PubMed] [Google Scholar]
- 31.Korff T, Augustin HG. J Cell Biol. 1998;143:1341–1352. doi: 10.1083/jcb.143.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerber H-P, Dixit V, Ferrara N. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 33.Hyman KM, Seghezzi G, Pintucci G, Stellari G, Kim JH, Grossi EA, Galloway AC, Mignatti P. Surgery. 2002;132:173–179. doi: 10.1067/msy.2002.125304. [DOI] [PubMed] [Google Scholar]
- 34.Dai C, Yang J, Liu Y. J Biol Chem. 2003;278:12537–12545. doi: 10.1074/jbc.M300777200. [DOI] [PubMed] [Google Scholar]
- 35.Yu L, Hebert MC, Zhang YE. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar P, Miller AI, Polverini PJ. J Biol Chem. 2004;279:43352–43360. doi: 10.1074/jbc.M405777200. [DOI] [PubMed] [Google Scholar]
- 37.Kimura N, Matsuo R, Shibuya H, Nakashima K, Taga T. J Biol Chem. 2000;275:17647–17652. doi: 10.1074/jbc.M908622199. [DOI] [PubMed] [Google Scholar]
- 38.Kim KY, Kim BC, Xu Z, Kim SJ. J Biol Chem. 2004;279:29478–29484. doi: 10.1074/jbc.M313947200. [DOI] [PubMed] [Google Scholar]
- 39.Undevia NS, Dorscheid DR, Marroquin BA, Gugliotta WL, Tse R, White SR. Am J Physiol. 2004;287:L515–L524. doi: 10.1152/ajplung.00044.2004. [DOI] [PubMed] [Google Scholar]
- 40.Sporn MB, Roberts AB. Nature. 1988;332:217–219. doi: 10.1038/332217a0. [DOI] [PubMed] [Google Scholar]
- 41.Maharaj AS, Saint-Geniez M, Maldonado AE, D'Amore PA. Am J Pathol. 2006;168:639–648. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grethe S, Ares MP, Andersson T, Porn-Ares MI. Exp Cell Res. 2004;298:632–642. doi: 10.1016/j.yexcr.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Krussel JS, Casan EM, Raga F, Hirchenhain J, Wen Y, Huang HY, Bielfeld P, Polan ML. Mol Hum Reprod. 1999;5:452–458. doi: 10.1093/molehr/5.5.452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}