

Abstract
Background
Familial tumoral calcinosis (FTC) is a rare autosomal recessive disease characterised by the development of multiple calcified masses in periarticular soft tissues; GALNT3 gene mutations have recently been described in an African American and in a Druse Arab family with FTC.
Objective
To report the clinical and histological features caused by a new GALNT3 mutation in a white family.
Results
Homozygosity for the nonsense mutation Lys463X was found in both affected siblings, who displayed a classic phenotype, the male also having testicular microlithiasis. He is the first subject described with testicular microlithiasis in FTC.
Conclusions
The high testicular expression of GALNT3 suggests that the gene alteration could act locally by causing deposition of calcium, and the testis may be an underestimated site of calcification in FTC. Autoimmune diseases are present in several members of the family. Although immune disorders have been described in FTC, autoimmunity does not segregate with the GALNT3 mutation in this family.
Keywords: tumoral calcinosis, tumoral calcinosis with hyperphosphataemia, testicular microlithiasis, GALNT3
Familial tumoral calcinosis (FTC; also named tumoral calcinosis with hyperphosphataemia, OMIM 211900) is a severe autosomal recessive metabolic disorder characterised by the development of calcified masses in soft tissues.1,2,3 Calcium deposits are mostly found in sites surrounding the hip, elbow, shoulder, and knee1,2,3; less common sites are the skin, intervertebral discs, extradural space, labia majora, and perineum.2,4 Diaphysitis with periostal reaction may develop in tubular bones,2 and dental abnormalities, angioid streaks of the retina, and arterial aneurysms have been reported occasionally.5,6
Biochemical abnormalities include hyperphosphataemia with normal serum calcium, enhanced renal phosphate reabsorption, and usually raised 1‐25(OH)2 vitamin D levels.1,3 The disease is difficult to treat pharmacologically and may be tragically debilitating; excision of the masses is often required.4
Mutations in GALNT3 gene, encoding the polypeptide N‐acetylgalactosaminyltransferase 3 (GalNAc‐T3), have been recently described in two families with FTC.7 We report the clinical, histological, and molecular features of an Italian family with a new GALNT3 mutation; the male affected sibling also showed testicular microlithiasis (TM), not previously described in the syndrome.
Case report
A six year old boy (II‐4 in fig 1A) was admitted to our Hospital because of a painful mass in the right elbow. His family was from Southern Italy and his parents were first‐degree cousins. Conventional X‐ray examination of the right arm showed a multilobular periarticular calcified lesion; three months later, a similar mass developed in the left elbow. Both masses were surgically excised and FTC was histologically diagnosed. When he was 14, in the course of a periodical clinical and radiological assessment, ultrasound showed diffuse bilateral increase of the testis echogenicity and the parenchyma scattered with tiny hyperechogenic foci, in absence of any congenital or clinically evident acquired testicular disease (fig 2 A). Left testis needle biopsy excluded an associated neoplasm but revealed diffuse calcifications; oligoazoospermia was subsequently documented by spermiogram.

Figure 1 Pedigree and molecular analysis of the family. (A) Pedigree. Clinical findings: I‐2, arthralgias; II‐1, vasculitis; II‐2, ITP (splenectomy at the age of 17); II‐4, calcified masses in both elbows, dense mandibular inclusions, calcification of choroid plexus, TM; II‐6, calcified masses in right elbow and left hip, acute diaphysitis at ulna, radius, metacarpal bones and tibia, ITP. I‐1, II‐3, II‐5, and II‐7 are healthy. (B) Electropherograms from the normal subject II‐2 (a), the heterozygous subject II‐1 (b), and the homozygous subject II‐6 (c). Position 13649, where an A→T substitution is present, is indicated by the bold characters. ITP, immune thrombocytopenic purpura; TM, testicular microlithiasis.

Figure 2 Features of the left testis in patient II‐4. Ultrasound shows diffuse intratesticular non‐shadowing echogenic foci (A); histology demonstrates calcifications in the seminiferous tubules (B), with compression of the tubular epithelial layers (C). Focally, calcifications appear surrounded by basement membrane only (D) (B: haematoxylin‐eosin stain, original magnification 20×; C: PAS stain, original magnification 10×; D: haematoxylin‐eosin stain, original magnification 20×).
A younger sister (II‐6) presented with a painful swelling of the right elbow with radiological features of tumoral calcinosis at the age of six. One year later, she developed chronic immune thrombocytopenic purpura (ITP) associated with additional serological markers of autoimmunity (antinuclear antibodies, lupus anticoagulant, and anti‐thyroid‐peroxidase autoantibodies). From the same age, the patient underwent repeated episodes of diaphysitis, localised at the ulna, tibia, metacarpal bones, and radius. At 10, she developed a bulky mass limiting the left hip movement and requiring excision (fig 3).
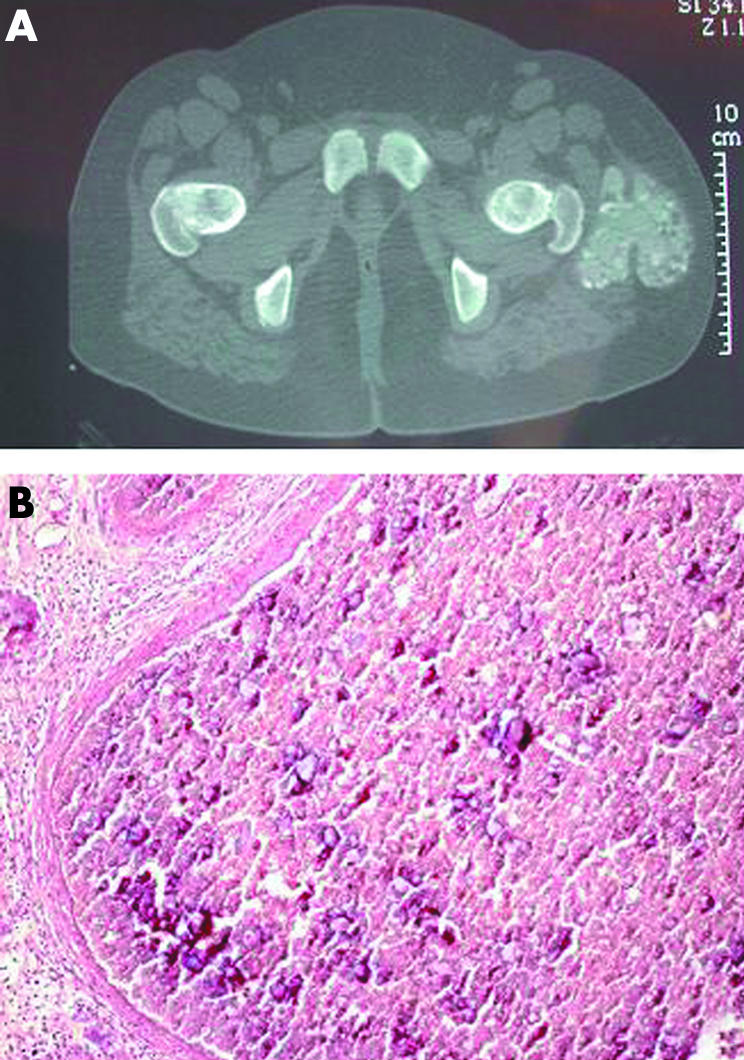
Figure 3 Left hip calcified mass in patient II‐6. Computed tomography (A) shows a bulky multilobar mass beside the left greater trocanther, approximately 9×7.5×5 cm; light microscopy (B) demonstrates cystic lesions containing fibrin and calcium deposits, lined by a fibrous cap (Slavin stage III1; haematoxylin‐eosin stain, original magnification 10×).
Both patients had hyperphosphataemia with normocalcaemia, normal parathyroid hormone, and slightly raised 1‐25(OH)2 vitamin D levels. Oral aluminium hydroxide, calcium carbonate, and low phosphorus diet had no effect on the serum phosphate levels or on the calcium deposits. Sevelamer hydrochloride lowered phosphate levels but did not induce remission of the symptoms.
The family's pedigree is shown in fig 1A. The mother (I‐2) had migrant arthralgias of undetermined aetiology. One sister (II‐1) suffered from a cutaneous rash resembling vasculitis and another (II‐2) had been affected by immune thrombocytopenia in childhood. They were free of clinical or laboratory markers of tumoral calcinosis.
Mutation analysis
Blood samples were taken from all members of the family after informed consent, and DNA was extracted by standard methods. GALNT3 mutation analysis of patient II‐6 was undertaken by polymerase chain reaction amplification of the 10 exons and the intron–exon boundaries, followed by sequencing using an ABI PRISM‐BigDye Terminator cycle sequencing ready reaction kit and an ABI PRISM 310 genetic analyser. A homozygous nonsense mutation was found in exon 6. It consisted of an A→T substitution at cDNA nucleotide 1387 causing the change of lysine 463 to a stop codon (c.1387A→T; p.Lys463X)8 (fig 1B). Exon 6 sequencing of the other members of the family showed homozygosity for the mutation in the affected sibling II‐4 and heterozygosity in both parents and in the healthy sister II‐1.
Pathological findings
The periarticular masses consisted of fibrous tissue, with scattered histiocytes and cystic cavities filled by both fibrin and calcium deposits (fig 3B), focally lined by osteoclast‐like giant cells or non‐foamy histiocytes, or both. Histiocytes showed large cytoplasmic vacuoles containing fibrin, calcified particles, and laminated calcospherites.
On testicular biopsy from patient II‐4, intratubular calcifications were scattered all over the specimen, often associated with dislocation and compression of the tubular epithelium (fig 2, panels B and C). Focally calcifications were interstitially located with a limiting basement membrane (fig 2D). Slight fibrous thickening of the tubular basement membrane and mild interstitial fibrosis were present; no granulomatous or giant cell reaction was detected.
Discussion
O‐linked glycosylation is a post‐translational modification of proteins and modulates a variety of cell functions. Its first step is the transfer of N‐acetylgalactosamine to a serine or threonine residue and is carried out by several UDP‐GalNAc:polypeptide N‐acetylgalactosaminyltransferases (GalNAc‐Ts) with different tissue specificities; of these, GalNAc‐T3 is highly expressed in the testis and the pancreas and, at lower levels, in the skin and the kidneys.7,9 Mutations in GALNT3 gene, encoding GalNAc‐T3, have recently been reported in two families with FTC7; one of the mutations—the splice site defect 1525+1G→A—was also described in two patients with hyperostosis‐hyperphosphataemia syndrome.10 We report an Italian family whose two FTC patients were found to be homozygous for the GALNT3 mutation Lys463X. This mutation is expected to result in either a non‐functional truncated enzyme or no protein because of nonsense mediated mRNA decay. In homozygotes, the total absence of wild‐type GalNAc‐T3 is supposed to affect the mineral homeostasis by decreasing O‐glycosylation of proteins that control circulating phosphate levels, such as fibroblast growth factor 23.11
The clinical, radiological, and histopathological features of FTC in the family were typical, except for the presence of testicular calcium deposits in the male patient. Calcifications were localised both in the lumen of seminiferous tubules and in the interstitium, lined by a basement membrane. This suggests progressive damage to the seminiferous epithelium, probably caused by mechanical injury, and possibly accounts for the oligoazoospermia.
TM, defined as the presence of calcium deposits in the seminiferous tubules associated with a typical ultrasound picture of diffuse non‐shadowing echogenic foci,12,13 is uncommon in childhood and mainly related to cryptorchidism, though it may also occur in systemic diseases such as McCune‐Albright syndrome.14 In adulthood it strongly suggests neoplasia. Subject II‐4 had shown the typical ultrasound and histological aspects of TM since the age of 14. Although TM has been described as an incidental finding in healthy young men,13 the association of bilateral TM with FTC is unlikely to be random, considering the very low prevalence of TM in children and the high expression of GALNT3 in the testis under normal conditions.
Recurring autoimmune diseases—namely ITP, vasculitis, and arthritis—were present in our family. Though some immune disorders, including vasculitis and arthritis, are associated with FTC,4,15 autoimmunity is shown not to segregate with the GALNT3 mutation. However, we cannot exclude the possibility that GalNAc‐T3 dysfunction plays a role in the development of autoimmunity in the family members I‐2, II‐1, and II‐6, who show both a GALNT3 mutation and immune disorders. Altered O‐linked glycosylation could impair functions of surface receptors of lymphocytes; alternatively, autoimmunity might be promoted by subclinical tissue inflammation owing to disturbed glycosylation and a tendency to calcification.16
In conclusion, we describe an Italian family with FTC, testicular microlithiasis, and a new GALNT3 mutation. This is the first description of testicular microlithiasis in FTC. The high expression of GALNT3 in the testis suggests that it may be an underestimated site of calcification in FTC. Ultrasound evaluation of the testes should thus be extended to males with FTC, and the functional consequences evaluated.
Take home message
Testicular microlithiasis was present in an Italian boy with familial tumoral calcinosis and a new GALNT3 mutation.
Ultrasound evaluation of the testes should be part of the evaluation of males with FTC.
Acknowledgements
This work was partially supported by an MURST and Banca del Piemonte grant to UR.
Abbreviations
FTC - familial tumoral calcinosis
ITP - immune thrombocytopenic purpura
TM - testicular microlithiasis
References
- 1.Slavin R E, Wen J, Kumar D.et al Familial tumoral calcinosis: a clinical, histopathologic, and ultrastructural study with an analysis of its calcifying process and pathogenesis. Am J Surg Pathol 199317788–802. [PubMed] [Google Scholar]
- 2.Martinez S, Vogler J B, Harrelson J M.et al Imaging of tumoral calcinosis: new observations. Radiology 1990174215–222. [DOI] [PubMed] [Google Scholar]
- 3.Pakasa N M, Kalengayi R M. Tumoral calcinosis: a clinicopathological study of 111 cases with emphasis on the earliest changes. Histopathology 19973118–24. [DOI] [PubMed] [Google Scholar]
- 4.Metzker A, Eisenstein B, Oren J.et al Tumoral calcinosis revisited – common and uncommon features. Report of ten cases and review. Eur J Pediatr 1988147128–132. [DOI] [PubMed] [Google Scholar]
- 5.Abraham Z, Rozner I, Rozenbaum M. Tumoral calcinosis: report of a case and brief review of the literature. J Dermatol 199623545–550. [DOI] [PubMed] [Google Scholar]
- 6.Adams W M, Laitt R D, Davies M.et al Familial tumoral calcinosis: association with cerebral and peripheral aneurysms formation. Neuroradiology 199941351–355. [DOI] [PubMed] [Google Scholar]
- 7.Topaz O, Shurman D L, Bergman R.et al Mutations in GALNT3, encoding a protein involved in O‐linked glycosylation, cause familial tumoral calcinosis. Nat Genet 200436579–581. [DOI] [PubMed] [Google Scholar]
- 8.International Human Genome Sequencing consortium cDNA sequence: NM_004482.2. Available at: htpp, // www.ncbi.nlm.nih.gov Accessed September 23, 2004
- 9.Bennet E P, Hassan H, Clausen H. cDNA cloning and expression of a novel human UDP‐N‐acetyl‐α‐D‐galactosamine:polypeptide N‐acetylgalactosaminyltransferase, GalNAc‐T3. J Biol Chem 199627117006–17012. [DOI] [PubMed] [Google Scholar]
- 10.Frishberg Y, Topaz O, Bergman R.et al Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis‐hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med 20058333–38. [DOI] [PubMed] [Google Scholar]
- 11.Benet‐Pagès A, Orlik P, Strom T M.et al A FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 200514385–390. [DOI] [PubMed] [Google Scholar]
- 12.Leenen A S, Riebel T W. Testicular microlithiasis in children: sonographic features and clinical implications. Pediatr Radiol 200232575–579. [DOI] [PubMed] [Google Scholar]
- 13.Drut R, Drut R M. Testicular microlithiasis: histologic and immunohistochemical findings in 11 pediatric cases. Pediatr Dev Pathol 20025544–550. [DOI] [PubMed] [Google Scholar]
- 14.Wasniewska M, De Luca F, Bertelloni S.et al Testicular microlithiasis: an unreported feature of McCune‐Albright syndrome in males. J Pediatr 2004145670–672. [DOI] [PubMed] [Google Scholar]
- 15.Monteagudo M, Lima J, Bravo M L.et al Arthritis mutilans, tumoral calcinosis, Raynaud's phenomenon and Sjogren's syndrome. Br J Rheumatol 199029303–305. [DOI] [PubMed] [Google Scholar]
- 16.Anderton S M. Post‐translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol 200416753–758. [DOI] [PubMed] [Google Scholar]