

Abstract
Structural alterations of the cellular prion protein (PrPC) seem to be the core of the pathogenesis of prion diseases. However, the physiological function of PrPC remains an enigma. Cell culture experiments have indicated that PrPC and in particular its N‐terminal octarepeat region together with the phosphatidylinositol 3‐kinase (PI3K)/Akt signaling pathways have a fundamental involvement in neuroprotection and oxidative stress reactions. We used wild‐type mice, PrP knockout (Prnp −/−) animals and transgenic mice that lack the octarepeat region (C4/−) and subjected them to controlled ischemia. We identified an increased cleavage and synthesis of PrPC in ischemic brain areas of wild‐type mice compared with sham controls. The infarct size in Prnp −/− animals was increased threefold when compared with wild‐type mice. The infarct size in C4/− animals was identical to Prnp −/− mice, that is, around three times larger than in wild‐type mice. We showed that the PrP in C4/− mice does not functionally rescue the Prnp −/− phenotype; furthermore it is unable to undergo β cleavage, although an increased amount of C1 fragments was found in ischemic brain areas compared with sham controls. We demonstrated that the N‐terminal octarepeat region has a lead function in PrPC physiology and neuroprotection against oxidative stress in vivo.
INTRODUCTION
The cellular prion protein (PrPC) is a copper‐binding protein 3, 17 located at the synapse (13) and has a primary role in the pathogenesis of prion diseases. A templated conformational transition of PrPC seems to be the cause of transmissibility and pathogenesis (25). PrPC has been shown to play a role in cellular antioxidative defense mechanisms 15, 27 and to protect human neurons in primary culture against Bax‐mediated cell death (2). Experiments in cerebellar granule cells (5) have shown a loss of neuroprotective activity of PrPC by a deletion of all five N‐terminal octarepeats (PrPΔ51–90). We have recently identified a functional link between PrPC expression and phosphatidylinositol 3‐kinase (PI3K) activation, a regulator of Akt phosphorylation and a protein kinase that plays a pivotal role in cell survival (33). Our findings are in agreement with published results that showed reduced Phospho‐Akt expression levels in Prnp −/− mouse brains following ischemia (38). We could also demonstrate that hippocampal cells that lack the octarepeat region showed reduced PI3K levels and decreased survival under stress conditions similar to PrPC knockout cells.
In neuroblastoma cells it could be demonstrated that the octapeptide repeats are required for the β cleavage and their absence (PrPΔoct) correlates with increased sensitivity of cells to oxidative stress (36). The cleavage of the molecule seems to play a major role in the biological properties of PrPC but succession of these proteolysis procedures as well as relations between the two cleavages is controversial and still remains to be elucidated and in particular, the relevance of the octapeptide region in this functional process has not been established in vivo.
Recently, brain injury models in mice have been used to investigate a potential neuroprotective role of PrPC in vivo 14, 21, 31, 37. There were significant differences between the volume of the lesion in PrP knockout (Prnp −/−) mice when compared with the wild‐type counterpart with larger area of injury in the former. Moreover, using in situ hybridization an increase of PrP mRNA levels and also PrP immunoreactivity were detected in penumbral neurons of the ischemic lesion (21). These results further support the hypothesis of the antioxidant function of PrPC in vivo.
With this work we specify the neuroprotective property of PrPC and the role of the octarepeat region in this function and contribute to understanding the succession and significance of cleavage processes in an in vivo ischemic model. We used transgenic C4/− mice that lack the octarepeat region of PrPC (6). We challenged C4/−, Prnp −/− and Prnp +/+ mice in a focal cerebral ischemia model. Prnp −/− and C4/− mice showed largely increased infarct volumes when compared with wild‐type littermates. The C4 protein (PrPΔ32–93) did not functionally rescue the Prnp −/− phenotype, the octapeptide region of PrPC therefore seems to play a key role in neuroprotection.
MATERIALS AND METHODS
Transgenic mice. We used homologous Prnp +/− recombination to produce Prnp +/+ and Prnp −/− littermates with a 129/Sv‐C57/Bl6 background. The role of the octarepeat region was investigated in C4/− mice lacking amino acids 32 to 93 of PrPC (30). In consideration of the Prnp −/− background of C4/− mice and the PrPC expression, we crossed hemizygous C4/− and Prnp −/− mice to obtain male (21–28 g) hemizygous C4/− and Prnp −/− littermates. Published polymerase chain reaction (PCR) protocols to identify the transgenes on the Prnp −/− background or wild‐type and Prnp −/− mice were used (30). In agreement with previous findings published by Shmerling and coworkers (30), we detected higher PrP expression in C4/− animals in comparison with wild‐type control mice, that is, approximately three times higher in homozygous and 1.5 times higher in hemizygous C4/− mice (data not shown). All C4 mice used in this study were hemizygous C4/−. The mice were assigned to four groups: G1 (Prnp −/−; n = 9), G2 (Prnp +/+; n = 7), G3 (C4/−; n = 8), G4 (Prnp −/−; n = 8). They were kept under standard diurnal conditions and allowed access to food and water ad libitum. The animal experiments were in accordance with animal protection standards and were approved by the government of Upper Bavaria (protocol number 209.1‐2531‐81/01). Every effort was made to reduce the number of animals used and to ensure they were free of pain and discomfort.
Surgical procedure. The intraluminal filament model was used for occlusion of the middle cerebral artery (MCAO) (35). After 1 h of ischemia reperfusion was induced by withdrawing the thread, the animals were then sacrificed after 24 h. Briefly, mice were anesthetized with 3% isoflurane (Forene, Abbott, Germany) for induction and maintained on 1.5% in a 70%/30% mixture of N2O/O2 administered via a face mask. The rectal temperature was maintained at 37 ± 0.5°C with a homeothermic blanket system. The regional cerebral blood flow (rCBF) was recorded in all animals using laser Doppler flowmetry (Perimed, Järfäla, Sweden) in which a fiberoptic probe placed over the MCA territory 2 mm posterior and 6 mm lateral of the bregma (12). After ventral midline neck incision, the left common and external carotid arteries were ligated. The internal carotid artery (ICA) was temporarily clipped with a microvascular clip (Aesculap, Tuttlingen, Germany), and a silicon‐coated 8‐0 nylon monofilament (Ethicon, Johnson&Johnson, Norderstedt, Belgium) was gently advanced into the ICA. The baseline rCBF values were reduced by >70%. The tip diameter of the thread was selected to match the body weight of the animals. After surgery, the laser Doppler probes were removed, and mice were allowed to wake up and return to their cages. Reperfusion was initiated by withdrawing the thread under short inhalation anesthesia. At the end of the reperfusion period, the mice were sacrificed in deep anesthesia by transcardial perfusion with cold isotonic saline solution containing bovine serum albumin (5 g/L), heparin (10 IU/L) and 2 mL nitroprusside sodium (100 µL, 18 mg in 10 mL saline) solution. Brains were removed, and the skull base was inspected for hemorrhage. In addition, four sham‐operated mice underwent the same surgical procedures, but in these mice the filament was only introduced into the common carotid artery; this does not lead to brain infarction. The neurological outcome was defined in all animals according to Garcia et al (7). It is defined by a sequence of six physiological tasks: (i) spontaneous activity, (ii) symmetry in the movement, (iii) forepaw outstretching, (iv) climbing, (v) body proprioception, and (vi) response to vibrissae touch. The overall score is 18 points. In all surgical procedures and following experiments the investigators were blinded as to the experimental groups individual mice belonged to.
Preparation of frozen sections. Frozen sections of 10‐µm thickness were collected on pre‐labeled glass slides at −20°C. Every 50th section was used for volumetric analysis; the remaining sections were stored at −80°C. For Western blotting and reverse transcriptase polymerase chain reaction (RT‐PCR), frozen coronal sections were prepared, Nissl‐stained and divided using a scalpel and a dissecting microscope into three regions, that is, necrotic infarct area (N), ipsilateral non‐necrotic hemisphere (I), and contralateral hemisphere (C) to detect different progressions of the damage in dependence on the injury distance (Figure 1A).
Figure 1.
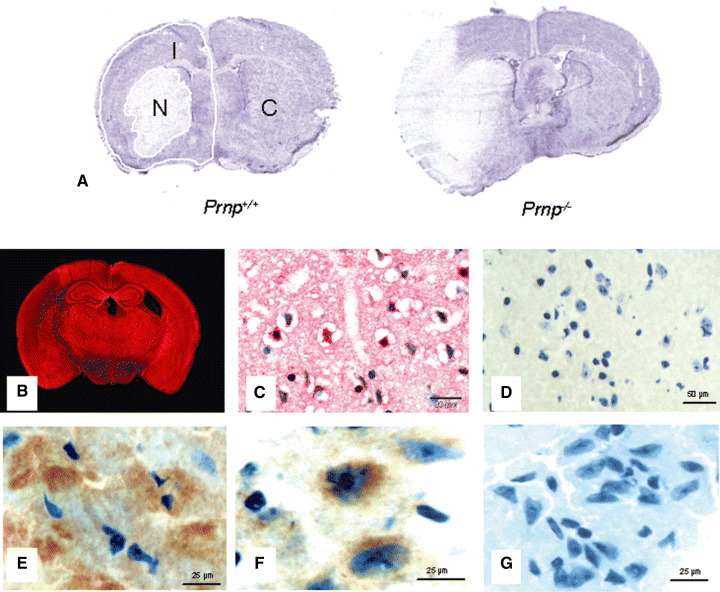
Histology after middle cerebral artery occlusion for 1‐h ischemia and 24‐h reperfusion time. A. Nissl‐stained sections at the level of the bregma, the maximal extent of the infarct in Prnp −/− and Prnp +/+ mice. Necrotic areas show a very faint staining. Frozen coronal sections were divided into three regions, that is, necrotic infarct area (N), ipsilateral non‐necrotic hemisphere (I) and contralateral hemisphere (C). B. Immunofluorescence for cellular prion protein (PrPC) in a Prnp +/+ mouse after cerebral ischemia using polyclonal antibody CDC1 and Fast Red as chromogen. There is strong staining for PrPC in all gray matter areas except the infarct zone on the left. C. Immunohistochemistry with polyclonal antibody CDC1. This formalin‐fixed and paraffin‐embedded section shows a number of strongly stained neurons in the area adjacent to the infarct, while other neurons and glial cells in this section appear negative. D. No reaction with the antibody directed against PrP was detected in frozen sections of the Prnp −/− littermates. E. Immunofluorescence for PrPC in Prnp +/+ mouse brains after cerebral ischemia using polyclonal antibody CDC1 and DAB as the chromogen. This frozen section shows again a number of strongly stained neurons in the area adjacent to the infarct. F. Immunofluorescence for PrPC in C4/− mouse brains after cerebral ischemia using polyclonal antibody 1A8 and DAB as the chromogen. Punctate PrP staining of intensely immunoreactive neuronal cells is seen especially in the ipsilateral area surrounding the infarct. G. No reaction with the antibody 1A8 directed against PrP was detected in the frozen sections of the Prnp −/− littermates.
Infarct definition and volumetry. Nissl staining was used to delineate infarct lesions. To calculate the infarct and hemispheric volumes, the method described by Kloss et al (16) was used. Briefly, Nissl‐stained consecutive sections with 500‐µm clearance were digitized using a flatbed scanner (Epson, Meerbusch, Germany). The files were imported into Optimas imaging software, the lesions and hemispheres delineated, and the size converted into the metric system. Next, partial volumes between two adjacent sections were computed using the formula for a conic section:
 |
with area 1,2,3, . . . n−1 being the partial area of the lesion of the serial sections. The total hemisphere volumes were calculated equally and finally the ratio of infarct volume vs. the total hemispheric volume was defined.
Quantitative RT‐PCR (QRT‐PCR). Immediately prior to RNA isolation, slides containing frozen sections were fixed in 75% ethanol for 45 s and Nissl‐stained. Subsequently, the slides were dehydrated in graded ethanol solutions for 5 s each and air‐dried. RNA isolation from Nissl‐stained sections was performed by using RNeasy Micro Kit (Qiagen, Hilden, Germany). Briefly, materials in particular brain regions (N, I and C) as described above were scraped and lysed with buffer RLT. Materials in a particular region from eight sections each animal were pooled and RNAs isolated according to the protocol recommended by Qiagen. The quantity of RNA was assessed using the NanoDrop® ND‐1000 Spectrophotometer (NanoDrop, Wilmington, DE, USA).
Four hundred nanograms of RNA was used to make single‐stranded cDNAs (Superscript II; Invitrogen, Karlsruhe, Germany) according to the manufacturer’s instructions. Using 2 µL diluted cDNA (1:10), real‐time quantitative PCR was carried out with Faststart Plus DNA Master SYBR Green I on LightCycler (Roche, Mannheim, Germany) under the following conditions: 95°C for 10 minutes for a hot start, followed by denaturing at 95°C for 10 s, annealing at 57°C for 5 s, and extension at 72°C for 10 s for 45 cycles. Primer sets used for PCRs were as follows: for β‐actin (9) (with modification), 5′ AAC CCT AAG GCC AAC CGT GAA AAG 3′ and 5′ CTA GGA GCC AGA GCA GTA ATC T 3′; for prion protein gene (Prnp), 5′ CCA ATT TAG GAG AGC C 3′ and 5′ GCT TCC CTG CCC GGG ATA CC 3′. Calibrator‐normalized relative quantification was performed by LightCycler Software version 4.0 (Roche).
Immunohistochemical staining procedures. Coronal frozen sections were pretreated including dehydration, short fixation in 0.25% glutaraldehyde and heat‐reliant epitope retrieval. Afterwards, nonspecific protein binding sites were blocked with 10% normal goat serum followed by incubation of the respective primary antibody. For PrP staining in Prnp +/+mice, an antiserum purified from a rabbit immunized against recombinant full‐length mouse PrP was used (CDC1), whereas 1A8, an antiserum purified from a rabbit immunized against the C‐terminal domain (amino acids 89–234) of mouse PrP was used in C4/− mice. The polyclonal antibody Actin I‐19 against β‐actin was obtained from Santa Cruz (Biotechnology, Heidelberg, Germany). The primary antibody was detected by a biotinylated secondary antibody and enhanced by streptavidin alkaline phosphatase complexes. Visualization of immunoreactivity followed by using Fast Red or DAB as the chromogen. Counterstaining followed with hemalaun. For immunofluorescence staining, a streptavidin CY3 conjugate was used instead of streptavidin alkaline phosphatase complexes. The nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride and slides were covered in aqueous solution. Staining of paraffin sections was performed after deparaffinizing the sections in xylene.
Immunoblotting. The process was performed as described in our previous data (18). According to the size, different regions (N, I and C) on the Nissl‐stained coronal frozen sections were solubilized by adding 5 µL of lysis buffer (with 2% SDS and 0.5 M DTT) on N, 10 µL on I and 15 µL on C. Identical regions were solubilized in the same volumes of buffer in sham‐operated animals. In five animals without surgery the brains were perfused to obtain calibration measurements. The solubilized tissue was transferred to reaction tubes and digested with PNGase according to the standard protocol of the manufacturer (Roche Diagnostics, Penzberg, Germany). Digestion was stopped by the addition of modified Laemmli sample buffer and boiling for 10 minutes. Five microliters of each sample was subjected to Western blotting. Proteins were separated on 12% SDS‐PAGE (NuPAGE, Invitrogen, Karlsruhe, Germany) and transferred to PVDF membranes (Millipore, Eschborn, Germany). After short blocking, the membranes were incubated with the primary antibodies, 6H4 (Prionics, Schlieren, Switzerland) and Actin I‐19 (Biotechnology, Heidelberg, Germany), separately, over night. After incubation with primary antibody, the membranes were incubated with secondary antibodies, coupled to alkaline phosphatase (DakoCytomation, Hamburg, Germany).
The complete process was performed with exact timing of the individual steps and at a constant temperature to reach maximum accuracy. For quantification of relative signal intensities, each Western blot contained a dilution series of a standard protein with known concentration. Intensity measurements were done with Total Lab V 2.01 software (Nonlinear Dynamics, Newcastle‐upon‐Tyne, UK). The Western blots of actin were used as a reference.
Statistical analysis. Statistical analysis was performed using the SPSS software system (SPSS for Windows, Version 9.0, SPSS, Inc., Chicago, IL, USA). The parametric T‐test for unpaired samples was used to determine significant differences between two groups. All P‐values were two sided and P < 0.05 was considered significant.
RESULTS
Characteristics of the ischemic brain injury in Prnp+/+ and Prnp−/− mice. Nissl‐stained sections from the brains subjected to 1‐h MCAO and 24‐h reperfusion were examined. Infarcts were clearly demarcated in all experimental groups. The damaged areas appeared as pale areas on gross inspection (Figure 1A). Severe swelling of the affected hemisphere with a corresponding midline shift was noticed. Clearly delineated lesions in the ischemic hemisphere were found in all animals after 24 h of reperfusion. Ischemic lesions extended from the forebrain and few millimeters past the bregma with the maximum extent of the lesion at the level of the bregma.
Lesion volumes, measured by summarizing delineated infarct areas in 500 µm steps, were 10.06 mm3 ± 4.8 mm3 in Prnp +/+ mice and 34.78 mm3 ± 12.99 mm3 in Prnp −/− mice (Figure 2A). The partial volumes of the hemispheres were measured from −3.0 mm to +7.0 mm behind the bregma, and were 50.19 mm3 ± 19.5 mm3 in the ipsilateral hemisphere and 50.89 mm3 ± 19.88 mm3 in the contralateral hemisphere in Prnp +/+ mice. In Prnp −/− mice they were 86.45 mm3 ± 18.4 mm3 in the ipsilateral and 79.20 mm3± 15.03 mm3 in the contralateral hemisphere. Thus there was a much more pronounced increase in volume in both hemispheres in Prnp −/− than in Prnp +/+ mice. The ratio of the infarct volume to the ipsilateral hemisphere volume showed an infarction of 39% ± 10% in Prnp −/− mice and 20% ± 3% in Prnp +/+ mice. These differences were significant between Prnp +/+ and Prnp −/− mice (P < 0.05).
Figure 2.
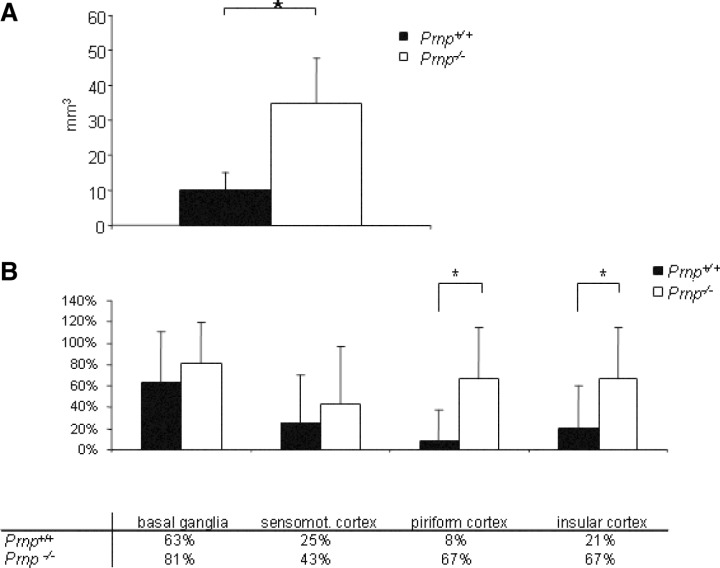
A. Lesion volumes of Prnp +/+ and Prnp −/− mice after 1 h of ischemia and 24‐h reperfusion. Prnp −/− mice (n = 9) show a more than three times larger lesion volume than Prnp +/+ mice (n = 7). Data are means ± standard deviation. *P < 0.05 (Student’s t‐test). B. Neuroanatomical pattern of necrosis in Prnp +/+ and Prnp −/− mice. The graph demonstrates the extent of the infarct over various anatomical regions in the ischemic hemisphere at the level of the bregma. The affected areas in the piriform and insular cortex were significantly larger in Prnp −/− than in Prnp +/+ mice (*P < 0.05). Error bars indicate standard errors of the mean; mean values are given in the table below.
Anatomical distribution, appearance, size and pattern of the infarct. During evaluation of the infarct size we noticed that various anatomical regions were affected in different degrees in Prnp +/+ and Prnp −/− mice. A more detailed comparison of sections at the level of the bregma (Figure 2B) showed that the basal ganglia were most consistently and the most severely affected; the area affected was 63% in Prnp +/+ mice while it was 81% in Prnp −/− mice. Twenty‐five percent of the motor cortex area was affected in Prnp +/+ and 43% in Prnp −/− mice. The highest diversity was found in the piriform cortex with 8.3% in Prnp +/+ and 67% in Prnp −/− mice followed by the insular cortex with 21% in Prnp +/+ and 67% in Prnp −/− mice. The differences in the piriform cortex and the insular cortex of Prnp +/+ and Prnp −/− mice were statistically significant (P < 0.05). Only 7.1% of the motor cortex was affected in Prnp −/− mice, whereas the cingulate cortex was unaffected in Prnp +/+ as well as in Prnp −/− mice. There were no pathologic findings in paraventricular nuclei. In conclusion, the greatest variability was seen in the piriform and the insular cortex.
Neurological evaluation. The neurological examination was carried at two different times, after 1 h of ischemia and at the end of the 24‐h reperfusion period. The correlation between the mean neurological score and mean number of necrotic neurons after focal cerebral ischemia has been described as highly significant (7); a decrease of the score stands for an increase of clinical symptoms. A mean neurological score of 18 points represents a healthy mouse without clinical symptoms. The mean neurological scores after 1 h of ischemia were significantly lower than those after 24 h of reperfusion in Prnp +/+ and Prnp −/− mice (Figure 3A). While after 1 h of ischemia no differences were noted between Prnp −/− and Prnp +/+ mice, after 24‐h reperfusion the scores of Prnp −/− mice were significantly lower compared with Prnp +/+ mice (P < 0.002).
Figure 3.
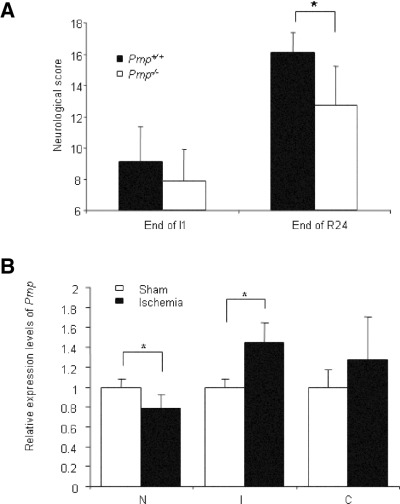
A. Neurological scores (±standard deviation) in Prnp +/+ and Prnp −/− mice subjected to transient middle cerebral artery occlusion after 1 h of ischemia (End of I1) and at the end of the 24‐h lasting reperfusion period (End of R24). No significant differences were seen after 1 h of ischemia, although there was a tendency for Prnp −/− mice to perform less well. After 24‐h reperfusion the scores of Prnp −/− mice were significantly lower than in Prnp +/+mice. The asterisk indicates significant difference (P < 0.002) as determined by t‐test. B. Quantitative reverse transcriptase polymerase chain reaction analysis of Prnp gene transcription after 1 h of transient focal cerebral ischemia and 24‐h reperfusion. The relative transcription levels of Prnp and β‐actin genes in the regions containing the necrotic infarct area (N) and the region free of infarct of the ipsilateral hemispheres (I) as well as the expression levels in the contralateral hemispheres (C) in ischemic animals are indicated by fold‐changes as compared with the corresponding regions of sham‐operated animals (n = 4 for all groups). The asterisk indicates a significant increase in Prnp transcription in the ipsilateral hemisphere I and a significant decrease in the necrotic area N compared with sham‐operated animals as determined by Student’s t‐test (P < 0.05). There is a tendency for increased Prnp transcription in the contralateral hemisphere C, which is statistically not significant.
RT‐PCR analysis of Prnp transcription after transient focal cerebral ischemia. Following 1‐h transient focal cerebral ischemia and 24‐h reperfusion in Prnp +/+ animals three distinct areas were subjected to investigation: (N) the region containing the necrotic infarct area, (I) the region free of infarct in the ipsilateral hemisphere and (C) the region free of infarct in the contralateral hemisphere. Transcription levels of Prnp from these areas were analyzed by QRT‐PCR and were compared with the corresponding ipsilateral (I) and the contralateral hemispheres (C) of sham‐operated animals (Figure 3B). RT‐PCR analysis revealed a significantly increased expression of Prnp in the infarct‐free region of the ipsilateral hemisphere after ischemia (P < 0.05). Prnp expression showed a slight but nonsignificant increase in the contralateral hemisphere. In contrast, PrPC mRNA levels were significantly decreased in the necrotic area (N) (P < 0.05), which may be associated with the massive cell death in this area.
Histology and immunohistochemistry. In the Nissl‐stained sections the nerve cells in the infarct region showed pale staining (Figure 1A). They appeared shrunken with small pyknotic nuclei. Vacuolar changes were also frequently observed. The polyclonal rabbit antibody directed against recombinant full‐length mouse PrP (CDC1) reacted strongly in neurons and sometimes glial cells in both gray and white matter (Figure 1B). In gray matter areas single strongly immunoreactive neurons were observed predominantly in the hippocampus and neocortex in MCAO and control animals. In the necrotic infarct area there was no PrPC immunostaining. In contrast, numerous neuronal cells in the area surrounding the infarct region in the ipsilateral hemisphere (I) showed a strong PrPC immunoreactivity, while other neurons in the immediate vicinity did not react (Figure 1C). Sections of Prnp −/− mice showed no reaction with the antibody directed against PrPC (Figure 1D).
Western blotting. Western blots were performed with tissue homogenates from brain sections at the level of the bregma. By using the primary antibody 6H4, which is directed against an epitope within the first alpha helix, full‐length unglycosylated PrPC (PrPC FL) was detected at an apparent molecular weight of 28.5 kDa. Two additional fragments were observed at about 21.1 and 16.4 kDa corresponding to the C2 and C1 fragments described by Mangé(20). Reprobing of Western blots with a primary antibody against β‐actin showed a band at 42.8 kDa.
In the samples extracted from the right and left hemispheres of sham‐operated mice (Figure 4A; lanes Sham C and Sham I) PrPC FL represented the strongest signal, whereas bands corresponding to the C1 fragment showed weaker staining. The C2 fragments were weaker but clearly discernible. The expression level of β‐actin was similar in both hemispheres. In contrast to this pattern, the necrotic infarct area (N) of an ischemic mouse brain showed weak staining for β‐actin. PrPC FL, C1 and the C2 fragments were barely visible. In comparison with PrPC FL the C1 appeared slightly stronger. Strong C1 bands were also seen in the ipsilateral hemisphere not affected by the infarct (I) and the contralateral hemisphere (C) of ischemic mice. These bands were not as intense as the bands corresponding to PrPC FL, but remarkably stronger than those representing the C1 fragment in sham‐operated mice.
Figure 4.
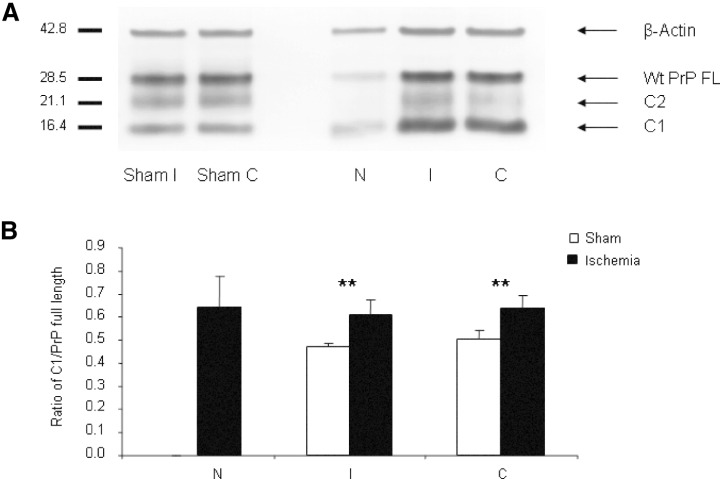
Expression of cellular prion protein (PrPC) and generation of cleavage products in Prnp+/+ mice. A. Western blots were performed with PNGase‐digested tissue homogenates from the necrotic infarct area (N), infarct‐free brain tissue of the ipsilateral hemisphere (I) and contralateral hemisphere (C). A strong band representing full‐length PrPC (PrPC FL) was detected at about 28.5 kDa. Additional bands of lower molecular weight can be observed at about 21.1 kDa, corresponding to the C2 fragment, and at 16.4 kDa, corresponding to the C1 fragment of PrPC. As a reference the blots were reprobed with an antibody directed against β‐actin. PrPC was found decreased in necrotic areas of ischemic mouse brains, it was increased in non‐necrotic areas of the ipsilateral and contralateral hemispheres. B. Densitometric analysis of the ratio of C1/PrPC full length reveals a significant relative increase of C1 in both the necrotic infarct area (N) and the infarct‐free area of the ipsilateral hemisphere (I), and of the contralateral hemisphere (C) in ischemic mouse brains (n = 7) in comparison with areas of sham‐operated controls (n = 4). Error bars indicate standard deviations. Significant differences between ischemic and sham controls are marked by an asterisk (**P < 0.01) as determined by Student’s t‐test.
The detected proteins were quantified via comparison with a dilution series (not shown). Figure 4B demonstrates the analysis of the ratio of C1 to PrPC FL in sham‐operated (white columns) and ischemic mice (dark columns). The ratio of the C1 fragment to PrPC FL was significantly increased in ischemic mice compared with sham‐operated animals in the ipsilateral hemisphere I and contralateral hemisphere C (P < 0.01).
C4/− (PrPΔ32–93) compared with Prnp−/− mice
Infarct volume. Lesion volumes measured by summarizing delineated infarct areas in 500 µm steps were 33.54 mm3± 14.93 mm3 in C4/− mice and 33.85 mm3± 16.13 mm3 in Prnp−/− mice after 24 h of reperfusion (Figure 5A). The volume of the hemispheres were measured from −3.0 mm to +7.0 mm behind the bregma, and reached 86.44 mm3 ± 29.28 mm3 ipsilaterally and 76.12 mm3 ± 24.66 mm3 contralaterally in C4/− mice. The brain sections of Prnp −/− mice showed a hemispheric volume of 82.31 mm3 ± 26.26 mm3 in the ipsilateral hemisphere and 71.44 mm3 ± 19.14 mm3 in the contralateral hemisphere. The ratio of ipsilateral to contralateral hemisphere was 113% ± 6% in C4/− mice and 114% ± 10% in Prnp −/− mice. These results signify a shift to the contralateral hemisphere in C4/− and Prnp −/− mice and stand for serious implications of the damage caused by transient MCAO in both strains in a similar manner. The ratio of the infarct area to the ipsilateral hemispheric volume showed infarction with 37% ± 6% in C4/− mice and 39% ± 10% in Prnp −/− mice. In conclusion, there were no differences in infarct volume and ratio of infarct volume to the ipsilateral hemisphere between C4/− and Prnp −/− mice.
Figure 5.
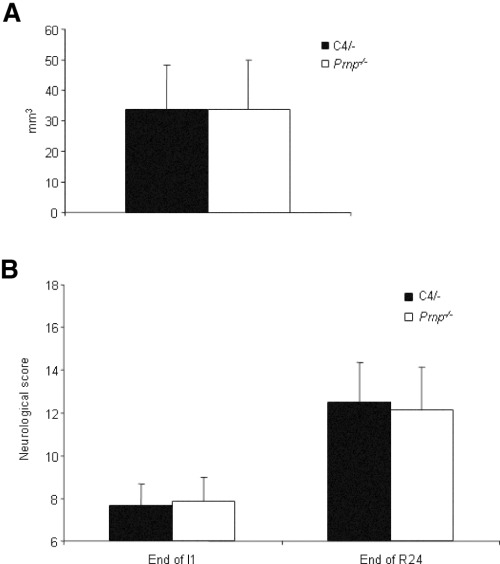
A. Lesion volumes of C4/− and Prnp −/− mice after 1 h of ischemia and 24‐h reperfusion. There are no significant differences between C4/− and Prnp −/− mice. Data are means ± standard deviation; n = 8 animals/group. B. Neurological scores (±standard deviation) in C4/− and Prnp −/− mice subjected to transient middle cerebral artery occlusion after 1 h of ischemia (End of I1) and at the end of the 24‐h lasting reperfusion period (End of R24). No significant differences in the neurological score were found at I1 and R24.
Neurological evaluation. The neurological examination of C4/− vs. Prnp −/− littermates was carried at two different time points, after 1 h of ischemia and at the end of 24‐h reperfusion. The mean neurological scores of C4/− and Prnp −/− mice after 1 h of ischemia were significantly lower than those after 24‐h reperfusion, no significant difference was seen between the C4/− mice and the Prnp −/− mice, neither after 1 h of ischemia nor at the end of 24‐h reperfusion (Figure 5B).
Histology and immunohistochemistry. In both C4/− and Prnp −/− mice, a remarkable swelling of the ischemic hemisphere with midline shift to the contralateral hemisphere was observed in a similar manner. After the 24‐h reperfusion period, clearly delineated lesions in the ischemic hemisphere were found in both groups. In C4/− and Prnp −/− mice the ischemic lesions began in the forebrain and ended a few millimetres behind the bregma. Furthermore, C4/− and Prnp −/− mice showed the maximal extent of the lesion at the level of the bregma. After focal cerebral ischemia the necrotic lesion area in the ipsilateral hemisphere of the C4/− mice showed a remarkably reduced immunoreactivity for PrP and β‐actin. The ipsilateral area surrounding the infarct presented a punctate PrP staining of intensely immunoreactive neuronal cells in a pale environment as seen in Prnp +/+ mice (Figure 1F). Similar to sections of Prnp +/+ mice, C4/− mice had indicated strongly PrP immunoreactive neurons predominantly in the hippocampal formation and cerebral cortex. An overall weak staining in the corpus callosum and basal ganglia was observed, while most immunoreactivity was found again in the hippocampus and piriform gyrus. In comparison, no reaction with the antibody directed against PrP was detected in the frozen sections of the Prnp −/− littermates (Figure 1G). β‐Actin immunoreactivity was observed in C4/− and Prnp −/− mice overall in an analogous manner. Both strains showed strong staining in the basal ganglia and outer layers of the cortex and weaker staining in the corpus callosum.
Western blotting analysis of PrPC after MCAO. Figure 6A shows Western blots of PNGase‐digested identical brain samples of ischemic C4/− mice (N, I and C) and corresponding areas in C4/− sham control animals with homogenates from brain sections at the level of the bregma. Antibody 6H4 was used as primary antibody, which is directed against an epitope within the first alpha helix of PrPC. Only two bands at about 20.1 and 16.4 kDa are seen corresponding to the full‐length C4 protein and the C1 fragment, whereas no isolated C2 fragment is discernible. In comparison with C4 FL the C1 fragments appeared slightly stronger in necrotic and in the ipsilateral hemisphere not affected by the infarct (I) after MCAO damage in C4/− mice but a C2 fragment is not detectable in C4/− mice. In the samples extracted from the ipsi‐ and the contralateral hemispheres of sham‐operated C4/− mice (Figure 6A; lanes Sham I and Sham C) C4 FL and bands corresponding to the C1 fragment do not differ in intensity but they are remarkably weaker compared with the ischemic areas I and C. There are differences of PrP full length and C1 signal intensities between ischemic Prnp +/+ and C4/− mice because of the 1.5 times higher PrP expression in C4/− mice (Figure 6B).
Figure 6.
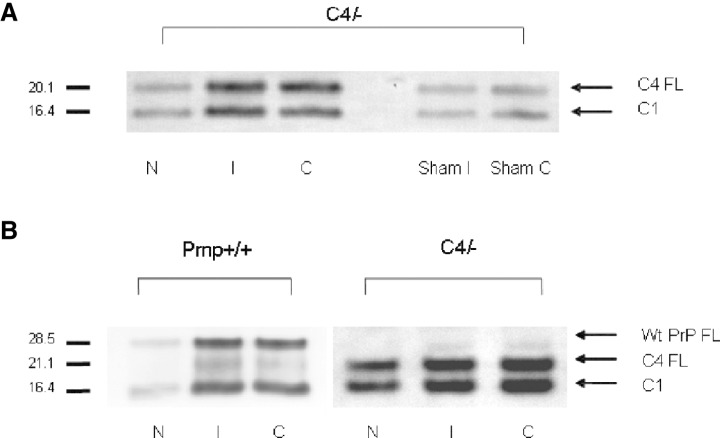
Expression of cellular prion protein (PrPC) and generation of cleavage products in C4/− and Prnp+/+ mice. The Western blots were performed with PNGase‐digested homogenates of sham‐operated C4/− mice and the three different brain regions of C4/− and Prnp +/+ mice after ischemic injury (N, I and C). A. Dominant bands of C4 full‐length (C4 FL) are detected at about 20.1 kDa in C4/− sham control mice. At about 16.4 kDa there are weaker bands that correspond to the C1 fragments in C4/− mouse brain homogenates. After middle cerebral artery occlusion (MCAO) damage in C4/− mice there is an increase in α cleavage in the three regions investigated (N, I, C). Two bands at about 20.1 and 16.4 kDa are seen corresponding to full‐length C4 protein and the C1 fragment. The signal intensities of C1 fragments in ischemic C4/− mice are remarkably stronger than those representing the C1 fragment in sham‐operated control mice. A C2 fragment is not discernible. B. A clear band of full‐length PrPC (PrPC FL) is detected at about 28.5 kDa in Prnp +/+ mice and about 20.1 kDa in C4/− mice. An additional band is seen at 21.1 kDa in the Prnp +/+ lanes corresponding to the C2 fragment. The C2 fragment of PrPC migrates approximately at the same molecular weight as the full‐length C4 protein (PrPΔ32–93). A C2 fragment is not discernible in C4/− mice. At about 16.4 kDa there are dominant bands that correspond to the C1 fragments in Prnp +/+ and C4/− mouse brain homogenates after MCAO damage.
DISCUSSION
In this study we first compared the lesion profile and extent of tissue damage in Prnp +/+ and Prnp −/− mouse brains after transient focal ischemia. As shown before, Prnp −/− mice have a tendency to larger infarct size when compared with Prnp +/+ animals 21, 31, 37. Our QRT‐PCR analysis of Prnp gene expression after 1‐h transient focal cerebral ischemia and 24‐h reperfusion indicated distinct local differences in Prnp +/+ mouse brains. We found a decrease in Prnp expression in the lesion area while the infarct‐free region around the lesion of the ipsilateral hemisphere showed a significant increase. A slight but nonsignificant increase of PrPC mRNA levels was found in the contralateral hemisphere. The decrease of PrPC mRNA levels in the necrotic area is readily explained by the large number of dying cells in this area. The up‐regulation of PrPC mRNA levels in the area adjacent to the infarct may be triggered by local oxidative damage and other metabolic derangements in this area; this would be in keeping with in vitro analyses showing a correlation between PrP expression and oxidative stress (3). The neuronal histology in the region of ischemic injury indicated oxidative damage and cell death. Intense PrPC immunoreactivity was seen in individual cells in the penumbra, while other neurons in this area showed no PrPC immunoreactivity.
The role of PrPC FL in neuroprotection and in particular in reversing the oxidative stress damage is well documented 15, 23, 27, 39. In this study we detected an increased degradation of PrPC FL to its breakdown product C1 in the area surrounding the infarct and in the contralateral hemisphere in Prnp +/+ mouse brains. This finding is in agreement with Mangé’s postulation of increased PrPC degradation under oxidative conditions (20). It is tempting to speculate that PrPC overexpression increases the chances for neuronal survival under the conditions of oxidative stress while lack of PrPC facilitates cell death. However, the cascade may be more complex; volumetric analyses of mouse brains overexpressing PrPC (tga20 line) did not show enhanced neuroprotection compared with wild‐type control animals after ischemia (31). Disproportionate availability of downstream ligands may have an impact on the neuroprotective ability of PrP. A number of proteins, for example, the neuronal cell adhesion molecule (29), the stress‐inducible protein 1 (19) or the laminin receptor and laminin receptor precursor protein 8, 11, have been postulated as potential interaction partners. It is also possible that PrP acts as a regulator of cytosolic signaling pathways. Increased activity of cell signaling factors involved in cell survival and apoptosis such as ERK‐1/‐2, STAT‐1 and caspase‐3 were found in Prnp −/− mice 31, 38. At present it is difficult to decide whether the extent and severity of ischemia are a cause or a consequence of altered signaling pathways.
In previous cell culture studies we demonstrated that oxidative stress together with PrPC elicited a signaling cascade involving PI3K (33). PI3K signaling increased the survival chances of PrPC‐expressing cells. PI3K has been shown to be sensitive to redox signaling by superoxide and hydrogen peroxide and to be activated by copper ions (24). This effect may be mediated or initiated by PrP. In our experiments this interaction could not be initiated by PrP lacking the octapeptide and flanking amino acids (PrPΔ32–93). Other studies (36) have demonstrated that the octapeptide repeat region of PrPC is required for reactive oxygen species‐mediated β cleavage of PrPC and cells lacking the octapeptide region are more susceptible to oxidative stress.
As our previous cell culture experiments pointed towards the neuroprotective effect of the N‐terminal octapeptide repeat region (5) we were interested to investigate this effect in vivo using the established focal ischemia model. Therefore, we subjected transgenic C4/− mice to controlled ischemia by MCAO and reperfusion. C4/− mice lack the N‐terminal octapeptide region (amino acids 51 to 91) and N‐terminally flanking amino acids (PrPΔ32–93) and have been described as phenotypically normal under standard conditions but develop atypical disease when infected with scrapie prions (6). We show that in the focal ischemia model animals lacking the octapeptide of PrP (PrPΔ32–93) behave like Prnp −/− mice. Thus, in our model the C4 protein (PrPΔ32–93) does not functionally rescue the Prnp −/− phenotype. However, the precise mechanism of the PrPC‐dependent neuroprotection remains to be elucidated.
On one hand studies have shown (28) that Prnp −/− neuronal cells expressing PrPC lacking the N‐terminal octapeptide repeat region (amino acids 53–94) show enhanced apoptosis and decreased superoxide dismutase activity. In accordance with these findings Bounhar et al (2) demonstrated that PrPC potently inhibits Bax‐induced cell death in human primary neurons and while the deletion of four octapeptide repeats of PrPC completely eliminates the neuroprotective effect of PrPC, PrP lacking the glycosylphosphatidylinositol anchor signal peptide remains anti‐apoptotic.
On the other hand the N‐terminal octapeptide region of PrPC has been shown to bind copper in a cooperative manner 1, 26, 34. The most obvious train of arguments would invoke the copper‐binding ability of PrPC and its involvement in cell signaling, oxidative stress and the contribution to cell survival under stress conditions (33). PrPC does not seem to be involved in copper transport at the synapse (10). Thus it would appear that PrPC is a sensor for oxidative stress, which may function in conjunction with copper binding to the N‐terminal octarepeat.
Sequence analysis by Chen et al (4) first detected a glycosylated, cell membrane‐anchored major product, designated C1 that is generated by a cleavage at position 111/112 (α cleavage), as well as an additional fragment longer than C1, termed C2 which is created by β cleavage. These cleaving processes seem to be copper dependent (32). Interestingly, our Western blotting analyses showed that in C4/− mice there is an increase of the α cleavage in ischemic brain regions compared with sham‐operated C4/− control animals. This finding indicates that the C1 fragment is not likely to be linked to the neuroprotective function of PrPC. Furthermore, in agreement with Watt and Hooper (36) no fragments at about 21.1 kDa corresponding to the C2 cleavage products were detectable in C4/− mice. As the β site is located in the octapeptide repeat region (22), the absence of a β cleavage in the octapeptide repeat region and its breakdown products in C4/− mice must be expected. In accordance with our data, McMahon et al (22) found a decrease of PrPC FL and an increase of amino‐terminally fragmented PrPC in cells under oxidative stress. Furthermore they reported that the octapeptide repeat region of PrPC undergoes a site‐specific copper‐ and pH‐dependent cleavage, which has vast implications for further proteolysis.
Mangé and coworkers (20) argued that PrPC is first cleaved at the β site in the octapeptide region and near the histidine in position 96, followed by a rapid second cleavage in the α site, which inactivates the biological active form of PrP cleaved at the β site. This is in line with data of our experiments, where bands corresponding to the C2 fragments appeared to decrease in Prnp +/+ ischemic mouse brains, whereas an increase of bands corresponding to the C1 fragments was visible compared with Prnp +/+ sham controls. In mice expressing PrPΔ32–93 remarkably stronger bands representing the C1 fragments were detectable in ischemic brain areas compared with C4/− sham controls as well. Although C4/− mice express 1.5 times more PrP than wild‐type mice and show an increase of C1 fragments under oxidative stress conditions, they fail to rescue the Prnp −/− phenotype. Therefore our findings show that mice expressing PrPΔ32–93 were able to provide C1 terminal fragments independent of previous cleavage at the β site in the octapeptide region but the C1 fragments have no protective role.
Taken together our data support the hypothesis that absence of cleavage at the β site, which is possibly facilitated by copper binding, is associated with loss of PrPC function.
ACKNOWLEDGMENTS
This study was supported by EU NeuroPrion (FOOD‐CT‐2004‐506579). We wish to thank Michael Schmidt, Getrud Kwiatkowski and Ramona Heptner for their excellent technical support. C4/− mice were a generous gift from Charles Weissmann.
Re‐use of this article is permitted in accordance with the Creative Commons Deed, Attribution 2.5, which does not permit commercial exploitation.
REFERENCES
- 1. Aronoff‐Spencer E, Burns CS, Avdievich NI, Gerfen GJ, Peisach J, Antholine WE, Ball HL, Cohen FE, Prusiner SB, Millhauser GL (2000) Identification of the Cu2+ binding sites in the N‐terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry 39:13760–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A (2001) Prion protein protects human neurons against Bax‐mediated apoptosis. J Biol Chem 276:39145–39149. [DOI] [PubMed] [Google Scholar]
- 3. Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, Von Bohlen A, Schulz‐Schaeffer W, Giese A, Westaway D, Kretzschmar H (1997) The cellular prion protein binds copper in vivo. Nature 390:684–687. [DOI] [PubMed] [Google Scholar]
- 4. Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio‐Gambetti L (1995) Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 270:19173–19180. [DOI] [PubMed] [Google Scholar]
- 5. Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, Chishti MA, Marvi M, Windl O, Ahrens R, Major F, Sy MS, Kretzschmar H, Fraser PE, Mount HT, Westaway D (2004) Genetic mapping of activity determinants within cellular prion proteins: N‐terminal modules in PrPC offset pro‐apoptotic activity of the Doppel helix B/B′ region. J Biol Chem 279:55443–55454. [DOI] [PubMed] [Google Scholar]
- 6. Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, Von Mering C, Aguzzi A, Weissmann C (2000) Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 27:399–408. [DOI] [PubMed] [Google Scholar]
- 7. Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26:627–634. [DOI] [PubMed] [Google Scholar]
- 8. Gauczynski S, Peyrin JM, Haik S, Leucht C, Hundt C, Rieger R, Krasemann S, Deslys JP, Dormont D, Lasmezas CI, Weiss S (2001) The 37‐kDa/67‐kDa laminin receptor acts as the cell‐surface receptor for the cellular prion protein. EMBO J 20:5863–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghazanfari FA, Stewart RR (2001) Characteristics of endothelial cells derived from the blood‐brain barrier and of astrocytes in culture. Brain Res 890:49–65. [DOI] [PubMed] [Google Scholar]
- 10. Giese A, Buchholz M, Herms J, Kretzschmar HA (2005) Mouse brain synaptosomes efficiently accumulate copper‐67 by two distinct processes independent of cellular prion protein. J Mol Neurosci 27:347–354. [DOI] [PubMed] [Google Scholar]
- 11. Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR (2000) Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res 76:85–92. [DOI] [PubMed] [Google Scholar]
- 12. Hata R, Mies G, Wiessner C, Fritze K, Hesselbarth D, Brinker G, Hossmann KA (1998) A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab 18:367–375. [DOI] [PubMed] [Google Scholar]
- 13. Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schurmann P, Windl O, Brose N, Kretzschmar H (1999) Evidence of presynaptic location and function of the prion protein. J Neurosci 19:8866–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoshino S, Inoue K, Yokoyama T, Kobayashi S, Asakura T, Teramoto A, Itohara S (2003) Prions prevent brain damage after experimental brain injury: a preliminary report. Acta Neurochir Suppl 86:297–299. [DOI] [PubMed] [Google Scholar]
- 15. Klamt F, Dal Pizzol F, Conte da Frota ML Jr, Walz R, Andrades ME, Da Silva EG, Brentani RR, Izquierdo I, Fonseca Moreira JC (2001) Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic Biol Med 30:1137–1144. [DOI] [PubMed] [Google Scholar]
- 16. Kloss CU, Thomassen N, Fesl G, Martens KH, Yousri TA, Hamann GF (2002) Tissue‐saving infarct volumetry using histochemistry validated by MRI in rat focal ischemia. Neurol Res 24:713–718. [DOI] [PubMed] [Google Scholar]
- 17. Kramer ML, Kratzin HD, Schmidt B, Romer A, Windl O, Liemann S, Hornemann S, Kretzschmar H (2001) Prion protein binds copper within the physiological concentration range. J Biol Chem 276:16711–16719. [DOI] [PubMed] [Google Scholar]
- 18. Krebs B, Kohlmannsperger V, Nolting S, Schmalzbauer R, Kretzschmar HA (2006) A method to perform Western blots of microscopic areas of histological sections. J Histochem Cytochem 54:559–565. [DOI] [PubMed] [Google Scholar]
- 19. Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR (2005) Interaction of cellular prion and stress‐inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci 25:11330–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mangé A, Beranger F, Peoc’h K, Onodera T, Frobert Y, Lehmann S (2004) Alpha‐ and beta‐ cleavages of the amino‐terminus of the cellular prion protein. Biol Cell 96:125–132. [DOI] [PubMed] [Google Scholar]
- 21. McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 165:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McMahon HE, Mangé A, Nishida N, Creminon C, Casanova D, Lehmann S (2001) Cleavage of the amino terminus of the prion protein by reactive oxygen species. J Biol Chem 276:2286–2291. [DOI] [PubMed] [Google Scholar]
- 23. Milhavet O, Lehmann S (2002) Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev 38:328–339. [DOI] [PubMed] [Google Scholar]
- 24. Ostrakhovitch EA, Lordnejad MR, Schliess F, Sies H, Klotz LO (2002) Copper ions strongly activate the phosphoinositide‐3‐kinase/Akt pathway independent of the generation of reactive oxygen species. Arch Biochem Biophys 397:232–239. [DOI] [PubMed] [Google Scholar]
- 25. Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qin K, Coomaraswamy J, Mastrangelo P, Yang Y, Lugowski S, Petromilli C, Prusiner SB, Fraser PE, Goldberg JM, Chakrabartty A, Westaway D (2003) The PrP‐like protein Doppel binds copper. J Biol Chem 278:8888–8896. [DOI] [PubMed] [Google Scholar]
- 27. Rachidi W, Vilette D, Guiraud P, Arlotto M, Riondel J, Laude H, Lehmann S, Favier A (2003) Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. J Biol Chem 278:9064–9072. [DOI] [PubMed] [Google Scholar]
- 28. Sakudo A, Lee DC, Saeki K, Nakamura Y, Inoue K, Matsumoto Y, Itohara S, Onodera T (2003) Impairment of superoxide dismutase activation by N‐terminally truncated prion protein (PrP) in PrP‐deficient neuronal cell line. Biochem Biophys Res Commun 308:660–667. [DOI] [PubMed] [Google Scholar]
- 29. Schmitt‐Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB (2001) Binding of neural cell adhesion molecules (N‐CAMs) to the cellular prion protein. J Mol Biol 314:1209–1225. [DOI] [PubMed] [Google Scholar]
- 30. Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, Von Mering C, Hangartner C, Aguzzi A, Weissmann C (1998) Expression of amino‐terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93:203–214. [DOI] [PubMed] [Google Scholar]
- 31. Spudich A, Frigg R, Kilic E, Oesch B, Raeber A, Bassetti CL, Hermann DM (2005) Aggravation of ischemic brain injury by prion protein deficiency: role of ERK‐1/‐2 and STAT‐1. Neurobiol Dis 20:442–449. [DOI] [PubMed] [Google Scholar]
- 32. Tsiroulnikov K, Chobert JM, Haertle T (2006) Copper‐dependent degradation of recombinant ovine prion protein. Phosphatidylinositol stimulates aggregation and copper‐driven disappearance of prion protein. FEBS J 273:1959–1965. [DOI] [PubMed] [Google Scholar]
- 33. Vassallo N, Herms J, Behrens C, Krebs B, Saeki K, Onodera T, Windl O, Kretzschmar HA (2005) Activation of phosphatidylinositol 3‐kinase by cellular prion protein and its role in cell survival. Biochem Biophys Res Commun 332:75–82. [DOI] [PubMed] [Google Scholar]
- 34. Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ (1999) Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc Natl Acad Sci USA 96: 2042–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vosko MR, Burggraf D, Liebetrau M, Wunderlich N, Jager G, Groger M, Plesnila N, Hamann GF (2006) Influence of the duration of ischemia and reperfusion on infarct volume and microvascular damage in mice. Neurol Res 28:200–205. [DOI] [PubMed] [Google Scholar]
- 36. Watt NT, Hooper NM (2005) Reactive oxygen species (ROS)‐mediated β‐cleavage of the prion protein in the mechanism of the cellular response to oxidative stress. Biochem Soc Trans 33:1123–1125. [DOI] [PubMed] [Google Scholar]
- 37. Weise J, Crome O, Sandau R, Schulz‐Schaeffer W, Bahr M, Zerr I (2004) Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett 372:146–150. [DOI] [PubMed] [Google Scholar]
- 38. Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz‐Schaeffer W, Zerr I, Bahr M (2006) Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase‐3 activation, and exacerbation of ischemic brain injury. Stroke 37:1296–1300. [DOI] [PubMed] [Google Scholar]
- 39. White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, Beyreuther K, Masters CL, Cappai R (1999) Prion protein‐deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol 155:1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]