

Abstract
Recent studies have shown that small noncoding RNAs, such as microRNAs and siRNAs, regulate gene expression at multiple levels including chromatin architecture, transcription, RNA editing, RNA stability, and translation. Each form of RNA-dependent regulation has been generally found to silence homologous sequences and collectively called RNAi. To further study the regulatory role of small RNAs at the transcriptional level, we designed and synthesized 21-nt dsRNAs targeting selected promoter regions of human genes E-cadherin, p21WAF1/CIP1 (p21), and VEGF. Surprisingly, transfection of these dsRNAs into human cell lines caused long-lasting and sequence-specific induction of targeted genes. dsRNA mutation studies reveal that the 5′ end of the antisense strand, or “seed” sequence, is critical for activity. Mechanistically, the dsRNA-induced gene activation requires the Argonaute 2 (Ago2) protein and is associated with a loss of lysine-9 methylation on histone 3 at dsRNA-target sites. In conclusion, we have identified several dsRNAs that activate gene expression by targeting noncoding regulatory regions in gene promoters. These findings reveal a more diverse role for small RNA molecules in the regulation of gene expression than previously recognized and identify a potential therapeutic use for dsRNA in targeted gene activation.
Keywords: gene regulation, promoter, Argonaute 2
Small dsRNAs were initially discovered as the trigger of RNAi, a mechanism by which homologous mRNA is degraded to result in posttranscriptional gene silencing (1, 2). dsRNA is also involved in transcriptional gene silencing by directing DNA methylation in plants (3, 4) and heterochromatin formation in fission yeast (5) and Drosophila (6). Only recently has transcriptional gene silencing been discovered to occur in mammals (7, 8). There are, however, a few cases in which small RNAs positively regulate cognate sequences. For example, a small RNA isolated from neural stem cells can activate the transcription of genes containing NRSE/RE1 sequences to stimulate neuronal differentiation in adult stem cells (9). In addition, a liver-specific microRNA was found to enhance viral replication by targeting the 5′ noncoding region of the viral genome (10). More than three decades ago, Britten and Davidson (11) proposed a theory in which so-called “activator” RNAs, transcribed from redundant genomic regions, activate a battery of protein coding genes. To further study the potential role that small RNAs have in gene transcription, we selectively targeted promoter regions with synthetic dsRNAs and identified several dsRNAs that readily activate gene expression at a transcriptional level.
Results
dsRNAs Targeting the Promoter of E-Cadherin Induce Gene Expression.
We first designed and synthesized two 21-nt dsRNAs targeting the E-cadherin promoter at sequence positions −302 (dsEcad-302) and −215 (dsEcad-215) relative to the transcription start site (Fig. 1A). Regions with either high GC content or low sequence complexity were excluded as dsRNA targets, such as the CpG island and the Alu repeat element in the E-cadherin promoter (Fig. 1A). We also designed a 21-nt control dsRNA (dsCon-1), which lacked significant homology to all known human sequences. We transfected these dsRNAs into two human prostate cancer cell lines, PC-3 and DU-145. In PC-3 cells we observed a profound induction of E-cadherin mRNA (Fig. 1B) and protein expression (Fig. 1C). A time-course and dose–response experiment revealed that E-cadherin induction occurred within 48 h of transfection and at dsRNA concentrations as low as 1 nM (Fig. 1 D and E). Morphologically, mock-transfected and dsCon-1-transfected cells maintained healthy growth after transfection, whereas cells transfected with E-cadherin dsRNA gradually lost viability after day 3 (Fig. 7, which is published as supporting information on the PNAS web site). This finding is consistent with previous studies in which forced expression of E-cadherin resulted in slower cell growth (12, 13). We also tested longer periods of transfection in PC-3 cells. Surprisingly, at days 10 and 13 after a single transfection of dsEcad-215, we detected a 14- and 3.8-fold increase in E-cadherin expression, respectively (Fig. 1F). Although DU-145 cells normally expressed high levels of E-cadherin, we also observed a moderate and consistent increase in E-cadherin expression after dsEcad-302 and -215 treatment (Fig. 8, which is published as supporting information on the PNAS web site). These results were reproducible in at least five independent experiments using dsRNAs synthesized from three separate batches. Overall, no changes in E-cadherin expression were detected in cells transfected with mock or dsCon-1 controls (Fig. 1 B–F).
Fig. 1.

dsRNAs targeting the E-cadherin gene promoter induce E-cadherin mRNA and protein expression. (A) A schematic representation of the E-cadherin promoter with its CpG island, Alu repeat element, transcription start site, and dsRNA targets. (B) PC-3 cells were transfected with 50 nM dsRNA for 72 h. mRNA expression of E-cadherin and GAPDH were analyzed by RT-PCR. Samples for each treatment are shown in duplicate. (C) E-cadherin, β-actin, and GAPDH protein levels were detected by Western blot analysis in PC-3 cells treated as in B. (D) PC-3 cells were treated with dsEcad-215 at the indicated concentrations for 72 h. E-cadherin and GAPDH expression was detected by Western blotting. (E) PC-3 cells were transfected with 50 nM dsEcad-215 for the indicated lengths of time. E-cadherin and β-actin expression was detected by Western blotting. (F) PC-3 cells were transfected with 50 nM dsCon-1 or dsEcad-215 for the indicated periods of time. Mock samples were transfected in the absence of dsRNA. Western blot analysis shows E-cadherin protein levels in PC-3 cells on days 10 or 13 after single transfections.
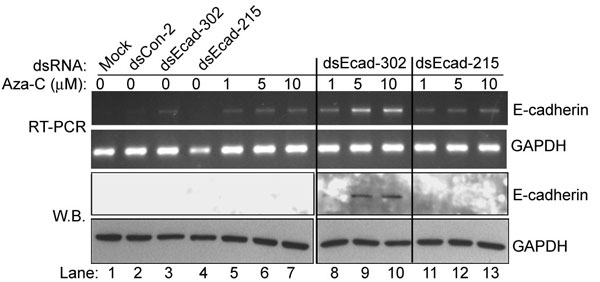
E-cadherin is epigenetically silenced in HeLa cells because of aberrant methylation of the CpG island within its promoter. To determine whether E-cadherin dsRNAs could activate E-cadherin expression from a silenced promoter, we transfected HeLa cells with dsEcad-302 and dsEcad-215. Initially, both dsRNA molecules failed to induce E-cadherin expression (lanes 3 and 4 in Fig. 9, which is published as supporting information on the PNAS web site). However, when cells were cotreated with low-doses (1–10 μM) of DNA demethylating agent 5-azacytidine (Aza-C) and dsRNA, E-cadherin expression elevated to levels much greater than treatments of Aza-C alone (Fig. 9, lanes 8–10). In fact, E-cadherin protein was detectable by Western blot analysis only after Aza-C and dsEcad-302 cotreatments (Fig. 9, lanes 9 and 10). These results suggest that preestablished methylation of the E-cadherin promoter inhibits gene induction by dsRNA.
A previous study by Ting et al. (8) showed that synthetic dsRNAs targeting the E-cadherin CpG island induced a silencing effect on E-cadherin expression in human cells. We subsequently decided to test three more 21-nt dsRNAs (dsEcad-185, -75, and -60) that targeted regions in the E-cadherin promoter similar to those targeted by Ting et al. (8) (Fig. 10A, which is published as supporting information on the PNAS web site). Consistent with the previous report (8), dsEcad-185, -75, and -60 did not induce gene expression, but rather caused subtle declines in E-cadherin levels when compared with mock transfections (Fig. 10 B and C). These data suggest that dsRNA-induced gene activation (RNAa) is sensitive to target location.
dsRNAs Targeting the Promoter of p21 and VEGF Induce Gene Expression.
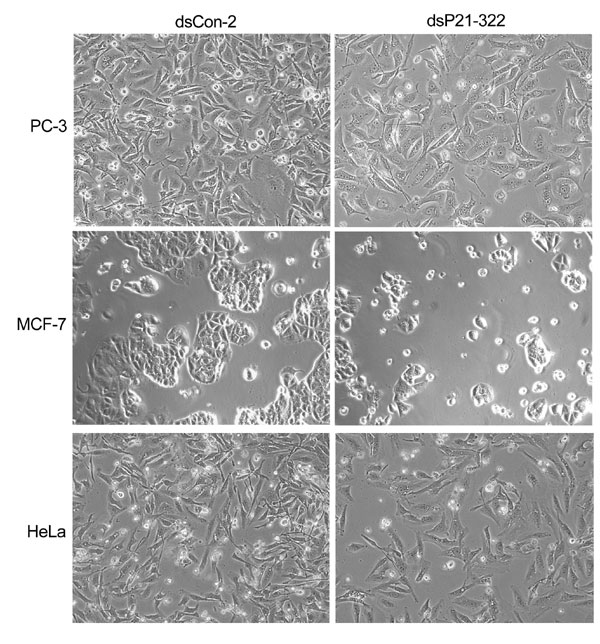
We further examined RNAa on two additional genes, p21WAF1/CIP1 (p21) and vascular endothelial growth factor (VEGF). Two dsRNAs were designed; one (dsP21–322) targeted the p21 promoter at position −322 (Fig. 2A), whereas the other (dsVEGF-706) targeted the VEGF promoter at position −706 (Fig. 3A). We transfected dsP21–322 and a control dsRNA (dsCon-2) into PC-3 cells, HeLa cells, and human breast cancer cell line MCF-7. The dsCon-2 control also lacked significant homology to all known human sequences and was created to further broaden our source of control dsRNA molecules. Forty eight hours after transfections, a slower growth rate was observed in all cell lines transfected with dsP21–322 in comparison to dsCon-2-transfected cells (Fig. 11, which is published as supporting information on the PNAS web site). At 72 h, dsP21–322 caused a significant induction in p21 mRNA expression (Fig. 2B). Compared with mock transfections, induction was 12.5-, 10.1-, and 2.4-fold in PC-3, HeLa, and MCF-7 cells, respectively (Fig. 2C). Induction of p21 was further confirmed by Western blot analysis (Fig. 2D). The elevated levels of p21 protein strongly correlated to the increase in p21 mRNA expression.
Fig. 2.

dsRNAs induce p21 expression in different human cell lines. Cells were transfected with 50 nM dsRNA for 72 h. mRNA and protein levels were analyzed by RT-PCR and Western blotting, respectively. (A) A schematic representation of the p21 promoter with its CpG island, SP1 sites, TATA signal, transcription start site, and the dsRNA target. (B) p21 and GAPDH mRNA expression levels in PC-3, MCF-7, and HeLa cells after mock, dsCon-2, or dsP21–322 transfections. (C) p21 mRNA expression levels were normalized to GAPDH. The results are presented as the mean ± SEM of two independent experiments (two sample repeats within each experiment). (D) Induction of p21 protein expression was confirmed by Western blot analysis in PC-3, MCF-7, and HeLa cells. GAPDH levels were also detected and served as a loading control. (E) Western blot analysis of p21 and GAPDH after mock, dsCon-2, or dsP21–322 transfections in HEK293, J82, LNCaP, and T24 cells.
Fig. 3.

dsRNAs induce VEGF expression in HeLa cells. Cells were transfected with 50 nM dsRNA for 72 h. mRNA levels were analyzed by RT-PCR. (A) A schematic representation of the VEGF promoter and the location of the dsRNA target. (B) mRNA expression of two VEGF isoforms (VEGF-189 and -165) and GAPDH in HeLa cells after mock, dsCon-2, or dsVEGF-706 transfections. (C) VEGF mRNA expression levels were normalized to GAPDH. The results are presented as the mean ± SEM of two independent experiments with each sample repeated twice.
Transfection of dsP21–322 was also performed in four additional human cell lines, including the embryonic kidney cell line HEK293, the prostate cancer cell line LNCaP, and two bladder cancer cell lines, J82 and T24. As shown in Fig. 2E, dsP21–322 transfection resulted in varying degrees of p21 induction in each cell line.
Transfection of dsVEGF-706 into HeLa cells resulted in VEGF induction (Fig. 3B). Compared with mock transfections, expression of two VEGF mRNA isoforms, VEGF-165 and -189, increased 4- and 3.7-fold in HeLa cells, respectively (Fig. 3C).
IFN Response Is Not Involved in dsRNA-Induced Transcriptional Activation.
Double-stranded RNA molecules have been shown to induce the expression of IFN-regulated genes by initiating the production of type I IFNs (14–16). This effect is mediated in part by the phosphorylation-dependent activation of the dsRNA-dependent protein kinase PKR. To rule out the possibility that gene induction may have occurred by a nonspecific IFN response, we analyzed the expression of two classic IFN-regulated genes, OAS1 and OAS3, as well as assessed PKR activation in cells transfected with dsRNAs. As shown in Fig. 12 A–C, which is published as supporting information on the PNAS web site, dsRNAs did not induce the expression of OAS1 or OAS3, nor did they promote PKR phosphorylation. Furthermore, type I IFN-α2a treatment had no effect on E-cadherin or p21 expression (Fig. 12D). Taken together, these results indicate that gene induction by dsRNA did not occur by a nonspecific IFN response.
dsRNA Sequence Requirement for RNAa.
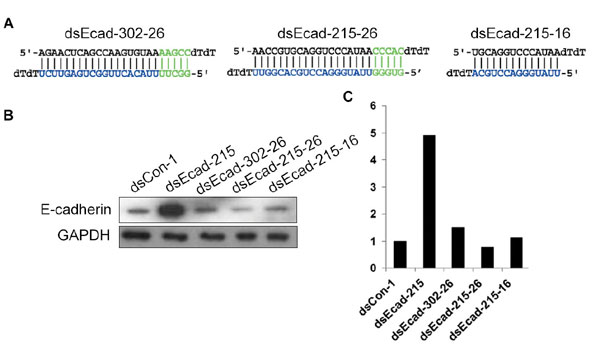
To evaluate the effect of dsRNA length on RNAa, we truncated 5 bp from the 3′ end (relative to the antisense strand) of the dsEcad-215 duplex resulting in a 16-nt dsRNA (dsEcad-215–16) (Fig. 13A, which is published as supporting information on the PNAS web site). We also extended dsEcad-215 and -302 to 26 nt (dsEcad-215–26 and dsEcad-302–26) (Fig. 13A). Interestingly, both the shortened and extended dsRNAs did not induce E-cadherin transcription (Fig. 13 B and C). These results suggest that dsRNAs of ≈21 nt in size are preferable for RNAa.
To characterize the sequence requirement and specificity for RNAa, we analyzed mutant dsRNAs based on dsEcad-215 (Fig. 4A) and dsP21–322 (Fig. 4D). Mutations to the first 5 bp of the dsEcad-215 duplex (dsEcad-215-G3′) created 5 mismatches between the 3′ end of the antisense RNA strand and the target DNA (Fig. 4A). Surprisingly, dsEcad-215-G3′ was still potent in inducing E-cadherin expression (Fig. 4 B and C). However, mutations to the last 5 bp of the dsEcad-215 duplex (dsEcad-215-G5′) (Fig. 4A) almost completely abolished E-cadherin activation (Fig. 4 B and C). We also tested three mutant dsRNAs derived from dsP21–322 in PC-3 and HeLa cells, including a 5-bp mutant at the 5′ end (dsP21–322-G5′, relative to the antisense strand), a 5-bp mutant at the 3′ end (dsP21–322-G3′), and a 5-bp mutant in the middle region (dsP21–322-m) (Fig. 4D). Consistent with results from the E-cadherin dsRNA mutation analysis, mutations to the 5′ end of the dsP21–322 antisense strand (dsP21–322-G5′) abolished its ability to activate p21 expression in both PC-3 and HeLa cells (Fig. 4E). Mutations to either the 3′ end or middle region retained, to varying extents, the ability to activate p21 (Fig. 4E). These results indicate that the 5′ portion of the antisense strand in dsEcad-215 and dsP21–322 is critical for initiating transcriptional activation, whereas mismatches to the 3′ end or middle region are tolerated.
Fig. 4.

Sequence specificity for RNAa. (A) Sequence for E-cadherin dsRNA dsEcad-215. The antisense (guide) strand is shown in blue. Mutation of the first and last 5 bp of the dsEcad-215 duplex resulted in dsEcad-215-G3′ and dsEcad-215-G5′, respectively. The mutated bases are in red. (B) PC-3 cells were transfected with 50 nM of the indicated dsRNA molecules for 72 h. Expression of E-cadherin and β-actin was assessed by Western blot analysis. (C) E-cadherin levels from corresponding Western blots were normalized to β-actin and are presented as the mean ± SEM of two independent experiments. (D) The sequence of dsP21–322 and its corresponding mutated dsRNAs. (E) PC-3 and HeLa cells were transfected with 50 nM of the indicated dsRNA molecules for 72 h. Expression levels of p21 and GAPDH were assessed by Western blot analysis.
RNAa Requires the Argonaute (Ago) 2 Protein.
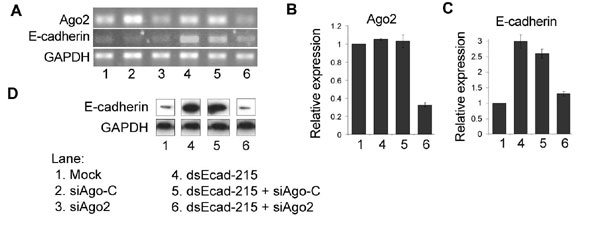
Because our findings indicate that RNAa and RNAi have similar requirements for trigger dsRNAs, such as the importance of the “seed” sequence and a preferred size of 21 nt, we decided to determine whether components of the RNAi pathway are also required for RNAa. We synthesized specific siRNAs that targeted Ago1–4 (siAgo1, siAgo2, siAgo3, and siAgo4) (17), as well as a control siRNA (siAgo-C). We carried out transfections in PC-3 cells using each of the Ago siRNAs alone or in combination with the p21-specific dsRNA dsP21–322. As shown in Fig. 5A and B, the expression of each of the Ago proteins was successfully knocked down by their corresponding siRNA. When each Ago siRNA was cotransfected with dsP21–322, only siAgo2 siRNA completely prevented p21 induction (Fig. 5 C–E). Knockdown of Ago1, 3, and 4 caused an ≈0.3-, ≈0.4-, and ≈0.3-fold reduction in dsRNA-induced p21 expression, respectively (Fig. 5 C–E). We also transfected PC-3 cells with siAgo2 alone or in combination with dsEcad-215. Similarly, E-cadherin induction was almost completely abolished in cells cotransfected with siAgo2 (Fig. 14, which is published as supporting information on the PNAS web site). Although other Ago family members may play supportive roles in RNAa, Ago2 is indispensable for RNAa.
Fig. 5.

RNAa requires the Ago2 protein. (A) PC-3 cells were transfected with 50 nM of the indicated dsRNA molecules for 72 h. mRNA expression of Ago1–4 was assessed by RT-PCR. Each Ago family member was knocked down by its corresponding Ago siRNA (siAgo1, 2, 3, or 4). (B) RT-PCR results were normalized to GAPDH expression levels and are presented as the mean ± SEM from two independent experiments. (C and D) PC-3 cells were transfected with 50 nM of each indicated dsRNA for 72 h. Both p21 and GAPDH mRNA (C) and protein (D) levels were determined by RT-PCR and Western blot analysis, respectively. (E) p21 mRNA levels were normalized to GAPDH and are presented as the mean ± SEM from at least two independent experiments (containing two sample repeats per experiment).
Loss of Histone 3 Methylation at Lysine-9 Is Associated with Gene Activation.
To address how promoter-targeting dsRNA molecules can increase gene expression, we conducted ChIP assays to determine the status of histone methylation at the E-cadherin promoter by using antibodies specific to dimethylated histone 3 at lysine-4 (H3m2K4) and lysine-9 (H3m2K9). Two promoter regions were mapped corresponding to the target of dsEcad-302 (region 1) and dsEcad-215 (region 2) (Fig. 6A). In mock and dsCon-1-transfected PC-3 cells, the E-cadherin promoter was associated with both H3m2K4 and H3m2K9 (Fig. 6 B and C). We observed significant decreases in H3m2K9 at dsRNA target sites after dsEcad-302 (74% decline at region 2) or dsEcad-215 (80% decline at region 2) transfections (Fig. 6 B and C). Although the decline was more pronounced in region 2, H3m2K9 also decreased in region 1 to a lesser extent (Fig. 6 B and C). We did not observe significant changes in H3m2K4 in any of the dsRNA treated cells (Fig. 6 B and C). Because dsEcad-215 caused the largest decline in H3m2K9, we also performed ChIP assays to determine the status of trimethylated histone 3 at lysine-9 (H3m3K9). As shown in Fig. 6 D and E, a loss of H3m3K9 occurred at the target site after dsEcad-215 transfection. Although it currently remains unclear how RNAa is linked to histone demethylation, these findings indicate that a decrease in H3-K9 methylation, a histone modification known to repress E-cadherin transcription (18), is associated with dsRNA-induced E-cadherin expression.
Fig. 6.

dsRNA-induced transcriptional activation is associated with loss of histone 3 methylation at lysine-9. PC-3 cells were transfected with 50 nM of the indicated dsRNA molecules for 72 h. ChIP assays were performed by using antibodies against H3m2K9, H3m2K4, and H3m3K9 to pull down associated DNA. The precipitated DNA was amplified by PCR using two primer sets specific for regions 1 and 2. Input DNA was amplified as a control. (A) A schematic representation of the E-cadherin promoter and the location of ChIP PCR regions 1 and 2 (double-arrowed lines). (B and C) PCR amplification of DNA precipitated with H3m2K9 and H3m2K4 antibodies. (D and E) PCR amplification of DNA precipitated with H3m3K9 antibody.
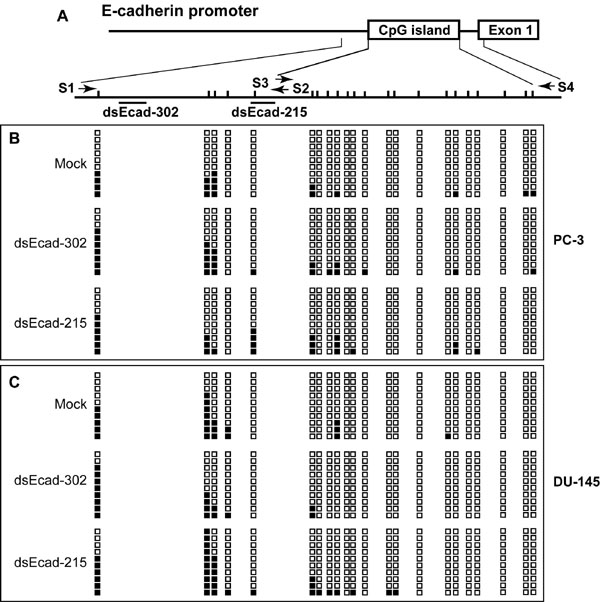
Changes in DNA Methylation Do Not Occur at dsRNA Target Sites.
We also analyzed the state of DNA methylation on the E-cadherin promoter using the bisulfite genomic sequencing technique. The E-cadherin CpG island and the dsRNA target regions are only sporadically methylated in PC-3 and DU-145 cells (Fig. 15, which is published as supporting information on the PNAS web site). dsEcad-302 and dsEcad-215 caused a slight increase in CpG methylation, which was confined to the dsRNA target regions (Fig. 15). However, the increase was not statistically significant suggesting that DNA methylation does not play a role in RNAa. This result is similar to previous reports investigating transcriptional silencing in which dsRNAs targeting promoter regions did not induce DNA methylation in mammalian cells (8, 19, 20).
Discussion
RNAi triggered by dsRNAs, such as siRNA and microRNA, is associated with the silencing of target mRNA sequences. In similar regards, several groups have also reported that targeting promoter regions with dsRNAs induces transcriptional silencing by triggering histone modification and/or DNA methylation (7, 8, 21). In these reports dsRNAs were intentionally designed to target CpG-rich regions or CpG islands within gene promoters and thus had unusually high GC content. In the present study we took a different approach to dsRNA design by closely following rational siRNA design rules (22). We scanned 1 kb of promoter sequence from each target gene and picked those candidates that did not reside in CpG islands and best adhered to the rules of functional siRNA design (i.e., low GC content, lack of repeated or inverted sequences, bias toward low internal stability at the sense strand 3′ terminus, etc.). As a result, our dsRNAs avoided CpG-rich regions. Transfection of dsRNA candidates led us to identify four dsRNAs that activated the expression of targeted genes E-cadherin, p21, or VEGF. The induction in gene expression after dsRNA treatments was generally quite robust; 2- to 10-fold increases in mRNA and protein levels were routinely detected. Transfected cells also acquired changes in phenotype associated with target gene overexpression, such as the observed growth arrest in cells transfected with dsRNAs targeting the E-cadherin and p21 promoter.
In association with these observations, several other lines of evidence suggest that gene induction by dsRNA was sequence-specific for targeted promoter sites. First, BLAST searches against the University of California (Santa Cruz) genome database were performed for each activator dsRNA to ensure it did not share homology with any sequence in the human genome other than its intended target site. Second, two control dsRNA molecules (dsCon-1 and dsCon-2), intentionally designed to lack significant homology to all known human sequences, did not impact the expression of any of the evaluated genes. Third, dsRNA mutation experiments revealed that the 5′ end of the antisense strand, or seed sequence, in the dsRNA duplex was critical for target gene induction. And fourth, dsRNA treatment did not induce an IFN response nor was E-cadherin or p21 expression susceptible to IFN exposure. Taken together, this evidence suggests that gene induction by promoter targeting dsRNAs is sequence-specific.
Classic RNAi by siRNA transfection is thought to be relatively short-lived lasting only 5–7 days (23). Interestingly, we observed long-lasting gene activation after a single transfection of dsRNA; E-cadherin induction remained detectable 13 days after dsEcad-215 transfection (Fig. 1F). ChIP analysis revealed that E-cadherin dsRNAs were able to induce epigenetic changes associated with active genes. This may provide an explanation for the longer lasting effects, as it is believed the epigenetic markers are inheritable across cell division (24).
The Ago family of proteins are key regulators of RNA silencing. Humans have a total of four closely related Ago proteins (Ago1–4) that interact with trigger dsRNA and function in target recognition; however, Ago2 is the only family member that possesses RNA cleaving activity (17, 25). Our dsRNA mutation analysis revealed that mismatches between the target sequence and dsRNA were still capable of inducing gene expression, thus suggesting that the cleavage of target sequences does not occur for RNAa. Ago2 is needed to process siRNA molecules into active forms by cleaving and discarding the passenger strand (26, 27). Perhaps Ago2 functions to convert our dsRNAs to active forms in a similar manner. Although we do not know whether our dsRNAs are interacting with complementary DNA or targeting cryptic promoter transcripts, Ago2 may also participate in target recognition to form a putative effector complex.
It is important to point out that not all of the dsRNAs complementary to promoter sequences activated gene expression. For instance, dsRNA molecules targeting p27, PTEN, and APC at locations −48, −294, and −45, respectively, failed to increase gene transcription (data not shown). In other cases, some cell lines were resistant to specific dsRNAs, while sensitive to others. Transfection of dsEcad-215 into HeLa cells initially failed to activate E-cadherin expression. Similarly, dsVEGF-706 was unable to induce VEGF expression in HEK293 cells (data not shown). Both cell lines, however, were susceptible to p21 induction by dsP21–322 indicating that it was not a global resistance to RNAa, but rather a specific condition of the target gene promoter that rendered it unresponsive. Aberrant DNA methylation in gene promoters may be one mechanism by which some promoters are resistant to RNAa. In support, we show that the activation of E-cadherin in HeLa cells by dsRNA is only possible in the presence of DNA-demethylating agent Aza-C. Target location may also contribute to the functional success of RNAa. For example, dsRNAs (dsEcad-185, -75, and -60) that targeted the CpG island in the E-cadherin promoter failed to induce gene expression.
In summary, by targeting the promoter region of three genes, we have identified several dsRNAs that induce gene transcription in a sequence-specific manner. This RNA mediated process requires the Ago2 protein and is associated with histone changes linked to gene activation. Although the exact mechanism is unknown at present, the identification of such a phenomenon may still have significant therapeutic potential. The use of RNAi is currently being implemented as a gene-specific approach for molecular medicine. By the same principle, the specific activation of silenced tumor suppressor genes (e.g., p21) or other common dysregulated genes (e.g., E-cadherin) by RNAa may further contribute to the growing therapeutic potential dsRNA-based drugs have in the treatment of cancer and other diseases. This information also reveals a new complication for RNAi in which siRNAs may undesirably induce the expression of off-target genes.
Materials and Methods
dsRNA Design and Synthesis.
For each gene, we retrieved 1 kb of promoter sequence and scanned it for dsRNA targets based on rational siRNA design rules (22). We picked those candidates that were located close to transcription start sites and best adhered to the rules of functional siRNA design. A BLAST search was performed against the University of California at Santa Cruz genome database (http://genome.ucsc.edu) to exclude those targets that shared homology to nonspecific human sequences. Two control dsRNAs (dsCon-1 and dcCon-2) were specifically designed to lack homology to all known human sequences. dsRNAs were chemically synthesized by Invitrogen (Carlsbad, CA) with dTdT 3′ overhangs. The Ago-specific siRNAs were synthesized as previously described (17). A control siRNA (siAgo-C) was also designed to lack homology to Ago genes. All dsRNA sequences are listed in Table 1, which is published as supporting information on the PNAS web site.
Cell Culture and Transfection.
PC-3, DU-145, and LNCaP were maintained in RPMI medium 1640 supplemented with 10% FBS, 2 mM l-glutamine, penicillin (100 units/ml), and streptomycin (100 μg/ml) in a humidified atmosphere of 5% CO2 maintained at 37°C. HeLa, J82, T24, HEK293, and MCF-7 cells were maintained in Eagle's minimum essential medium supplemented with 2 mM l-glutamine, Earle's balanced salt solution, penicillin, streptomycin, and 10% FBS. MCF-7 and HeLa cells were supplemented with an additional 1 mM sodium pyruvate and 0.01 mg/ml bovine insulin. The day before transfection cells were plated in growth medium without antibiotics at a density of 50–60%. Transfections of dsRNA were carried out by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and lasted for 72 h (exceptions occurred during time course experiments).
Nucleic Acid Extraction.
Total cellular RNA was extracted by using TriReagent (Molecular Research Center, Cincinnati, OH) or the RNeasy RNA isolation kit (Qiagen, Valencia, CA). Genomic DNA was isolated by using TriReagent.
Western Blot Analysis.
Cultured cells were washed with PBS and lysed with M-PER protein extraction buffer (Pierce, Rockford, IL). Cell lysates were centrifuged for 10 min and supernatants were collected. Equivalent amounts of protein were resolved on 7.5–15% SDS/PAGE gels and transferred to polyvinylidene difluoride membranes by voltage gradient transfer. The resulting blots were blocked overnight with 5% nonfat dry milk. Specific proteins were detected with appropriate antibodies by using enhance chemiluminescence detection (SuperSignal West Dura Lumino/Enhancer Solution; Pierce). Immunoblotting antibodies used were: anti-β-actin (Sigma, St. Louis, MO), anti-GAPDH (Chemicon, Temecula, CA), anti-E-cadherin (Zymed, South San Francisco, CA), anti-p21 (Upstate Biotechnology, Lake Placid, NY), anti-PKR and anti-phospho-PKR (Cell Signaling Technology, Boston, MA).
Bisulfite Modification of DNA, PCR Amplification, and Cloning.
Primers for bisulfite genomic sequencing PCR (Table 1) were designed by using the online program MethPrimer developed in our laboratory (28). Bisulfite modification of DNA (1 μg) was performed by using the CpGenome DNA modification kit (Chemicon). The bisulfite-modified DNA was amplified as previously described (29). PCR products were cloned into the pCR2.1 plasmid (Invitrogen) and transformed into TOP10 cells (Invitrogen). Ten positive clones from each reaction were picked at random and grown overnight in 2 ml of LB medium. Plasmid DNA was then isolated and sequenced for analysis.
ChIP Assay.
The ChIP assays were performed by using a ChIP assay kit (Upstate Biotechnology) according to the manufacturer's instructions. Antibodies that recognized H3m2K9, H3m3K9, and H3m2K4 (Upstate Biotechnology) were used to detect specific changes in histone methylation patterns. Immunoprecipitated DNA was reverse cross-linked, purified, and analyzed by PCR for 25–32 cycles. PCR primers used for ChIP analysis are described in Table 1.
Semiquantitative RT-PCR.
One microgram of total RNA was reverse transcribed by using SuperScript reverse transcriptase (Invitrogen) and oligo(dT) primers. The resulting cDNA samples were amplified by PCR using primers specific for E-cadherin, p21, VEGF, Ago1–4, GAPDH, OAS1, or OAS3 (Table 1). Amplification of GAPDH served as a loading control.
Supplementary Material
Acknowledgments
We thank G. R. Deng, R. Erickson, and K. Greene for critical reading of the manuscript and Y. Shi for helpful discussion. This work was supported by National Institutes of Health Grants R01 AG21418 and R01 CA1018447 and Veterans Affairs Research Enhancement Award Program and Merit Review grants.
Abbreviations
- Ago
Argonaute
- Aza-C
5-azacytidine
- RNAa
dsRNA-induced gene activation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 3.Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJ. EMBO J. 2000;19:5194–5201. doi: 10.1093/emboj/19.19.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sijen T, Vijn I, Rebocho A, van Blokland R, Roelofs D, Mol JN, Kooter JM. Curr Biol. 2001;11:436–440. doi: 10.1016/s0960-9822(01)00116-6. [DOI] [PubMed] [Google Scholar]
- 5.Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 6.Pal-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SC. Science. 2004;303:669–672. doi: 10.1126/science.1092653. [DOI] [PubMed] [Google Scholar]
- 7.Morris KV, Chan SW, Jacobsen SE, Looney DJ. Science. 2004;305:1289–1292. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 8.Ting AH, Schuebel KE, Herman JG, Baylin SB. Nat Genet. 2005;37:906–910. doi: 10.1038/ng1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. Cell. 2004;116:779–793. doi: 10.1016/s0092-8674(04)00248-x. [DOI] [PubMed] [Google Scholar]
- 10.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 11.Britten RJ, Davidson EH. Science. 1969;165:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 12.Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE, Herlyn M. Am J Pathol. 2000;156:1515–1525. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stockinger A, Eger A, Wolf J, Beug H, Foisner R. J Cell Biol. 2001;154:1185–1196. doi: 10.1083/jcb.200104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Nat Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 15.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Nat Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 16.Kim DH, Longo M, Han Y, Lundberg P, Cantin E, Rossi JJ. Nat Biotechnol. 2004;22:321–325. doi: 10.1038/nbt940. [DOI] [PubMed] [Google Scholar]
- 17.Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Mol Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 19.Park CW, Chen Z, Kren BT, Steer CJ. Biochem Biophys Res Commun. 2004;323:275–280. doi: 10.1016/j.bbrc.2004.08.096. [DOI] [PubMed] [Google Scholar]
- 20.Svoboda P, Stein P, Filipowicz W, Schultz RM. Nucleic Acids Res. 2004;32:3601–3606. doi: 10.1093/nar/gkh697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castanotto D, Tommasi S, Li M, Li H, Yanow S, Pfeifer GP, Rossi JJ. Mol Ther. 2005;12:179–183. doi: 10.1016/j.ymthe.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 23.Dykxhoorn DM, Novina CD, Sharp PA. Nat Rev Mol Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 24.Egger G, Liang G, Aparicio A, Jones PA. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 26.Rand TA, Petersen S, Du F, Wang X. Cell. 2005;123:621–629. doi: 10.1016/j.cell.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 27.Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 28.Li LC, Dahiya R. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 29.Li LC, Chui R, Nakajima K, Oh BR, Au HC, Dahiya R. Cancer Res. 2000;60:702–706. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}