

Abstract
Diagnosis and differential diagnosis of cutaneous lymphoproliferative disorders is one of the most difficult areas in dermatopathology, and biopsies are often taken to rule out a cutaneous lymphoma in patients with “unclear” or “therapy‐resistant” skin lesions. Histopathological features alone often enable a given case to be classified to a diagnostic group (eg, epidermotropic lymphomas), but seldom allow a definitive diagnosis to be made. Performing several biopsies from morphologically different lesions is suggested, especially in patients with suspicion of mycosis fungoides. Immunohistochemistry is often crucial for proper classification of the cases, but in some instances is not helpful (eg, early lesions of mycosis fungoides). Although molecular techniques provide new, powerful tools for diagnosing cutaneous lymphoproliferative disorders, results of molecular methods should always be interpreted with the clinicopathological features, keeping in mind the possibility of false positivity and false negativity. In many cases, a definitive diagnosis can be made only on careful correlation of the clinical with the histopathological, immunophenotypical and molecular features.
The histopathological diagnosis of cutaneous lymphoproliferative disorders is one of the most vexing problems in dermatopathology. The protean manifestations of these diseases, the confusing terminology and the frequent changes in classification schemes all contribute to the difficulties in this field. Hence, many dermatopathologists find it frustratingly difficult to classify a given case correctly. In this context, it should be remembered that in many instances a correct diagnosis is possible only on correlation of the histopathological findings with the clinical features, and that lack of clinical information results in insecure diagnoses. Although it may be frustrating for surgical pathologists, dermatopathologists and haematopathologists not to be able to classify a case correctly without proper correlation with the clinical features, there is at present no other way to achieve a correct diagnosis.
Classification of cutaneous lymphomas has been inconsistent in the past, with different schemes adopted in different countries. Recently, the World Health Organization (WHO) and the European Organization for Research and Treatment of Cancer (EORTC)—Cutaneous Lymphoma Project Group proposed a classification scheme that reflects the current state of the art in the field of cutaneous lymphomas (box 1).1 This classification reflects what most specialist skin lymphoma centres were already doing routinely every day, and provides a valid basis for a uniform terminology worldwide. It should be remembered that a common terminology is a prerequisite for meaningful comparison of data obtained from different centres. Thus, the WHO–EORTC classification should be used consistently for patients with cutaneous lymphomas.
Box 1 WHO—EORTC classification of primary cutaneous lymphomas.
| Cutaneous T cell and NK cell lymphomas |
| Mycosis fungoides |
| MF variants and subtypes |
| Folliculotropic MF |
| Pagetoid reticulosis |
| Granulomatous slack skin |
| Sézary syndrome |
| Adult T cell leukaemia/lymphoma |
| Primary cutaneous CD30 lymphoproliferative disorders |
| Primary cutaneous anaplastic large‐cell lymphoma |
| Lymphomatoid papulosis |
| Subcutaneous panniculitis‐like T cell lymphoma |
| Extranodal NK/T cell lymphoma, nasal type |
| Primary cutaneous peripheral T cell lymphoma, unspecified |
| Primary cutaneous aggressive epidermotropic CD8 T cell lymphoma (provisional) |
| Cutaneous γ/δ T cell lymphoma (provisional) |
| Primary cutaneous CD4 small/medium‐sized pleomorphic T cell lymphoma (provisional) |
| Cutaneous B cell lymphomas |
| Primary cutaneous marginal zone B cell lymphoma |
| Primary cutaneous follicle centre lymphoma |
| Primary cutaneous diffuse large B cell lymphoma, leg type |
| Primary cutaneous diffuse large B cell lymphoma, other |
| Intravascular large B cell lymphoma |
| Precursor haematological neoplasm |
| CD4/CD56 haematodermic neoplasm (blastic NK cell lymphoma) |
MF, mycosis fungoides; NK, natural killer; WHO–EORTC, World Health Organization–European Organization for Research and Treatment Classification.
Later, I will discuss the main entities of cutaneous lymphomas, and my approach to diagnosis and differential diagnosis. I will concentrate on primary cutaneous lymphomas—that is, those lymphomas that arise in the skin without systemic manifestations at presentation. In this context, it should be underlined that any non‐Hodgkin's lymphoma (and even Hodgkin's lymphoma) arising at extracutaneous sites can give specific cutaneous manifestations, and that accurate staging is often required to exclude secondary cutaneous spread of an extracutaneous lymphoma.
Cutaneous T cell Lymphomas
Mycosis fungoides
Mycosis fungoides is the most common cutaneous lymphoma, representing almost 50% of cases.1,2 It is also a disease with many variants and subtypes, both clinical and histopathological (box 2).3 Knowledge of these variants is necessary to make specific diagnoses in cases that differ from the conventional presentation.
Box 2 Clinicopathological variants of mycosis fungoides.
| Acanthosis nigricans‐like mycosis fungoides |
| Angiocentric/angiodestructive mycosis fungoides |
| Bullous (vesiculobullous) mycosis fungoides |
| Dyshidrotic mycosis fungoides |
| Erythrodermic mycosis fungoides |
| Follicular (pilotropic) mycosis fungoides |
| Granulomatous mycosis fungoides |
| Granulomatous slack skin |
| Hyperpigmented mycosis fungoides |
| Hypopigmented mycosis fungoides |
| Ichtyosis‐like mycosis fungoides |
| Interstitial mycosis fungoides |
| Invisible mycosis fungoides |
| Mucinous mycosis fungoides |
| Mycosis fungoides palmaris et plantaris |
| Mycosis fungoides with eruptive infundibular cysts |
| Mycosis fungoides with follicular mucinosis |
| Mycosis fungoides with large‐cell transformation |
| Pagetoid reticulosis (Woringer–Kolopp type) |
| Papular mycosis fungoides |
| Papuloerythroderma Ofuji |
| Perioral dermatitis‐like mycosis fungoides |
| Pigmented purpura‐like mycosis fungoides |
| Poikilodermatous mycosis fungoides (poikiloderma vascolare atrophicans) |
| Pustular mycosis fungoides |
| Small‐plaque parapsoriasis |
| Syringotropic mycosis fungoides |
| Unilesional (solitary) mycosis fungoides |
| Verrucous/hyperkeratotic mycosis fungoides |
| Zosteriform mycosis fungoides |
The diagnosis of mycosis fungoides in early stages rests mainly on clinicopathological correlation, and in many cases cannot be differentiated from benign reactive infiltrates without knowledge of the clinical picture. Clinical features that are helpful in the differential diagnosis of mycosis fungoides from benign inflammatory dermatoses are location on sun‐protected areas such as the buttocks, presence of lesions with different morphological aspects, and history of long‐standing lesions that do not tend to regress without treatment (fig 1). It should be remembered that biopsy of the lesions will often be carried out after various treatments, that change the histopathological features of the disease. In doubtful cases, it is a good rule to suggest taking several biopsies from different lesions.

Figure 1 Mycosis fungoides, early lesion (patch stage). Irregular erythematous patch on sun‐protected skin of the buttocks.
In some cases microscopic examination of early lesions can fail to show the typical aspects of the disease, and may be characterised only by a superficial patchy‐lichenoid infiltrate located in a fibrotic, thickened papillary dermis. These features alone are suspect, but not diagnostic of mycosis fungoides. However, the application of repeatable histopathological criteria usually allows the diagnosis, even in early patches of the disease.3,4,5,6,7,8,9,10 The main histopathological criteria for diagnosis of early lesions of mycosis fungoides include band‐like or patchy‐lichenoid infiltrate of lymphocytes in the papillary dermis; thickened, fibrotic papillary dermis; coarse bundles of collagen in the papillary dermis; epidermotropic T lymphocytes with nuclei that are larger than those of T lymphocytes in the dermis; epidermotropic T lymphocytes aligned along the basal layer of the epidermis; epidermotropic lymphocytes with a clear halo around the nuclei; and intraepidermal collections of T lymphocytes (so‐called Pautrier's microabscesses; fig 2). It must be underlined, however, that Pautrier's microabscesses are rare in early lesions of mycosis fungoides.4 Epidermotropism may also be missing altogether in early patches of the disease.4

Figure 2 Mycosis fungoides, early lesion (patch stage). (A) Intraepidermal lymphocytes with only scant lymphoid infiltrates in the dermis; and (B) psoriasiform epidermal hyperplasia with a few atypical lymphocytes in the epidermis and a band‐like lymphoid infiltrate in the papillary dermis.
Immunohistochemical studies are not helpful in the diagnosis of early mycosis fungoides. The most common phenotype is α/β, CD3 and CD4. Although there are claims that subtle phenotypic abnormalities, such as loss of CD7 expression, may be helpful in the diagnosis, in my experience phenotypic investigations are virtually useless in the differential diagnosis of early lesions of mycosis fungoides from inflammatory dermatoses. In a few cases, particularly those characterised by prominent epidermotropism, neoplastic T lymphocytes have a cytotoxic phenotype, but at present there are no data showing that the disease in these patients has a more aggressive behaviour (fig 3).3,11

Figure 3 Mycosis fungoides, early lesion (patch stage) with cytotoxic phenotype. (A) Lymphoid infiltrate with marked epidermotropism; and (B) positivity of the intraepidermal lymphocytes for the cytotoxic marker T cell intracellular antigen 1.
Molecular analyses show a monoclonal population of T lymphocytes in about 50–60% of the cases, thus showing a low sensitivity. Although microdissection of the tissue specimens has been shown to increase sensitivity, this method is very time‐consuming and cannot be used routinely.12,13 Specificity, too, is far from being optimal, as many reactive skin conditions may display a monoclonal population of T lymphocytes (eg, lichen planus, lichen sclerosus). Thus, we do not routinely use immunohistochemical stainings or polymerase chain reaction (PCR) assays to diagnose early manifestations of mycosis fungoides. Of course, the finding of monoclonality in the context of a lesion suspect for mycosis fungoides is a further criterion for diagnosis (though not the most important one).
Recent data on differentiation of mycosis fungoides from inflammatory dermatoses using DNA microarrays should be confirmed on larger numbers of cases.14 In addition, microarrays are not yet a routine diagnostic technique in the study of cutaneous lymphomas.
In plaque and tumour stages, the differential diagnosis of mycosis fungoides is with other cutaneous T cell lymphomas, and in particular aggressive cytotoxic T cell or natural killer cell lymphomas. In these cases, accurate clinical history, complete phenotype and genotypic analyses are necessary for a correct classification (see later).
Many of the variants of mycosis fungoides are characterised by clinical features that differ from those of the conventional lesions of the disease, but that are histopathologically indistinguishable from those of common mycosis fungoides (eg hypopigmented mycosis fungoides). In some cases dermatopathologists will, however, receive specimens with histopathological features completely different from those of patches, plaques or tumours of conventional mycosis fungoides. For example, follicular mycosis fungoides is characterised by dense lymphoid infiltrate arranged around the hair follicles, with or without deposition of mucin. I consider so‐called “generalised idiopathic” follicular mucinosis as a variant of mycosis fungoides.15 There are no clinical, histopathological or molecular criteria that allow a distinction to be made between “benign” alopecia mucinosa and mycosis fungoides associated with follicular mucinosis.15 Another histopathological variant of the disease that is very difficult to diagnose is interstitial mycosis fungoides, where the infiltrate simulates that observed in granuloma annulare, interstitial type. It is useful to stain for T cell markers and for CD68 in interstitial mycosis fungoides as the cells are positive for T cell markers, whereas most of the interstitial cells in granuloma annulare are CD68 positive.
Finally, it is virtually impossible to distinguish between mycosis fungoides and Sézary syndrome through histopathological observations. Even in patients with erythroderma, erythrodermic mycosis fungoides can be distinguished from Sézary syndrome only by laboratory investigations including CD4/CD8 count in the peripheral blood, presence and number of circulating Sézary cells, and presence of a dominant T cell clone in the blood and skin. It may be that “atypical” histopathological features are less pronounced in Sézary syndrome than in early mycosis fungoides.16 In the new WHO–EORTC classification, the term Sézary syndrome is restricted to cases that do not have a history of mycosis fungoides.1 Distinction of mycosis fungoides from adult T cell lymphoma or leukaemia is also virtually impossible on histopathological grounds, and only the presence of human T cell lymphoma or leukaemia virus‐I (HTLV‐I) can confirm the diagnosis. Although in the past there have been claims that HTLV‐I could also be found in lesions of mycosis fungoides, it is now believed that this virus has no role in the aetiology and pathogenesis of mycosis fungoides, and clonal integration in neoplastic cells confirms the diagnosis of adult T cell lymphoma or leukaemia.
Primary cutaneous CD30 lymphoproliferative disorders
One of the most frequently asked questions on cutaneous lymphomas is how to differentiate lymphomatoid papulosis (LyP) from cutaneous anaplastic CD30‐positive large‐cell lymphoma (ALCL) histopathologically. That question, unfortunately, has no clear‐cut answer. Although it is true that clinically typical cases of LyP can easily be distinguished from typical cases of ALCL (fig 4), a huge area of overlap exists, and these two entities are really better understood as a continuum. In fact, their features may be blurred clinically: LyP may begin with a solitary tumour followed by an eruption of the more common waxing and waning papules and small nodules, and ALCL may present with tumours surrounded by small papules (satellites), resembling so‐called regional LyP.1,3,17 Spontaneous resolution is the rule in LyP, but may occur in ALCL too. In short, the two ends of the spectrum are usually easy to diagnose with confidence, but in many instances a clear‐cut differentiation cannot be achieved. For these reasons, in the new WHO–EORTC classification, LyP and cutaneous ALCL are put into a single group of cutaneous CD30 lymphoproliferative disorders.1

Figure 4 Cutaneous CD30 lymphoproliferative disorders. (A) Lymphomatoid papulosis. Note multiple papules, some with ulceration, on both arms; and (B) anaplastic large‐cell lymphoma. Solitary ulcerated nodule on the thigh.
Typical lesions of LyP present histopathologically with a wedge‐shaped infiltrate with variable numbers of large, atypical lymphocytes that are arranged as solitary units and in small clusters, but not in large sheets (fig 5).17 Epidermal association is variable and may simulate mycosis fungoides. These papular lesions are easily distinguished from ALCL. On the other hand, patients with LyP sometimes present with larger tumours characterised by sheets of atypical lymphocytes that are histopathologically indistinguishable from those of ALCL (so‐called type C LyP).17 In a similar way, ALCL may present with clusters of neoplastic CD30 cells admixed with strong inflammatory infiltrates containing many neutrophils and eosinophils (the so‐called inflammatory type of ALCL), similar to the picture of LyP (fig 6).3 In short, in borderline cases the term “cutaneous CD30 lymphoproliferative disorder—spectrum LyP/ALCL” should be used, mentioning that a definitive diagnosis can be reached only on clinicopathological correlation.

Figure 5 Lymphomatoid papulosis. (A) Wedge‐shaped lymphoid infiltrate with epidermal hyperplasia; (B) note atypical lymphocytes; and (C) positivity of the atypical cells for CD30.

Figure 6 Anaplastic large‐cell lymphoma. (A) Exophytic tumour with dense, nodular lymphoid infiltrates; (B) note several large atypical cells admixed with many neutrophils and eosinophils; and (C) sheets of cells positive for CD30.
The expression of CD30 is a prerequisite for the diagnosis of ALCL and LyP, but is by no means restricted to these two entities. Besides other malignant lymphomas (CD30 positivity may be observed in tumour‐stage mycosis fungoides as well as in some cytotoxic natural killer cell or T cell lymphomas, and even, rarely, in diffuse large B cell lymphomas), many reactive conditions are characterised by variable amounts of large CD30 lymphocytes, including arthropod bites, drug eruptions and viral infections. A typical pitfall is represented by cutaneous lesions due to herpes simplex ½, varicella zoster virus.18 It is not unusual, in these cases, to observe dense lymphoid infiltrates with many atypical cells positive for CD30, and these cells are sometimes arranged in small clusters. I have received in consultation several cases of herpes infection with a suspect histopathological diagnosis of cutaneous lymphoma. Typical cytopathic features of herpes infection may be subtle, but it is worth searching for them, and we should keep in mind that they may be confined to the hair follicles and sebaceous glands. PCR analysis for herpes virus DNA is a helpful technique for confirming the diagnosis in suspect cases.
Although the t(2;5) is found only rarely in cases of primary cutaneous ALCL (and ALK‐1 is usually not expressed by neoplastic cells in cutaneous cases), it has been suggested that it may be observed more frequently in paediatric cases, and a positive result does not necessarily imply a secondary cutaneous manifestation of extracutaneous ALCL.19
Subcutaneous panniculitis‐like T cell lymphoma
Subcutaneous panniculitis‐like T cell lymphoma (SPTCL) is now a well‐defined entity, but was one of the most confusing groups of cutaneous lymphoma in the recent past.3 Confusion was due to many factors, mainly inconsistency in classification of cases, inadequate phenotypic and genotypic criteria of published cases, and confused terminology.3,20,21 The WHO–EORTC classification defines SPTCL as a tumour confined to the subcutaneous fat composed of αβ CD8 cytotoxic lymphocytes.1 The disease has an intermediate prognosis. In contrast, aggressive cases of γ/δ cutaneous T cell lymphoma and natural killer cell or T cell lymphoma, nasal‐type, used to be included in this group, with more rare cases, all characterised by prominent involvement of the subcutaneous fat tissue.20,21 In addition, many cases of true SPTCL were (and most likely still are) diagnosed as lupus erythematosus panniculitis, as distinction may be extremely difficult, both clinically and histopathologically (fig 7).3,22 In short, three main rules apply here:

Figure 7 Subcutaneous panniculitis‐like T cell lymphoma. Subcutaneous nodules on the upper leg.
prominent involvement of the subcutis is necessary but not sufficient for a diagnosis of SPTCL;
complete phenotypic analyses should be carried out before rendering a diagnosis of SPTCL; and
be aware of mainly lobular “panniculitis” with massive necrosis and granulomatous changes, as it may mask an SPTCL.
In cases of suspect SPTCL, complete phenotyping should be carried out, including at least antibodies for the α/β chain of the T cell receptor (antibodies for the γ/δ chains are not available for paraffin wax‐embedded tissue), CD3, CD4, CD8, CD20, CD30, CD56, CD57, TIA‐1 and granzyme B (fig 8). SPTCL should be differentiated not only from benign panniculitis but also from more aggressive lymphomas that may present with identical morphological features. An antibody useful in differentiating SPTCL from lupus erythematosus panniculitis is MIB‐1/Ki‐67. In SPTCL, the cells show an increased proliferation and it is often possible to highlight the so‐called “rimming” of adipocytes by proliferating lymphocytes, whereas in lupus erythematosus panniculitis the proliferation is less marked, and lymphocytes around adipocytes are mostly negative for MIB‐1.3 In this context, the rimming of the adipocytes by neoplastic lymphocytes is not pathognomonic of SPTCL, as it can be observed in any lymphoma in the subcutaneous tissues, even B cell lymphomas.23 Other useful differential diagnostic features of lupus erythematosus panniculitis are the presence of plasma cells and lymphoid follicles, sometimes with germinal centres.

Figure 8 Subcutaneous panniculitis‐like T cell lymphoma. (A) Prominent lymphoid infiltrate in the fat lobuli; (B) rimming of the adipocytes by lymphocytes; (C) positivity of the lymphocytes for CD8; and (D) for the proliferation marker MIB‐1.
In situ hybridisation for Epstein–Barr virus should always be carried out, and positive cases can be better diagnosed as natural killer cells or T cell lymphoma, nasal type. Finally, PCR assays are used to detect clonality of the T lymphocytes in these cases (found in about 70–85% of cases), as cases of lupus erythematosus panniculitis are consistently negative.
Cutaneous aggressive cytotoxic natural killer cell/T cell lymphomas
This group of diseases is the source of many problems in histopathological diagnosis and differential diagnosis. It includes cutaneous γ/δ T cell lymphoma, primary cutaneous natural killer cell/T cell lymphoma, nasal type and aggressive CD8 T cell lymphoma.1,20 Most cases of primary cutaneous peripheral T cell lymphoma, unspecified, also have a cytotoxic phenotype.20 Luckily (both for patients and dermatopathologists), it is also a rare group, and accounts for <5% of primary cutaneous lymphomas.1
The main differential diagnosis of cutaneous aggressive cytotoxic natural killer cell/T cell lymphomas is with the tumour stage of mycosis fungoides. Clinical and histopathological features are virtually indistinguishable (fig 9). In addition, mycosis fungoides in tumour stage may acquire a cytotoxic phenotype. In short, distinction can be made only on careful clinical history, ruling out that patients had previous lesions of conventional mycosis fungoides. In the past, these cases were considered to be aggressive variants of mycosis fungoides, and only recently have phenotypic criteria allowed them to be separated from mycosis fungoides. In this context, the histopathological picture of so‐called “pagetoid reticulosis” can be observed both in cases of mycosis fungoides and in those of aggressive cytotoxic lymphoma. Solitary pagetoid reticulosis (Woringer–Kolopp) is classified today as a variant of mycosis fungoides (usually with cytotoxic phenotype),1 whereas cases of generalised pagetoid reticulosis (Ketron–Goodman) represent for the most part examples of aggressive cytotoxic lymphomas.3,20
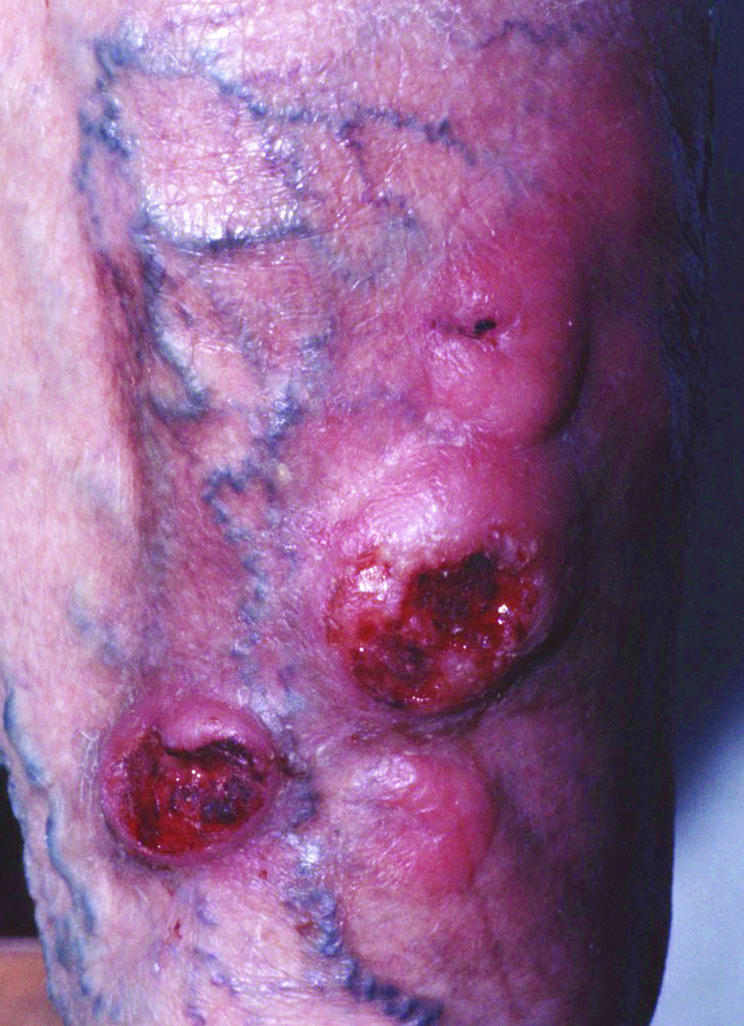
Figure 9 Cutaneous γ/δ T cell lymphoma. Reddish, ulcerated plaques and tumours on the leg, indistinguishable from tumour‐stage mycosis fungoides.
One crucial aspect of cutaneous aggressive cytotoxic natural killer cell/T cell lymphomas is that cell morphology plays no part, neither for specific classification of cases nor for determination of prognosis.3,20 All of these entities may present with infiltrates of small, medium or large cells, and the behaviour is aggressive irrespective of the size of the cells (fig 10). In this context, phenotypic investigations have a crucial role, as proper classification can be achieved only on complete phenotypic analyses. In addition, PCR analysis of the TCR gene rearrangement should always be carried out. A monoclonal band is commonly found in cases with a T cell phenotype, whereas no clonality can be detected in those with true natural killer cell lineage (most of the cases of natural killer cell/T cell lymphoma, nasal type). Finally, these cases should not be classified without carrying out in situ hybridisation for Epstein–Barr virus (fig 11).

Figure 10 Cutaneous CD8 epidermotropic T cell lymphoma. (A) Lymphoid infiltrate with marked epidermotropism; and (B) positivity for CD8.

Figure 11 Natural killer cell or T cell lymphoma, nasal type with association of the subcutaneous tissue. In situ hybridisation for Epstein–Barr virus/shows a positive signal in most neoplastic cells.
These lymphomas should be suspected particularly in the following settings:
The clinician refers that the patient has rapidly progressive plaques and ulcerated tumours without history of mycosis fungoides.
Histological examination shows a markedly epidermotropic infiltrate, and/or prominent involvement of subcutaneous fat tissue.
There is marked angiotropism or angiodestruction.
An infiltrate of atypical lymphocytes is associated with prominent necrosis (this pattern can also be observed in ALCL).
There is strong positivity for CD56 (it should be underlined, though, that CD56 is by no means specific for any lymphoma entity, and that negativity does not rule out an aggressive cytotoxic lymphoma).
Criteria for exact classification of these cases have been recently outlined.1,20 In this context, some overlap may occur among different entities, and sometimes a given case cannot be classified with certainty into one of the diagnostic categories. In fact, epidermotropic CD8 T cell lymphoma and cutaneous γ/δ T cell lymphoma are only provisional categories in the WHO–EORTC classification.1
Small or medium pleomorphic T cell lymphoma
Small or medium pleomorphic T cell lymphoma is considered to be a provisional entity in the new WHO–EORTC classification.1 Most cases reported in the past as idiopathic T cell pseudolymphoma or as pseudolymphomatous folliculitis in fact represent examples of small or medium pleomorphic T cell lymphoma.
Clinically, small or medium pleomorphic T cell lymphoma presents with reddish solitary nodules; ulceration is very rare (fig 12). Most cases are located on the head and neck region.3 Histological examination shows dense, nodular‐diffuse lymphoid infiltrates of the entire dermis (fig 13). Cytomorphological studies show predominance of small to medium‐sized T helper lymphocytes (CD3+, CD4+, CD8− and TIA‐1−). It should be underlined that a prominent B cell component is the rule, sometimes leading to the misdiagnosis of cutaneous B cell lymphoma. PCR analyses of the TCR gene rearrangement are useful in this context, as they usually show the presence of a monoclonal population of T lymphocytes, and polyclonality of the B cell component.

Figure 12 Small or medium pleomorphic T cell lymphoma. Solitary reddish nodule on the cheek.

Figure 13 Small or medium pleomorphic T‐cell lymphoma. Predominant small lymphocytes with a few medium‐sized atypical cells.
In doubtful cases, I use the term “atypical lymphoid proliferation—spectrum T cell pseudolymphoma or small or medium pleomorphic T cell lymphoma”, with an addendum explaining that this last entity has not yet been well characterised, but that prognosis seems to be very good in most cases.
Cutaneous B cell lymphomas
Marginal zone B‐cell lymphoma
Primary cutaneous marginal zone B cell lymphoma (PCMZL) is one of the most common types of cutaneous B cell lymphoma (fig 14).1,2,3 Similar cases used to be classified as cutaneous immunocytoma.3 In addition, it seems likely that cases reported as primary cutaneous plasmacytoma also represent PCMZLs with prominent plasma cell differentiation.1 In this context, it should be added that in the past many cases of PCMZL were considered to be pseudolymphomas of the skin, and that only accurate phenotypic and genotypic analyses and follow‐up data have allowed these cases to be reclassified correctly.24

Figure 14 Marginal zone B cell lymphoma. Grouped reddish papules and small nodules on the upper arm.
Clearly, in most cases of PCMZL neoplastic lymphocytes constitute only a minority of the infiltrate, which is dominated by reactive T lymphocytes and B lymphocytes (often with formation of reactive germinal centres), commonly admixed with eosinophils.3,24 In addition, the neoplastic population in most cases is not monomorphous, but comprises cells with different morphology (marginal zone cells, lymphoplasmacytoid cells and plasma cells). The neoplastic cells are often arranged at the periphery of large nodules of reactive lymphocytes, giving a characteristic biphasic appearance to the lesions (dark in the centre of the nodule, paler at the periphery; fig 15). The most important stainings for confirmation of the diagnosis of PCMZL, besides routine stainings for T lymphocytes and B lymphocytes, are those for the immunoglobulin light chains, for MIB‐1/Ki‐67, and for Bcl‐6. It is often easy to demonstrate monoclonality by immunohistochemistry; on the other hand, negativity for Bcl‐6 in the marginal zone cells (centrocyte‐like cells) helps in the distinction from cutaneous follicle centre lymphoma.25 Staining for MIB‐1/Ki‐67 shows the characteristic arrangement of the proliferating cells at the margin of the nodules (fig 16). Routine molecular analyses detect monoclonality in slightly more than half of the cases. Thus, lack of monoclonality does not rule out PCMZL.

Figure 15 Marginal zone B cell lymphoma. (A) Large lymphoid nodules with reactive germinal centres; and (B) marginal zone cells with larger nuclei and clear cytoplasm at the periphery of the nodules.

Figure 16 Marginal zone B cell lymphoma. Staining for MIB‐1/Ki‐67 highlights the proliferating cells at the margin of the nodule.
In countries endemic for Borrelia burgdorferi infection, PCR analyses may show the presence of Borrelia DNA in roughly 20% of cases of PCMZL.26,27 Although the percentage is small, these patients can be managed by non‐aggressive antibiotic treatments, thus rendering this analysis useful as a routine. Molecular analyses by the fluorescence in situ hybridisation technique show chromosomal abnormalities in <50% of cases, and cannot yet be considered to be a routine diagnostic investigation.28,29
Distinction from reactive infiltrates may be very difficult in some cases. I sometimes use the term “atypical lymphoid proliferation” to refer to lesions in which the diagnosis is not clear cut. Patients are put on short‐term controls, and eventual biopsies of new lesions are examined.
Cutaneous follicle centre lymphoma
Primary cutaneous follicle centre lymphoma (PCFCL) can present with a purely follicular, purely diffuse or mixed pattern of growth. The follicular and mixed patterns are morphologically indistinguishable from nodal follicular lymphoma. On the other hand, some cases with a diffuse pattern of growth would be classified as diffuse large B cell lymphoma in organs other than the skin, but should be classified as PCFCL at a cutaneous site.1
It is usually possible to make a diagnosis of PCFCL, follicular type, on morphological criteria alone, based on the atypical features of the follicles (lack of or reduced mantle zone, lack of or reduced number of tingible body macrophages, lack of polarisation of the follicle; fig 17).25,30 Many of these cases occur in the head and neck regions. The cells are constantly positive for Bcl‐6 (and usually positive for CD10) and do not express Bcl‐2, a major difference with nodal follicular lymphomas.1,30,31,32 When present, Bcl‐2 expression is usually confined to a minority (<30%) of the neoplastic cells, and only rarely shows the strong positive characteristic of nodal follicular lymphoma.3,33 The finding of Bcl‐2 positivity in a follicular lymphoma at cutaneous sites should raise the strong suspicion of a secondary cutaneous lymphoma.

Figure 17 Cutaneous follicle centre lymphoma, follicular. (A) Atypical follicle showing lack of polarisation and tingible body macrophages; and (B) negativity of a lymphoid follicle for Bcl‐2.
Cases with a mixed follicular‐diffuse pattern show features similar to those discussed earlier. Cases presenting with a purely diffuse pattern of growth are characterised by predominance of medium‐large cleaved cells (large centrocytes) and may be misdiagnosed as cutaneous diffuse large B cell lymphoma. Although positivity for Bcl‐6 is the rule, CD10 is often negative in these cases. The main criteria for differential diagnosis from diffuse large B cell lymphoma, leg type, include predominance of cleaved cells over round cells, and lack of Bcl‐2 and MUM‐1 expression (in PCFCL, MUM‐1 is either negative or expressed by a small minority of neoplastic cells; fig 18).34 The clinical aspect of these cases is also helpful, as most of them present with the features of so‐called Crosti's lymphoma (tumours and nodules on the trunk, usually the back, surrounded by erythematous papules and patches; fig 19).

Figure 18 Cutaneous follicle centre lymphoma, diffuse. (A) Dense, diffuse lymphoid infiltrates without follicular structures; (B) predominance of large cleaved cells; (C) positivity for Bcl‐6; and (D) staining for MUM‐1 shows positivity of only a small minority of the cells.
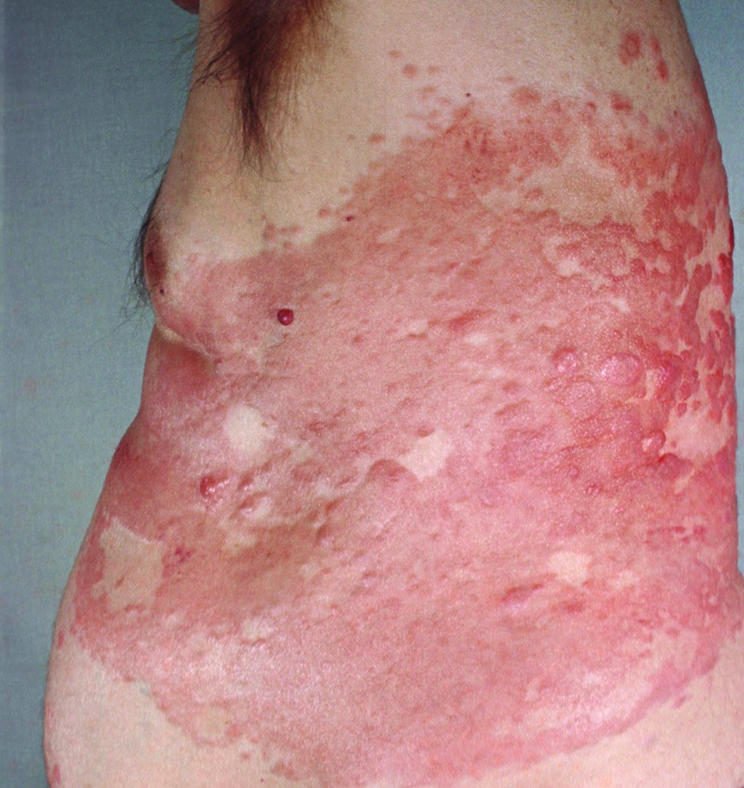
Figure 19 Cutaneous follicle centre lymphoma, diffuse. Erythematous plaques and nodules on the trunk (picture of so‐called Crosti's lymphoma).
An important differential diagnosis of PCFCL is lymphocytoma cutis induced by B burgdorferi infection. Germinal centres in Borrelia lymphocytoma are often atypical, completely lacking a mantle zone (tingible body macrophages, though, are abundant).35 The confluence of such naked follicles can even simulate the picture of a large B cell lymphoma.36 In >80% of cases Borrelia lymphocytoma involves the nipple, earlobe or genital region.35 Thus, at these body sites Borrelia lymphocytoma should be ruled out before rendering a diagnosis of cutaneous lymphoma.
In some cases, PCFCL of the diffuse type shows predominance of spindled and bizarre cells, simulating the picture of a spindle‐cell neoplasm.37 These cases can be accurately diagnosed by a combination of morphological and phenotypical (CD20 and Bcl‐6) criteria.
Standard molecular analyses are of limited value in the diagnosis and differential diagnosis of PCFCL. Data obtained with DNA microarrays showed that the genetic signature of PCFCL, diffuse type, differs from that of diffuse large B cell lymphoma, leg type.38 These analyses, however, are far from being routine in the evaluation of cutaneous lymphomas.
Primary cutaneous diffuse large B cell lymphoma, leg type
This entity got its name because most cases (>80%) occur on the legs (fig 20).1,3,34,39 Sections of primary cutaneous diffuse large B cell lymphoma, leg type, catch your eye because of the dense, monomorphous infiltrates of large round cells (immunoblasts and centroblasts; fig 21). Dermatopathologists should be aware that primary cutaneous diffuse large B cell lymphoma, leg type, may show focal epidermotropism with “Pautrier's”‐like intraepidermal collections of atypical lymphocytes.3 These cases should not be mistaken histopathologically for a T cell lymphoma.
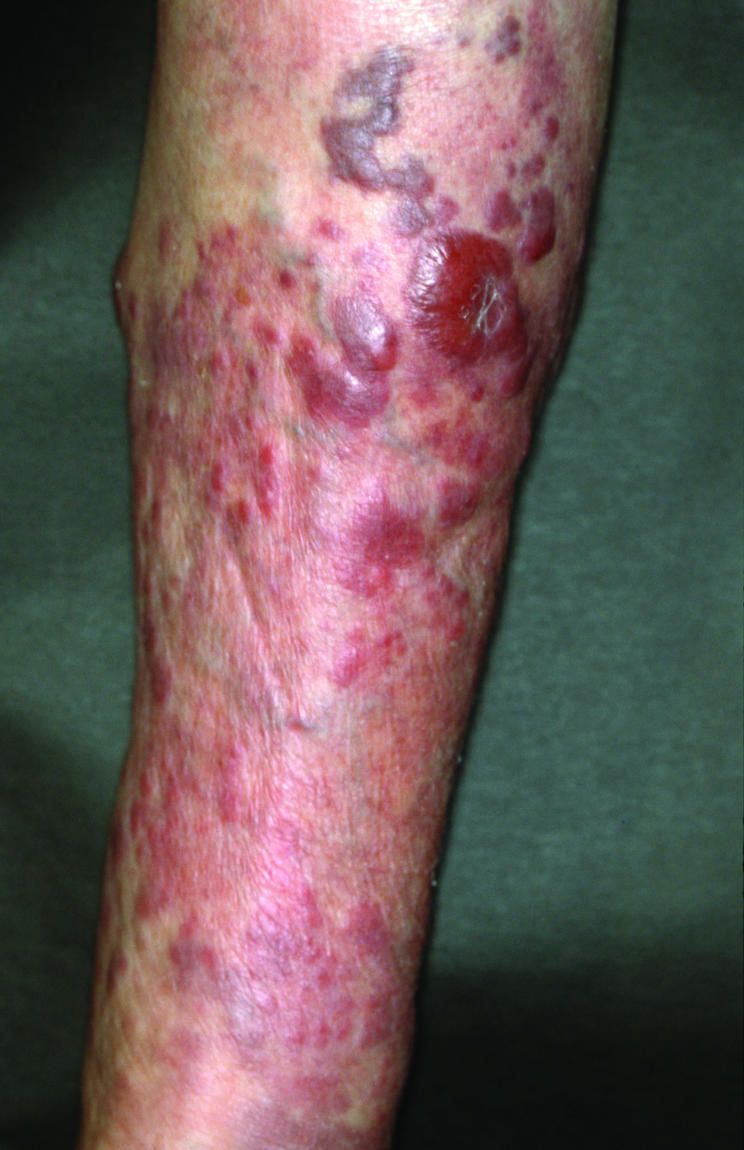
Figure 20 Diffuse large B cell lymphoma, leg type. Multiple reddish nodules and plaques on the lower leg.

Figure 21 Diffuse large B cell lymphoma, leg type. (A) Dense, diffuse infiltrate of lymphoid cells; (B) predominance of large round cells (immunoblasts); (C) positivity of all neoplastic cells for Bcl‐2; and (D) MUM‐1.
Besides cell morphology, stainings for B cell and T cell markers as well as for Bcl‐2 and MUM‐1 should be carried out to confirm the diagnosis. Primary cutaneous diffuse large B cell lymphoma, leg type, shows strong positivity for Bcl‐2 and MUM‐1, in contrast with PCFCL, diffuse type (fig 21).34 Bcl‐6 does not provide differential diagnostic clues, as it is positive in most cases of both PCFCL and primary cutaneous diffuse large B cell lymphoma, leg type. Cases with prominent round‐cell morphology and negative staining for Bcl‐2 are classified as diffuse large B cell lymphoma, other, but may represent a phenotypic (Bcl‐2) variation of primary cutaneous diffuse large B cell lymphoma, leg type, or a morphological (round‐cell) variation of PCFCL, diffuse type.34
Although in the past it has been suggested that Bcl‐2 expression is related to prognosis in cutaneous large B cell lymphomas,40 it seems that the prognostic value disappears completely once the cases are classified correctly.34 The number of lesions, location on the leg and round‐cell morphology, too,41 are devoid of any prognostic value once the cases are classified properly.34 However, it seems that cases of PCFCL, diffuse type, arising on the legs may have a worse prognosis than those arising at other body sites.34
Analyses by fluorescence in situ hybridisation show a wide spectrum of chromosomal abnormalities in primary cutaneous diffuse large B cell lymphoma, leg type.42 Most of these aberrations are similar to those observed in nodal diffuse large B cell lymphomas.
Primary cutaneous diffuse large B cell lymphoma, other
Only cases that do not fit into the previous categories, mainly Bcl‐2‐negative examples of diffuse large B cell lymphomas with round‐cell morphology are termed as primary cutaneous diffuse large B cell lymphoma, other.34 These cases are a small minority of cutaneous B cell lymphomas. Other rare cases such as plasmablastic lymphomas seen in patients with HIV or in the setting of iatrogenic immunosuppression should be classified according to criteria used for haematological neoplasms.3,43 For practical purposes, these cases are rarely encountered in the normal routine.
Intravascular large B cell lymphoma is also rare.1,3 The morphological diagnosis is relatively easy, as neoplastic cells are arranged in clusters stuffing dilated blood vessels in the dermis and superficial subcutis. These cases should always be analysed by immunohistology, as in rare cases the neoplastic cells are not B lymphocytes but T or natural killer cells, and morphology alone does not enable these variants to be differentiated. It is interesting to note that intravascular large B cell lymphoma occurs often at the site of cherry haemangiomas and that the diagnosis in some of these cases has been made by accidental biopsy of the haemangioma.44,45
CD4/CD56 haematodermic neoplasm (blastic natural killer cell lymphoma)
This is a controversial entity closely related to myelogenous leukaemia, but with a peculiar and repeatable phenotype.1,3 It was previously termed “blastic NK cell lymphoma”. The disease often presents in the skin, and in about half of the patients is confined to it at presentation (fig 22).20

Figure 22 CD4/CD56 haematodermic neoplasm. Characteristic “bruise‐like” tumour.
The histopathological specimens are characterised by dense, monomorphous infiltrates of medium‐sized cells (fig 23). The epidermis is always spared and the subcutaneous tissues are consistently involved. The main differential diagnoses, on morphology, are lymphoblastic lymphoma and myelogenous leukaemia. A complete phenotyping is required to classify these cases correctly. Neoplastic cells are positive for CD4, CD56, CD123, terminal deoxynucleotidyl transferase (TdT) (often focally), and negative for T cell and B cell markers, CD1a, CD10, CD30, CD57, TIA‐1 and myeloperoxidase (fig 23). The combination of CD4 and CD56 positivity is so striking that it has been used to term this entity; useful clues for diagnosing are positivity for TdT and the negativity for TIA‐1 in the context of CD56 positivity (stainings for CD4 on paraffin wax sections may be equivocal). Molecular analyses of the TCR and IgH gene rearrangement are negative in these cases, and positivity rules out the diagnosis of CD4/CD56 haematodermic neoplasm.

Figure 23 CD4/CD56 haematodermic neoplasm. (A) Monomorphous infiltrate of medium‐large blastic cells positive for (B) terminal deoxynucleotidyl transferase, (C) CD4 and (D) CD56.
Other lymphomas of the skin
Extracutaneous (usually nodal) lymphomas may present with secondary spread to the skin. In many instances, morphological aspects are similar to those of the cutaneous counterparts (eg, ALCL and follicle centre lymphoma); thus, complete staging investigations belong to the routine management of these patients. Some cases, on the other hand, do not have a “primary cutaneous” counterpart. The most important are B cell chronic lymphocytic leukaemia and B lymphoblastic lymphoma. Lesions of B cell chronic lymphoblastic leukaemia are characterised by dense, homogenous infiltrates of small lymphocytes.46 It is possible to confirm the diagnosis by immunohistology, showing an aberrant CD20/CD5/CD43 positive phenotype. Molecular analyses show the presence of a monoclonal population of B lymphocytes in most cases. B cell chronic lymphocytic leukaemia may present at sites of cutaneous inflammation (eg, at sites of previous herpes simplex 1/2 or herpes zoster, or at sites of Borrelia infection).47,48 B lymphoblastic lymphoma may include the skin secondarily, and in some patients may even present in the skin without manifest systemic disease (staging investigations negative).49 Morphologically, there are dense, diffuse infiltrates of monomorphous medium‐sized cells, focally arranged in a “mosaic stone” pattern. The so‐called “starry sky” pattern can also be observed, but is less frequent in the skin than in the lymph nodes. Phenotypic analyses show positivity for CD20, CD10 and TdT, and in some cases for CD34. For practical purposes, it is useful to carry out a staining for TdT in cases presenting morphologically with monomorphous proliferations of medium‐sized lymphoid cells. No reactive conditions are evident where you can expect positivity with this marker, and a positive staining clearly points to a precursor lymphoma or leukaemia.
In conclusion, diagnosis and differential diagnosis of cutaneous lymphoproliferative disorders rests on careful correlation of histopathological features with the clinical aspects. Whenever you encounter a tumoral lymphoid lesion with a T phenotype, make sure to rule out mycosis fungoides before making a diagnosis of some rare type of cutaneous T cell lymphoma, and remember that mycosis fungoides in tumour stage can present with every possible morphological–phenotypical pattern. We must be wary of false negative results when carrying out immunohistology: external positive controls, in my experience, do not suffice, and internal positive controls should always be checked for. In skin biopsies, they are available for most antibodies used routinely (eg, normal nerves are stained by CD56 antibody). A staining for κ or λ should not be interpreted as negative unless we find at least a few positive cells in our “negative” section. Positivity for a given marker does not allow diagnosis: CD56, for example, is positive in some cases of conventional early mycosis fungoides, in many γ/δ T cell lymphomas, in natural killer cell/T cell lymphoma, nasal type, and in some cases of LyP and ALCL, as well as in CD4/CD56 haematodermic neoplasm, and the immunohistochemical results need to be evaluated carefully with a panel of antibodies. Molecular analyses of the TCR and IgH genes are a valuable tool, but should never be evaluated out of the clinicopathological context. Finally, new molecular methods such as fluorescence in situ hybridisation and microarrays, though very promising, do not yet belong to the routine histopathological diagnosis of cutaneous lymphoproliferative disorders.
Abbreviations
ALCL - anaplastic CD30‐positive large‐cell lymphoma
EORTC - European Organization for Research and Treatment of Cancer
HTLV - human T cell lymphoma or leukaemia virus
LyP - lymphomatoid papulosis
PCFCL - primary cutaneous follicle centre lymphoma
PCMZL - primary cutaneous marginal zone B cell lymphoma
PCR - polymerase chain reaction
SPTCL - subcutaneous panniculitis‐like T cell lymphoma
TIA - T cell intracellular antigen
TdT - terminal deoxynucleotidyl transferase
WHO - World Health Organization
Footnotes
Competing interests: None declared.
References
- 1.Willemze R, Jaffe E S, Burg G.et al WHO‐EORTC classification for cutaneous lymphomas. Blood 20051053768–3785. [DOI] [PubMed] [Google Scholar]
- 2.Fink‐Puches R, Zenahlik P, Böck B.et al Primary cutaneous lymphomas: applicability of current classification schemes (EORTC, WHO) based on clinicopathologic features observed in a large group of patients. Blood 200299800–805. [DOI] [PubMed] [Google Scholar]
- 3.Cerroni L, Gatter K, Kerl H.An illustrated guide to skin lymphoma 2nd edn. Malden: Blackwell Publishing, 2004
- 4.Massone C, Kodama K, Kerl H.et al Histopathologic features of early (patch) lesions of mycosis fungoides. A morphologic study on 745 biopsy specimens from 427 patients. Am J Surg Pathol 200529550–560. [DOI] [PubMed] [Google Scholar]
- 5.Ming M, LeBoit P E. Can dermatopathologists reliably make the diagnosis of mycosis fungoides? If not, who can? Arch Dermatol 2000136543–546. [DOI] [PubMed] [Google Scholar]
- 6.Santucci M, Biggeri A, Feller A C.et al Accuracy, concordance, and reproducibility of histologic diagnosis in cutaneous T‐cell lymphoma. Arch Dermatol 2000136497–502. [DOI] [PubMed] [Google Scholar]
- 7.Santucci M, Biggeri A, Feller A C.et al Efficacy of histologic criteria for diagnosing early mycosis fungoides. An EORTC Cutaneous Lymphoma Study Group investigation. Am J Surg Pathol 20002440–50. [DOI] [PubMed] [Google Scholar]
- 8.Vonderheid E C, Bernengo M G, Burg G.et al Update on erythrodermic cutaneous T‐cell lymphoma: report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 20024695–106. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez J L, Ackerman A B. The patch stage of mycosis fungoides. Criteria for histologic diagnosis. Am J Dermatopathol 197915–26. [PubMed] [Google Scholar]
- 10.Shapiro P E, Pinto F J. The histologic spectrum of mycosis fungoides/Sézary syndrome (cutaneous T‐cell lymphoma). A review of 222 biopsies, including newly described patterns and the earliest pathologic changes. Am J Surg Pathol 199418645–667. [DOI] [PubMed] [Google Scholar]
- 11.Wain E M, Orchard G E, Mayou S.et al Mycosis fungoides with a CD56+ immunophenotype. J Am Acad Dermatol 200553158–163. [DOI] [PubMed] [Google Scholar]
- 12.Cerroni L, Arzberger E, Ardigó M.et al Monoclonality of intraepidermal T lymphocytes in early mycosis fungoides detected by molecular analysis after laser‐beam‐based microdissection. J Invest Dermatol 20001141154–1157. [DOI] [PubMed] [Google Scholar]
- 13.Yazdi A S, Medeiros L J, Puchta U.et al Improved detection of clonality in cutaneous T‐cell lymphomas using laser capture microdissection. J Cut Pathol 200330486–491. [DOI] [PubMed] [Google Scholar]
- 14.Tracey L, Villuendas R, Dotor A M.et al Mycosis fungoides shows concurrent deregulation of multiple genes involved in the TNF signaling pathway: an expression profile study. Blood 20031021042–1050. [DOI] [PubMed] [Google Scholar]
- 15.Cerroni L, Fink‐Puches R, Bäck B.et al Follicular mucinosis. A critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol 2002138182–189. [DOI] [PubMed] [Google Scholar]
- 16.Diwan A H, Prieto V G, Herling M.et al Primary Sézary syndrome commonly shows low‐grade cytologic atypia and an absence of epidermotropism. Am J Clin Pathol 2005123510–515. [DOI] [PubMed] [Google Scholar]
- 17.El Shabrawi‐Caelen L, Kerl H, Cerroni L. Lymphomatoid papulosis. Reappraisal of clinicopathologic presentation and classification into subtypes A, B, and C. Arch Dermatol 2004140441–447. [DOI] [PubMed] [Google Scholar]
- 18.Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol 20063050–58. [DOI] [PubMed] [Google Scholar]
- 19.Hinshaw M, Trowers A B, Kodish E.et al Three children with CD30+ cutaneous anaplastic large cell lymphomas bearing the t(2;5)(q23;q35) translocation. Ped Dermatol 200421212–217. [DOI] [PubMed] [Google Scholar]
- 20.Massone C, Chott A, Metze D.et al Subcutaneous, blastic natural killer (NK), NK/T‐cell, and other cytotoxic lymphomas of the skin. A morphologic, immunophenotypic, and molecular study of 50 patients. Am J Surg Pathol 200428719–735. [DOI] [PubMed] [Google Scholar]
- 21.Massone C, Lozzi G P, Egberts F.et al The protean spectrum of non‐Hodgkin lymphomas with prominent involvement of subcutaneous fat. J Cut Pathol 200633418. [DOI] [PubMed] [Google Scholar]
- 22.Massone C, Kodama K, Salmhofer W.et al Lupus erythematosus panniculitis (lupus profundus): clinical, histopathological, and molecular analysis of nine cases. J Cut Pathol 200532396–404. [DOI] [PubMed] [Google Scholar]
- 23.Lozzi G P, Massone C, Citarella L.et al Rimming of adipocytes by neoplastic lymphocytes is a histopathologic feature not restricted to subcutaneous T‐cell lymphoma. Am J Dermatopathol 2006289–12. [DOI] [PubMed] [Google Scholar]
- 24.Cerroni L, Signoretti S, Höfler G.et al Primary cutaneous marginal zone B‐cell lymphoma: a recently described entity of low‐grade malignant cutaneous B‐cell lymphoma. Am J Surg Pathol 1997211307–1315. [DOI] [PubMed] [Google Scholar]
- 25.Leinweber B, Colli C, Chott A.et al Differential diagnosis of cutaneous infiltrates of B lymphocytes with follicular growth pattern. Am J Dermatopathol 2004264–13. [DOI] [PubMed] [Google Scholar]
- 26.Cerroni L, Zöchling N, Pütz B.et al Infection by Borrelia burgdorferi and cutaneous B‐cell lymphoma. J Cut Pathol 199724457–461. [DOI] [PubMed] [Google Scholar]
- 27.Goodlad J R, Davidson M M, Hollowood K.et al Primary cutaneous B‐cell lymphoma and Borrelia burgdorferi infection in patients from the highlands of Scotland. Am J Surg Pathol 2000241279–1285. [DOI] [PubMed] [Google Scholar]
- 28.Streubel B, Lamprecht A, Dierlamm J.et al T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood 20031012335–2339. [DOI] [PubMed] [Google Scholar]
- 29.Streubel B, Simonitsch‐Klupp I, Müllauer L.et al Variable frequencies of MALT lymphoma‐associated genetic aberrations in MALT lymphomas of different sites. Leukaemia 2004181722–1726. [DOI] [PubMed] [Google Scholar]
- 30.Cerroni L, Arzberger E, Pütz B.et al Primary cutaneous follicle center cell lymphoma with follicular growth pattern. Blood 2000953922–3928. [PubMed] [Google Scholar]
- 31.Cerroni L, Volkenandt M, Rieger E.et al bcl‐2 protein expression and correlation with the interchromosomal 14;18 translocation in cutaneous lymphomas and pseudolymphomas. J Invest Dermatol 1994102231–235. [DOI] [PubMed] [Google Scholar]
- 32.Goodlad J R, Krajewski A S, Batstone P J.et al Primary cutaneous follicular lymphoma. A clinicopathologic and molecular study of 16 cases in support of a distinct entity. Am J Surg Pathol 200226733–741. [DOI] [PubMed] [Google Scholar]
- 33.Kim B K, Surti U, Pandya A.et al Clinicopathologic, immunophenotypic, and molecular cytogenetic fluorescence in situ hybridization analysis of primary and secondary cutaneous follicular lymphomas. Am J Surg Pathol 20052969–82. [DOI] [PubMed] [Google Scholar]
- 34.Kodama K, Massone C, Chott A.et al Primary cutaneous large B‐cell lymphomas: clinicopathologic features, classification, and prognostic factors in a large series of patients. Blood 20051062491–2497. [DOI] [PubMed] [Google Scholar]
- 35.Colli C, Leinweber B, Müllegger R.et al Borrelia burgdorferi‐associated lymphocytoma cutis: clinicopathologic, immunophenotypic, and molecular study of 106 cases. J Cut Pathol 200431232–240. [DOI] [PubMed] [Google Scholar]
- 36.Grange F, Wechsler J, Guillaume J C.et al Borrelia burgdorferi‐associated lymphocytoma cutis simulating a primary cutaneous large B‐cell lymphoma. J Am Acad Dermatol 200247530–534. [DOI] [PubMed] [Google Scholar]
- 37.Cerroni L, El‐Shabrawi‐Caelen L, Fink‐Puches R.et al Cutaneous spindle‐cell B‐cell lymphoma. A morphologic variant of cutaneous large B‐cell lymphoma. Am J Dermatopathol 200022299–304. [DOI] [PubMed] [Google Scholar]
- 38.Hoefnagel J J, Dijkman R, Basso K.et al Distinct types of primary cutaneous large B‐cell lymphoma identified by gene expression profiling. Blood 20051053671–3678. [DOI] [PubMed] [Google Scholar]
- 39.Vermeer M H, Geelen F A M J, van Haselen C W.et al Primary cutaneous large B‐cell lymphomas of the legs. A distinct type of cutaneous B‐cell lymphoma with an intermediate prognosis. Arch Dermatol 19961321304–1308. [PubMed] [Google Scholar]
- 40.Grange F, Petrella T, Beylot‐Barry M.et al Bcl‐2 protein expression is the strongest independent prognostic factor of survival in primary cutaneous large B‐cell lymphomas. Blood 20041033662–3668. [DOI] [PubMed] [Google Scholar]
- 41.Grange F, Bekkenk M W, Wechsler J.et al Prognostic factors in primary cutaneous large B‐cell lymphomas: a European multicenter study of 145 cases. J Clin Oncol 2001193602–3610. [DOI] [PubMed] [Google Scholar]
- 42.Wiesner T, Streubel B, Huber D.et al Genetic aberrations in primary cutaneous large B‐cell lymphoma. A fluorescence in situ hybridization study of 25 cases. Am J Surg Pathol 200529666–673. [DOI] [PubMed] [Google Scholar]
- 43.Jaffe E S, Harris N L, Stein H, Vardiman J W. eds. World Health Organization Classification of Tumours. Tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press, 2001
- 44.Cerroni L, Zalaudek I, Kerl H. Intravascular large B‐cell lymphoma colonizing cutaneous hemangiomas. Dermatology 2004209132–134. [DOI] [PubMed] [Google Scholar]
- 45.Cerroni L. Hemangiomas with a bad surprise. Dermatology 200420979–80. [DOI] [PubMed] [Google Scholar]
- 46.Cerroni L, Zenahlik P, Höfler G.et al Specific cutaneous infiltrates of B‐cell chronic lymphocytic leukaemia. A clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol 1996201000–1010. [DOI] [PubMed] [Google Scholar]
- 47.Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B‐cell chronic lymphocytic leukaemia arising at the site of herpes zoster and herpes simplex scars. Cancer 19957626–31. [DOI] [PubMed] [Google Scholar]
- 48.Cerroni L, Höfler G, Bäck B.et al Specific cutaneous infiltrates of B‐cell chronic lymphocytic leukaemia (B‐CLL) at sites typical for Borrelia burgdorferi infection. J Cut Pathol 200229142–147. [DOI] [PubMed] [Google Scholar]
- 49.Chimenti S, Fink‐Puches R, Peris K.et al Cutaneous involvement in lymphoblastic lymphoma. J Cut Pathol 199926379–385. [DOI] [PubMed] [Google Scholar]