

Abstract
CheY is a response regulator protein involved in bacterial chemotaxis. Much is known about its active and inactive conformations, but little is known about the mechanisms underlying long-range interactions or correlated motions. To investigate these events, molecular dynamics simulations were performed on the unphosphorylated, inactive structure from Salmonella typhimurium and the 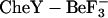 active mimic structure (with
active mimic structure (with 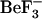 removed) from Escherichia coli. Simulations utilized both sequences in each conformation to discriminate sequence- and structure-specific behavior. The previously identified conformational differences between the inactive and active conformations of the strand-4-helix-4 loop, which are present in these simulations, arise from the structural, and not the sequence, differences. The simulations identify previously unreported structure-specific flexibility features in this loop and sequence-specific flexibility features in other regions of the protein. Both structure- and sequence-specific long-range interactions are observed in the active and inactive ensembles. In the inactive ensemble, two distinct mechanisms based on Thr-87 or Ile-95 rotameric forms, are observed for the previously identified g+ and g− rotamer sampling by Tyr-106. These molecular dynamics simulations have thus identified both sequence- and structure-specific differences in flexibility, long-range interactions, and rotameric form of key residues. Potential biological consequences of differential flexibility and long-range correlated motion are discussed.
removed) from Escherichia coli. Simulations utilized both sequences in each conformation to discriminate sequence- and structure-specific behavior. The previously identified conformational differences between the inactive and active conformations of the strand-4-helix-4 loop, which are present in these simulations, arise from the structural, and not the sequence, differences. The simulations identify previously unreported structure-specific flexibility features in this loop and sequence-specific flexibility features in other regions of the protein. Both structure- and sequence-specific long-range interactions are observed in the active and inactive ensembles. In the inactive ensemble, two distinct mechanisms based on Thr-87 or Ile-95 rotameric forms, are observed for the previously identified g+ and g− rotamer sampling by Tyr-106. These molecular dynamics simulations have thus identified both sequence- and structure-specific differences in flexibility, long-range interactions, and rotameric form of key residues. Potential biological consequences of differential flexibility and long-range correlated motion are discussed.
INTRODUCTION
CheY, a 128-residue protein (1), is a member of a large family of proteins involved in signal transduction in prokaryotes and eukaryotes. It plays a key role in the control of the bacterial movements in response to environmental chemotactic stimuli (2), functioning as a response regulator and transmitting chemical stimuli to the bacterial flagella via a signal transduction cascade. The activation of CheY occurs via phosphorylation of the conserved Asp-57 residue by the histidine kinase CheA. When phosphorylated, CheY binds to the flagellar protein FliM causing the flagella to adopt a clockwise rotation mode (3–5). The default counterclockwise rotation mode is rapidly restored by the dephosphorylation (deactivation) of CheY.
The structures of CheY from different organisms (Escherichia coli, Salmonella typhimurium, and Sinorhizobium meliloti) have been solved by x-ray crystallography and NMR spectroscopy (6–9). All of these structures show the same general architecture, in which the protein is arranged in an α/β parallel motif with a five-stranded parallel β-sheet surrounded by five helices (Fig. 1 a). The β-strands and α-helices, which alternate along the sequence, are connected by loops. The site of covalent phosphate attachment, Asp-57, is located in a pocket at the C-terminus of several central β-strands and consists of a highly negatively charged cluster of three aspartic acid residues (Asp-12 and Asp-13 within the loop from strand-1 to helix-1; and Asp-57 at the C-terminus of strand-3) and a lysine residue (Lys-109 at the C-terminus of strand-5) (Fig. 1 b). The fifth residue in the active site, Thr-87, located at the C-terminus of strand 4, is highly conserved among receiver domains as either a threonine or serine (10). The metal ion (Mn2+ or Mg2+), which is essential for phosphorylation/dephosphorylation, is coordinated by two aspartic acid side chains (Asp-13 and Asp-57), the carbonyl main-chain oxygen atom of Asn-59 and three water molecules (7,11).
FIGURE 1.

CheY structure shown with specific side chains, secondary structures, and location of amino acid sequence differences between the two structures. (a) Ribbon representation of the active form of E. coli CheY (1fqw, 8) showing secondary structure elements within the crystal structure (β-strands, yellow; α-helices, red; other structures, white). Helices and strands are labeled with the α-helix or β-strand number, A1-A5 and B1-B5. The relative position of 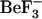 (pink) and divalent metal (dark pink) binding sites are shown as van der Waals radii. In addition, three residues that differ in identity (Gly-76, Tyr-51, Val-54 compared to Ser-76, Phe-51, Ile-54 in E. coli and S. typhimurium, respectively) are shown as cyan van der Waals spheres. (b) Close-up view of the site of phosphorylation, with the 1fqw backbone shown as a ribbon representation (white),
(pink) and divalent metal (dark pink) binding sites are shown as van der Waals radii. In addition, three residues that differ in identity (Gly-76, Tyr-51, Val-54 compared to Ser-76, Phe-51, Ile-54 in E. coli and S. typhimurium, respectively) are shown as cyan van der Waals spheres. (b) Close-up view of the site of phosphorylation, with the 1fqw backbone shown as a ribbon representation (white), 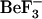 (the phosphate mimic, pink) and metal-binding (dark pink) sites as van der Waals spheres. Residues involved in
(the phosphate mimic, pink) and metal-binding (dark pink) sites as van der Waals spheres. Residues involved in 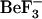 binding (Asp-57, Thr-87, and Lys-109) and metal binding (Asp-13 and Asn-59) are labeled and shown as dark blue side chains. The location of Tyr-106 (dark blue) relative to these residues is also shown. (c) Ribbon representation of backbone-aligned crystal structures of active (1fqw) and inactive (2che) cheY structures. Those regions showing the largest differences in conformation are emphasized in red (1fqw) and yellow (2che) ribbons and blue (1fqw) and black (2che) side chains. Key differences are in the positions of the Thr-87 and Lys-109 side chains, the χ-1 rotameric forms of Tyr-106 (t and buried versus g+ and solvent accessible in the 1fqw and 2che structures, respectively), helix-1, helix-5, and the sheet-4-helix-4 loop. All figures were prepared in VMD v1.8.3 (20).
binding (Asp-57, Thr-87, and Lys-109) and metal binding (Asp-13 and Asn-59) are labeled and shown as dark blue side chains. The location of Tyr-106 (dark blue) relative to these residues is also shown. (c) Ribbon representation of backbone-aligned crystal structures of active (1fqw) and inactive (2che) cheY structures. Those regions showing the largest differences in conformation are emphasized in red (1fqw) and yellow (2che) ribbons and blue (1fqw) and black (2che) side chains. Key differences are in the positions of the Thr-87 and Lys-109 side chains, the χ-1 rotameric forms of Tyr-106 (t and buried versus g+ and solvent accessible in the 1fqw and 2che structures, respectively), helix-1, helix-5, and the sheet-4-helix-4 loop. All figures were prepared in VMD v1.8.3 (20).
Although there is a great deal of structural information about the inactive, unphosphorylated form of CheY (7), there is relatively little information available about the active phosphorylated form. This is, in part, due to the autophosphatase activity of CheY, which makes the phosphorylated form unstable (half-life of ∼10 s) (12). Recently, CheY from E. coli has been crystallized with a phosphoryl group mimic (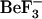 ) resulting in crystal structures of the protein in its active form (8,13).
) resulting in crystal structures of the protein in its active form (8,13).
Comparison between the active, 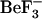 -bound CheY structure from E. coli (Protein Data Bank accession code 1fqw) (8) and the inactive CheY structure from S. typhimurium (Protein Data Bank (PDB) accession code 2che) (7) has proven useful for exploring the structural differences between phosphorylated and unphosphorylated CheY (14). The specific structural differences observed in side-chain and backbone conformations between the active (phosphorylated) and inactive forms are shown in Fig. 1 c. Analysis of the active form indicates that
-bound CheY structure from E. coli (Protein Data Bank accession code 1fqw) (8) and the inactive CheY structure from S. typhimurium (Protein Data Bank (PDB) accession code 2che) (7) has proven useful for exploring the structural differences between phosphorylated and unphosphorylated CheY (14). The specific structural differences observed in side-chain and backbone conformations between the active (phosphorylated) and inactive forms are shown in Fig. 1 c. Analysis of the active form indicates that 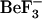 complexes with the side chains of Asp-57, Thr-87, and Lys-109 (Fig. 1 b), and with the Asn-59 backbone (8). In the inactive form, which does not contain a phosphate or its analog, the conformations of these residues are different (Fig. 1 c), with Thr-87 and Lys-109 side chains pointing away from the site of phosphate modification (7).
complexes with the side chains of Asp-57, Thr-87, and Lys-109 (Fig. 1 b), and with the Asn-59 backbone (8). In the inactive form, which does not contain a phosphate or its analog, the conformations of these residues are different (Fig. 1 c), with Thr-87 and Lys-109 side chains pointing away from the site of phosphate modification (7).
Two of the residues involved in  binding are also involved in metal binding (Asp-57 and Asn-59); thus, it might be expected that any conformational changes in the
binding are also involved in metal binding (Asp-57 and Asn-59); thus, it might be expected that any conformational changes in the 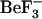 binding site would involve changes in the metal-binding site. A comparison between the metal-binding residues for the active and inactive structures indicates that this is not the case, as there is little variation in the conformation of these residues. Indeed, the function of the conserved metal ion binding site appears to position the metal ion within the active site for involvement in the phosphoryl transfer reaction to Asp-57 (7).
binding site would involve changes in the metal-binding site. A comparison between the metal-binding residues for the active and inactive structures indicates that this is not the case, as there is little variation in the conformation of these residues. Indeed, the function of the conserved metal ion binding site appears to position the metal ion within the active site for involvement in the phosphoryl transfer reaction to Asp-57 (7).
More pronounced differences between the active and inactive conformations are observed in helix-4, helix-5, and particularly in the loops preceding each of them, loops that contain Thr-87 and Lys-109, respectively (Fig. 1 c). A conformational change at helix-4 (residues 92–101) is involved in the chemotactic signaling pathway (7,9,14). As chemotactic signaling involving CheY is mediated by phosphorylation at Asp-57, one might postulate either a direct or indirect mechanism coupling the helix-4 conformation to the conformation of the site of phosphorylation. Comparison of the active and inactive structures does not support this because there is only a small difference in the helix-4 orientation relative to the rest of the protein, although there is a large difference in the conformation of the loop preceding helix-4 (Fig. 1 c).
A significant difference between the active and inactive crystal structures is the burial of the Tyr-106 side chain in the active conformation and its solvent accessibility in the inactive crystal structure. Previous studies have shown that the coupling of the helix-4 orientation with phosphate binding is achieved through the rearrangement of Thr-87 and Tyr-106 (9,15,16). This is known as Y-T coupling and involves the burial of the Tyr-106 side chain upon CheY phosphorylation. Experimental (9), structural (15), and NMR chemical shift data (14) show that this interplay between Thr-87 and Tyr-106 is key for signaling regulation in CheY. The difference in the side-chain conformation of Tyr-106 is a specific rotation of the χ-1 dihedral angle (with the χ-1 dihedral angle defined by atoms N, Cα, Cβ, and Cγ in the tyrosine, or any other, residue). In the active conformation, Tyr-106 is in the t (or trans) rotamer, with the dihedral angle at 180°. In the inactive conformation, Tyr-106 is in the g+ (or gauche+) rotamer, with the χ-1 dihedral angle at 60° (compare blue, active, and black, inactive, Tyr-106 side chains in Fig. 1 c).
Both x-ray crystallography and NMR spectroscopy provide structural information about the mean structures and variance parameters of a limited number of conformational states corresponding to those ground states accessible upon binding of different ligands. Such structural data can only suggest the nature of ensembles associated with those conformational states, but provide no information concerning the possible mechanisms by which they interchange. Subject to the limitations of sampling times and molecular mechanics approximations, molecular dynamics can, in principle, provide more detailed information about both ground state ensembles and mechanisms of conformational change. To investigate the order of events involved in CheY relaxation, the conformational changes sampled by CheY in the inactive conformation, and the sequence- and structure-specific correlated motions, we performed molecular dynamics (MD) simulations initiated from the inactive (S. typhimurium structure (7)) and the active forms of CheY (E. coli structure (8)). Although these are functionally equivalent proteins (17), the starting crystal structures are derived from different species and exhibit three sequence differences: F51Y, I54V, and S76G (S. typhimurium to E. coli). As force-field parameters for 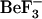 and aspartyl-phosphate are unavailable, we did not study the dynamics of the active form directly, rather, we removed the bound
and aspartyl-phosphate are unavailable, we did not study the dynamics of the active form directly, rather, we removed the bound 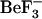 to study the relaxation of CheY from the active to inactive form. Because the timescale for the relaxation of CheY from the active to the inactive form is too long to investigate using current MD simulation techniques, this study focuses on the dynamics of the inactive form and comparison of those to the early events in the relaxation of CheY. These early events are the most difficult to observe experimentally. Simulations of the inactive forms provide information on conformations that are accessible to the inactive form, but which may facilitate activation. Additionally, we performed simulations using each CheY structure with each sequence to distinguish conformational changes that are caused by sequence differences from those caused by the removal of
to study the relaxation of CheY from the active to inactive form. Because the timescale for the relaxation of CheY from the active to the inactive form is too long to investigate using current MD simulation techniques, this study focuses on the dynamics of the inactive form and comparison of those to the early events in the relaxation of CheY. These early events are the most difficult to observe experimentally. Simulations of the inactive forms provide information on conformations that are accessible to the inactive form, but which may facilitate activation. Additionally, we performed simulations using each CheY structure with each sequence to distinguish conformational changes that are caused by sequence differences from those caused by the removal of 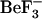 . Herein, we report the analysis of these MD simulations and describe correlated motion and long-range interactions that are specific to either the starting sequence or to the starting structure. A potential ordering of events involved in the initial steps of CheY relaxation is described. Our results suggest that species-specific differences in the flexibility and long-range communication, not obvious from the positions of amino acid differences but observable by molecular dynamics, may subtly affect basal CheY activity.
. Herein, we report the analysis of these MD simulations and describe correlated motion and long-range interactions that are specific to either the starting sequence or to the starting structure. A potential ordering of events involved in the initial steps of CheY relaxation is described. Our results suggest that species-specific differences in the flexibility and long-range communication, not obvious from the positions of amino acid differences but observable by molecular dynamics, may subtly affect basal CheY activity.
MATERIALS AND METHODS
System preparation
Four MD simulations were performed (Table 1), two based upon the wild-type protein crystal structures and sequences and two variants involving modifications to the wild-type crystal structures that changed the sequence from one organism to that of the other. To prepare the structures, first, all water molecules and counterions (with the exception of the divalent metal ion, Mg+2 or Mn+2, which is essential for CheY function) were deleted from the crystal structures. For the variant proteins, the sequences were changed from one sequence to the other by renaming the appropriate residues and deleting the uncommon atoms. (Changing the S. typhimurium [2che] sequence to the E. coli [1fqw] sequence requires changing F51Y, I54V, and S76G, and vice-versa.) The positions of these differences in the structure are illustrated in Fig. 1 a. Unresolved or missing atoms, including hydrogens, were added to the resulting structures using the CHARMM package (18). The assignment of histidine side-chain protonation states was performed based upon hydrogen bonding patterns.
TABLE 1.
Proteins on which simulations were performed in this study
| Simulation name | PDB accession code for starting structure* | Sequence | 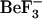 |
Tyr-106 rotamer | Starting conformation |
|---|---|---|---|---|---|
| 1fqwEc_Ec | 1fqw (E. coli) | E. coli | Removed | t | Active |
| 1fqwEc_St | 1fqw (E. coli) | S. typhimurium | Removed | t | Active |
| 2cheSt_St | 2che (S. typhimurium) | S. typhimurium | None | g+ | Inactive |
| 2cheSt_Ec | 2che (S. typhimurium) | E. coli | None | g+ | Inactive |
To relax steric overlaps and optimize the hydrogen atom positions, each structure was minimized for 1500 cycles using heavy atom harmonic position restraints of 30.0 kcal mol−1 Å−2, decreasing by 10 kcal mol−1 Å−2 after every 500 cycles of minimization utilizing the CHARMM force field and PARAM22 parameter set (19). The resulting systems were solvated, using the solvate command within the VMD package (20), in a box of TIP3P water molecules (21). The box size was defined by a minimum distance cutoff of 12 Å from the wall of the box to the nearest solute atom. The solvated systems were then neutralized by adding sodium ions (2 Na+ ions in each system) using the autoionize command within VMD. The protein structure resulting from the system preparation stage is referred to as the “reference structure”, which typically exhibited a 0.042–0.086 Å all-atom root mean square deviation (RMSD) from the initial crystal structures and is used throughout this work for comparison to structures obtained from the simulation.
Equilibration simulations
The four systems resulting from the preparation steps were minimized for 100 cycles and then subjected to 224 ps of MD simulation using Berendsen pressure regulation with isotropic position scaling (22). During these MD simulations, the temperature was reassigned from a Boltzmann distribution every 1000 cycles, in 25 K increments, from an initial temperature of 0 K to a target temperature of 300 K. To utilize a 2.0-fs integration time step, the SHAKE algorithm was utilized for all bonds containing hydrogen atoms (23,24). The charge interactions were dealt with using the particle mesh Ewald method, utilizing ∼1 Å resolution grid (25). All equilibration calculations were performed within the NAMD package (26), utilizing the CHARMM 22 parameter set (19).
Production simulations
Following equilibration, production simulations were run for 10 ns on each system, under approximate NVE conditions (constant number of atoms (N), constant volume of system (V), and approximate energy conservation (E) due to integration and nonbonded cutoff truncation), utilizing the particle mesh Ewald method for the treatment of electrostatic interactions. The initial coordinates, velocities, and system dimensions were taken from the final state of the corresponding equilibration simulations. All calculations were performed within the NAMD package (26), utilizing the CHARMM22 parameter set (19).
Calculations for structural analysis
In-house software was developed to accomplish each of the following analyses.
Root mean squared deviation in atomic positions. Calculating the RMSD between atomic positions for two protein conformers involves an initial superposition step to optimally align the structures, and a further RMSD calculation for the subset of atoms of interest. Throughout this study, we have used software developed in-house to align (based upon the quarternion method; (27)) and calculate the RMSD of atomic positions. Structural superposition was performed for all protein atoms and then subset RMSDs (such as the Cα RMSD) were calculated based upon this alignment. In each simulation, the reference structure used for the calculation of positional RMSDs was the final structure obtained from the system preparation stage.
Root mean squared deviation in contact distances (Rd). One of the problems associated with calculating a positional RMSD is the error associated with performing the initial superposition. If calculations are performed in contact space, as is the case with Rd (utilizing the distances between atoms), superposition is not required, thus eliminating this source of error. The following equation was used to calculate Rd:
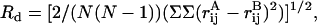 |
where N is the number of atoms and 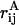 is the interatomic distance between atoms i and j in structure A, the outer sum being over all atoms in the protein and the inner sum over all atoms before that in the outer sum (28). In this study, software developed in-house was used to calculate the Cα atom Rd for the structures generated during the simulations, relative to the reference structure obtained from the system preparation phase.
is the interatomic distance between atoms i and j in structure A, the outer sum being over all atoms in the protein and the inner sum over all atoms before that in the outer sum (28). In this study, software developed in-house was used to calculate the Cα atom Rd for the structures generated during the simulations, relative to the reference structure obtained from the system preparation phase.
Radius of gyration (Rg). This measure provides an indication of changes in the “compactness” of the protein during simulations:
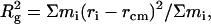 |
where mi is the mass of atom i, ri is the Cartesian position vector of atom, and rcm is the center of mass of the molecule and the sum is over all protein atoms. A decrease in Rg during a simulation has been interpreted as indicative of an increase in the packing density of the protein.
Calculated Cα B-factors. The thermal parameter (B) is related to the magnitude of atomic fluctuations by:
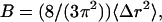 |
where Δr2 is the mean-square radial displacement (Δx2 + Δy2 + Δz2), and 〈 〉 denotes a time and space average from the mean structure. The B-factors were obtained first by calculating the average structure for the simulation trajectory (utilizing an all-atom RMSD alignment to the reference structure, then taking the mean coordinate of each atom), followed by the calculation of 〈Δr2〉 from this “mean” conformation. We report only the B-factors for the Cα atoms, which can be compared directly to those obtained from x-ray crystallography (29), allowing us to compare backbone flexibility of the protein from the simulations to those observed from crystallography.
Correlated motions. Coupling between the atomic displacements in protein simulation trajectories can be investigated by examining the correlation of the displacement of the residue atom centroids (30):
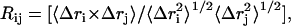 |
where Δri is the instantaneous displacement of the geometric center of the atoms of residue i from its mean position and 〈 〉 denotes a time average. The matrix obtained contains correlation coefficients with values between −1.0 and +1.0, indicating the degree and manner to which the fluctuations in the positions of residue i and j are coupled. A correlation coefficient approaching 1.0 indicates that the fluctuations in position of residue i and j are strongly coupled, and that they move in a similar fashion. A correlation between ∼−0.3 and ∼+0.3 indicates little coupling between the motions of residues, whereas a correlation approaching −1.0 indicates that motion is strongly correlated between the residues, but that the fluctuations in positions are in opposite directions.
Residue-residue Cα variance matrix. The residue-residue Cα variance matrix provides information about the variation in Cα interatomic distances (contacts) during simulations:
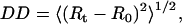 |
where 〈 〉 denotes a time average, and the subscript t and 0 denote the distance matrix at times t and 0, respectively. During this study the reference Cα contact matrix (R0) was calculated using the reference structure resulting from the system preparation stage. Flexible regions of the protein are clearly indicated as regions with higher variances in their contacts.
Secondary structure. Secondary structure changes throughout the simulation were monitored by using DSSP (31) to define the secondary structure of the conformations generated throughout the simulations.
Relative solvent accessible surface area. The solvent accessible surface area for each residue and its components were monitored throughout the simulations using the NACCESS package (32,33). The relative solvent accessibility (calculated relative to maximum solvent accessibility of residue X in the Ala-X-Ala trimer in extended conformation) (34) for the residue-backbone and residue-side chains were calculated.
Cluster analysis. To determine the most occupied conformations during the time course of these simulations, clustering was performed. The members of each cluster and the conformation that best represents those in the cluster were output for visual analysis. The clustering was performed using the RMSD values calculated across all residues and RMSD values calculated across only helix-4 residues. Utilizing RMSDs of specific residue sets for clustering allows us to focus on specific structural differences in these regions. Conformations obtained during the simulations were partitioned into differing numbers of clusters using average link clustering (35). The number of clusters that resulted in a minimum in the Pi value (a measure of cluster compactness) (36) was then used to identify the clusters. The conformation best representing the conformations in a given cluster was obtained by finding the cluster member possessing the minimum RMSD from all other members of that cluster. This method has an advantage over averaging the coordinates of the conformations in a cluster; representative conformations obtained by this method should always be physically reasonable.
RESULTS
Simulations were initiated from both the active (1fqw) and inactive (2che) structures. In those simulations starting from the 1fqw (active) structure, 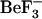 was removed before starting the simulations. Thus, simulations initiated from the inactive structure explore the ensemble of conformations available to the inactive form of CheY (on the nanosecond timescale), while simulations starting from the active structure explore the ensemble of structures available to the active form of CheY and the initial conformations involved in the relaxation from active to inactive form. This relaxation process is long with respect to the simulations in these studies. Many features that characterize the active form are not lost during our 10-ns simulations (Fig. 2); however, we are sampling the initial steps in the relaxation process and we can compare dynamics and correlated motions observed in the two ensembles. Specific aspartyl phosphate parameters are not available and require quantum mechanical calculations to determine correctly. Our initial calculations suggest that the aspartyl phosphate group in proteins has a much weaker C-O-P bond than in isolation; thus, we chose not to perform simulations on the active conformation using phosphate parameters “borrowed” from other phosphate complexes.
was removed before starting the simulations. Thus, simulations initiated from the inactive structure explore the ensemble of conformations available to the inactive form of CheY (on the nanosecond timescale), while simulations starting from the active structure explore the ensemble of structures available to the active form of CheY and the initial conformations involved in the relaxation from active to inactive form. This relaxation process is long with respect to the simulations in these studies. Many features that characterize the active form are not lost during our 10-ns simulations (Fig. 2); however, we are sampling the initial steps in the relaxation process and we can compare dynamics and correlated motions observed in the two ensembles. Specific aspartyl phosphate parameters are not available and require quantum mechanical calculations to determine correctly. Our initial calculations suggest that the aspartyl phosphate group in proteins has a much weaker C-O-P bond than in isolation; thus, we chose not to perform simulations on the active conformation using phosphate parameters “borrowed” from other phosphate complexes.
FIGURE 2.

RMSD plots during the course of the simulation indicate simulation stability and times of minor conformational fluctuations. All-atom (black) and Cα (dark gray) root mean-squared position deviation (RMSD) and the RMSD in Cα distances (Rd, light gray), plotted during the time course of each simulation of: (a) 1fqwEc_Ec; (b) 2cheSt_Ec; (c) 1fqwEc_St; and (d) 2cheSt_St. In all cases, the RMSD is calculated versus the reference structure resulting from the system preparation stage (see Methods). Regions noted in the text are annotated by black arrows (maxima) and a dashed line (plateau).
1fqw and 2che crystal structures originate from two organisms and are thus not identical in sequence. To distinguish between conformational changes resulting from the removal of 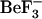 and those resulting from differences in sequence, we performed four MD simulations (Table 1); two of these are based upon both wild-type protein sequences and two involve modifications to the protein sequences. Such sequence modifications are denoted by a simulation name with either a “_Ec” or a “_St” suffix. The prefix for each simulation name can be 1fqwEc or 2cheSt, indicating the starting structure used and the organism from which the sequence was obtained. For example, the simulation that started from the 1fqw crystal structure (active) using the E. coli sequence is 1fqwEc_Ec, whereas the simulation starting with the 1fqw structure, but sequence mutated to the S. typhimurium sequence, is denoted as 1fqwEc_St.
and those resulting from differences in sequence, we performed four MD simulations (Table 1); two of these are based upon both wild-type protein sequences and two involve modifications to the protein sequences. Such sequence modifications are denoted by a simulation name with either a “_Ec” or a “_St” suffix. The prefix for each simulation name can be 1fqwEc or 2cheSt, indicating the starting structure used and the organism from which the sequence was obtained. For example, the simulation that started from the 1fqw crystal structure (active) using the E. coli sequence is 1fqwEc_Ec, whereas the simulation starting with the 1fqw structure, but sequence mutated to the S. typhimurium sequence, is denoted as 1fqwEc_St.
Conformational variability indicates simulations are stable
The progress of the simulations was monitored by calculating the RMSD in atomic positions between each trajectory structure and the reference structure (defined in Methods). This RMSD oscillates around 1.8–2.0 Å and 1.2–1.4 Å (for all atom and Cα RMSD, respectively) (Fig. 2), indicating that the simulations sample conformations which, overall, are similar to the reference structures and that the simulations are stable, at least over the 10-ns period of the production runs reported here. Mean-squared deviation in contact distances (Rd) was also calculated as a measure of overall conformational change during the simulations. Common maxima are frequently observed in both plots of Cα RMSD and the Rd versus time (Fig. 2), indicating that the changes occurring at these times involve the protein's backbone conformation. In the Rd plot for the 1fqwEc_St simulation, there is a pronounced elevated plateau from 2.0 to 3.5 ns (dashed line, Fig. 2 c), which is not observed in the 1fqwEc_Ec simulation, but which correlates with an increase in the local deviation in Cα position during this time period for residues 107–115 (C-terminus of sheet-5 to N-terminus of helix-5) (data not shown). There is a small backbone conformational change around these residues in this simulation that is not sampled in the other simulations. Structures observed once could indicate actual conformations sampled less often with respect to the timescale of the simulation, but we cannot interpret the biological significance of a conformational change observed only once during our simulations.
The radius of gyration calculations (data not shown) indicate very slightly higher variability in Rg for the simulations based on the active structures (1fqw-initiated) relative to the inactive (2che-initiated) ones. The Rg for 1fqwEc_Ec and 1fqwEc_St varies from 13.6 to 14.1 Å, whereas for 2cheSt_St and 2cheSt_Ec it varies from 13.6 to 13.9 Å. This very slight (but repeatable) variation is expected as the active conformation adjusts to the absence of the 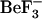 phosphate mimic during the 1fqw-based simulations initiated from the active conformation. Overall, the Rg plots indicate that the conformations sampled in these four simulations occupy similar volumes in space and that there is little variation in these volumes. This result again suggests that the simulations are stable and behaving as expected for globular proteins. In addition, the ensemble derived in the absence of
phosphate mimic during the 1fqw-based simulations initiated from the active conformation. Overall, the Rg plots indicate that the conformations sampled in these four simulations occupy similar volumes in space and that there is little variation in these volumes. This result again suggests that the simulations are stable and behaving as expected for globular proteins. In addition, the ensemble derived in the absence of 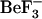 provides an appropriate representation of the active ensemble and the very first steps in relaxation.
provides an appropriate representation of the active ensemble and the very first steps in relaxation.
The protein secondary structure calculated throughout the simulations indicates that the overwhelming majority of regular secondary structure remains intact throughout the 10-ns production trajectories (data not shown), further indicating that the simulations are stable. Where there are changes in the secondary structure, these are typically in regions of the protein at the ends of regular secondary structures, adjacent to loops. All of the simulations show fraying at the C-termini of helices 2, 3, and 5. In addition, the 2cheSt_St simulation exhibits significant fraying at the N-terminus of helix-4.
Comparison of experimental and calculated B-factors show that differences in starting structure and starting sequence affect flexibility in different regions
Comparison of B-factors from simulations to those obtained from crystallography allows us to compare the local dynamics to structural diversity in the crystal structure (Fig. 3). The CheY crystal structures from S. typhimurium and E. coli exhibit B-factor peaks at similar regions of the protein, namely at residues: 14, 26, 31, 47, 76, and 89 (S. typhimurium) and 15, 26, 31, 46, 63, 76, 91, and 93 (E. coli). All of our simulations show B-factor peaks around residues 16, 31, 48, 63, 76, and 89, which correspond to residues that are in loop regions or at helix termini. Comparison of these residues indicates general consistency between observations in the crystal and fluctuations observed during the MD simulation.
FIGURE 3.

Calculated and experimental Cα atom B-factors indicate the backbone relative mobility of specific residues in the crystal structures and simulations. Overall, the calculated B-factors (blue and green traces) compare well with those observed in the crystal structures (red and black traces). Important differences between the simulations initiated from the active structure (a) compared with the inactive structure (b) are identified by arrows. Specifically, higher B-factors observed for residues 45–50 and 85–95 in the simulations utilizing the S. typhimurium sequence (bold arrows) and for residues 76–79 and 97–102 in the simulations initiated from the inactive structure (dotted arrows). DSSP-derived secondary structure assignment for the reference structure is annotated by the colored blocks above each plot (red = α-helix, yellow = extended β-strand, blue = bend). Ribbon representations of the active (1fqw, c) and inactive (2che, d) crystal structures, showing residues involved in sequence-based differences in B-factors (dark blue ribbon) and initial structure-based differences (red ribbon). The locations of the three amino acid differences between the two structures are indicated by cyan van der Waals spheres. The locations of Asn-31, which is flexible in both crystal structures and simulations; Met-63, which is in a region that is flexible in all simulations; and Ala-97, which is in a region that is flexible in simulations initiated from the inactive conformation, are labeled and shown by colored spheres. Panels c and d were generated in VMD v1.8.3 (20).
Two differences between simulations and crystal B-factors should be noted. In all of our simulations, high B-factors are observed around residue 63 (magenta side chain, Fig. 3, c and d). In the CheY crystal structures, a peak in the B-factors at this residue is only observed in the active, but not the inactive, form. Met-63 is found in the loop between strand-3 and helix-3 (magenta residue, Fig. 3, c and d), six residues beyond the modifiable Asp-57. The simulations suggest that the region of this loop around Met-63 is flexible in all conformations, which is not apparent from the crystal structure B-factor comparison. In the second observed difference, the CheY simulation initiated from the active structure with the E. coli sequence (1fqwEc_Ec) shows larger B-factors around residue 31 (orange residue, Fig. 3, c and d) than those in the other simulations, indicating that significant changes in backbone conformation are occurring in the loop between helix-1 and strand-2 during this simulation. This large B-factor observed only in one simulation suggests conformational flexibility in this region that might be sampled only rarely.
Flexibility differences correlating with initial structure at residues 76–79 and 97
Comparison of B-factors across simulations indicates two specific B-factor effects, the first around residue 97 and the second at residues 76–79, that correlate with the identity of the starting structure (dotted arrows, Fig. 3, a and b). The flexibility around Ala-97 (in helix-4) is slightly increased in the simulations initiated from the inactive structure (2che) compared to those initiated from the active structure. Ala-97 is part of a group of residues in helix-4 that form the cavity into which the Tyr-106 side chain is buried in the active protein conformation (Fig. 3, c and d, red backbone). The additional flexibility we observe around Ala-97 is likely due to lower packing density that exists because of the cavity formed when Tyr-106 is solvent accessible (and in the g+ rotamer) in the inactive structure (see black conformation for Tyr-106 side chain in Fig. 1 c). Data showing a large NMR chemical shift induced by 129Xe binding in apo- (inactive) CheY relative to phosphorylated CheY is indicative of a hydrophobic cavity between helix-4 and strand-5 (37), which is consistent with the presence of an actual cavity in the inactive structure in solution. Furthermore, NMR hydrogen/deuterium exchange data show lack of amide protection for most helix-4 residues in inactive CheY, indicating that this region is flexible (38), likely because of the cavity. Thus, observation of higher B-factors for Ala-97 in the MD simulations initiated from the inactive structure is consistent with these experimental data.
Higher B-factors are also observed for residues 76–79 (loop between helix-3 and strand-4; Fig. 3, c and d, red backbone) in the simulations initiated from the inactive structure. This region has been noted by Cho and colleagues (14) to be ill-defined in the NMR structure of the inactive form of CheY, consistent with the simulation results. Our simulations show that this flexibility is only observed in the inactive ensemble, and not in the ensemble of structures from the simulation of the active conformation. We note that residues 76–79 are not near the Tyr-106 cavity in the structure, the cavity and the loop being ∼20 Å apart (red backbone to red backbone, Fig. 3, c and d). Because we observe the flexibility only in simulations started from the inactive structure, the Tyr-106 cavity is the likely explanation; consequently, the flexibility must be communicated to the helix-3-strand-4 loop through long-range interactions. These simulations clearly show that sequence changes (Ser-76 in S. typhimurium and Gly-76 in E. coli, cyan residues in Fig. 3, c and d) in this loop have little impact on loop flexibility. The main determinant of loop flexibility is a structural difference between the active and inactive forms of CheY. Our simulations indicate that the cavity not filled by Tyr-106 in the inactive conformation allows increased flexibility specifically in adjacent residues, around Ala-97, and distant residues, 76–79.
Flexibility differences correlating with protein sequence at residues 45–52 and 85–92
On the other hand, differences in the S. typhimurium (2che) and E. coli (1fqw) sequences, and not the starting conformations, are responsible for some of the mobility differences observed in our simulations (solid arrows, Fig. 3). In simulations utilizing the S. typhimurium sequence, the B-factors between residues 45–52 and 85–92 (helix-2 and following loop and strand-4 to helix-4 loop, respectively; Fig. 3, c and d, blue ribbons) are larger relative to the equivalent simulations using the E. coli sequence (solid arrows, Fig. 3). One of these regions, at residues 45–52, encompasses a sequence difference: residue 51 is a phenylalanine in the S. typhimurium sequence and a tyrosine in the E. coli sequence. The results of several simulations indicate that sequence differences are the cause of the flexibility difference; however, the structural explanation for this flexibility difference is not obvious—the single difference at position 51, the hydroxyl group in Tyr-51 in the E. coli sequence, does not appear to be involved in a hydrogen bond with another atom in the E. coli crystal structure, nor during the simulations.
The other region exhibiting increased B-factors due to sequence difference is residues 85–92, the strand-4 to helix-4 loop, whose conformation has been shown to be critical in the function of this protein (8,14). This loop also exhibits increased B-factors in the simulations utilizing the S. typhimurium sequence. The difference in flexibility is seen in all simulations starting with the S. typhimurium sequence, so is likely a real feature and not a problem with limited sampling. This observation is difficult to explain from sequence differences: it is distant from the sites of sequence variation between S. typhimurium and E. coli (Fig. 3, c and d, compare blue ribbons as regions of flexibility differences and cyan side chains as sites of sequence differences). Sequence differences would have to affect the flexibility of this region through long-range interactions. With larger fluctuations also observed at residues 45–52 in the same simulations, it is logical to conclude that these two distantly located sites are involved in some form of long-range communication. Observations such as these, with increased fluctuations in two distinct regions, provide interesting starting points for studying the mechanism of long-range communication in proteins. In addition, the species-specific differences in the flexibility of the important strand-4-helix-4 loop may subtly affect basal CheY activity in these organisms.
Overall, B-factor comparisons indicate that our simulations agree quite well with experimental data. The simulations identify specific differences in flexibility that correlate with starting structure and others that correlate with sequence-based differences between E. coli and S. typhimurium CheY proteins. Both the structure-based and sequence-based differences result in dynamic differences in areas of the protein both encompassing, and distant from, the location of the specific structural or sequence variations, indicating the presence of long-range interactions distinct from the sites of phosphorylation and binding, throughout this protein.
Variability of contact distance identifies distinct conformational changes that are specific to the initial structure
To investigate the variability in the tertiary contact distances, the α-carbon distance variance for every Cα pair compared to its distance in the starting reference structure was calculated (Fig. 4). Note that this calculation is between α-carbons, so a deviation indicates a shift in backbone, not just side chains, observed during the simulation. As expected, there is little deviation, less than 1 Å, for between most α-carbons in all simulations (dark blue regions, Fig. 4, a and b). Also as expected, there is little variability in the Cα contacts for three of the metal-binding residues (Asp-13, Asp-57, and Asn-59), with Thr-87 being the single exception. The involvement of Thr-87 in the first steps of relaxation is not surprising; it is a key residue involved in phosphate binding and exhibits a different side-chain location in the active and inactive structures (Fig. 1 c).
FIGURE 4.

Mean deviation of Cα interresidue distances with ribbon drawings indicating regions of largest mean deviations from the different starting sequence and structure combinations. Simulations initiated from panel a the E. coli sequence (1fqwEc_Ec, above diagonal; 2cheSt_Ec, below diagonal) and panel b the S. typhimurium sequence (2cheSt_St, above diagonal; 1fqwEc_St, below diagonal) (dark blue = 0.0 Å, bright red ≥ 4.0 Å). Large variations for the distances involving Met-85, Thr-87, and Glu-89, components of the strand-4-to-helix-4 loop, are observed in most simulations (white rectangles), as discussed in the text. (c, d, e, and f) CheY ribbon structures using color to indicate regions that exhibit large deviations that are indicated by the data in panels a and b. To provide orientation Tyr-106 and Thr-87 are shown as blue side chains and Asp-57 is represented by green van der Waals spheres in all structures in (c–f). The strand-4-to-helix-4 loop, site of the largest changes is colored cyan in panel c, with the Met-85 and Glu-89 side chains colored orange. In simulations from the inactive (2che) structure, deviations with Glu-89 are localized to three regions indicated by white circles on both graphs and located around residues 12, 36, and 59, the black van der Waals spheres in panel c. To indicate regions of largest structural deviation, the colored ribbons in panels d, e, and f correlate with the white, pink, orange, and yellow ellipses in the graphs. The most significant deviation in simulations initiated from the active structure is between residues 21 and 32 and 106–113 (pink ellipses in both graphs; pink ribbons in d). There are also variations in the Cα contacts for helix-1 with helices 3, 4 and 5 for the simulations starting from the active conformation (orange ellipses (a and b); orange and red ribbons in e). In the simulations initiated from the inactive structure, changes in contacts between helix-1 and helices-2 and -3 (yellow ellipses in a and b; red and yellow ribbons in f). DSSP-derived secondary structure assignment for the reference structure is annotated by the colored blocks above and to the right of the plots (red = α-helix, yellow = extended β-strand, blue = bend).
Conformations at the strand-4 to helix-4 loop are specific to the initial structure
The most significant observation from these mean deviation plots is the distinction between simulations starting from the active and inactive conformations. We focus first on the changes in the strand-4 to helix-4 loop, residues 85–92, a key loop in the protein's function (8,14) and the site of some of the largest conformational changes observed in these simulations. At first glance, it appears that this loop changes its Cα contacts considerably with many of the residues in the protein, particularly in three of the four simulations (1fqwEc_Ec, 1fqwEc_St, and 2cheSt_St; Fig. 4, white rectangles). A more detailed analysis indicates specific differences based on starting structure. In simulations from the active structure, Met-85 and Thr-87 α-carbons exhibit large changes in distance from many residues of the protein (1fqwEc_Ec, Fig. 4 a, above diagonal; 1fqwEc_St, Fig. 4 b, below diagonal) indicating that this part of the loop (containing these two residues) is moving with respect to the rest of the structure. Thus, the initial relaxation steps in the region upon phosphate mimic removal specifically occur around residues 85 and 87, at the C-terminus of strand-4 and the N-terminus of this loop (side chains shown in Fig. 4 c). This is consistent with NMR evidence showing that significant structural changes occur in CheY upon phosphorylation. Large chemical shift changes are observed for a number of residues, including Met-85, Thr-87, Val-107, and Lys-109 (37,39).
In contrast, the loop conformations explored by the protein in the inactive structure are smaller and are focused around Glu-89 (white circles in both Fig. 4 a, below diagonal, and Fig. 4 b, above diagonal). These deviations are most readily observed in the 2cheSt_St simulation; the 2cheSt_Ec simulation shows smaller changes in this region, but the pattern is similar. In contrast to the observations of Met-85 and Thr-87, where the deviation of Cα distances is large across many residues in the simulations initiated from the active structure, Glu-89 exhibits Cα deviations with three groups of residues located around Asp-12, Ala-36, and Asn-59 (Fig. 4 c, black side chains). These residues are located in loops on the “front face” of the molecule, as viewed from the orientation in Figs. 1 a and 4 c. The origin of the specific deviation between these residues in the inactive conformation is not readily apparent from observation of the structure.
Thus, these deviation calculations indicate different movement in the strand-4 to helix-4 loop that is specific to the starting conformation; furthermore, the identification of distinct interactions during the equilibrium simulation of the inactive structure suggests long-range interactions between the active site loop and the other loops that are located across this face of the protein.
Conformational changes specific to the initial structure in regions outside the strand-4-helix-4 loop
Cα mean deviation in interresidue distances during the simulations indicates that there are variations in tertiary structure outside of the strand-4-helix-4 loop that are also dependent on the initial conformations (compare ellipses in Fig. 4 above diagonal (a) to below diagonal (b), and vice versa). In both simulations initiated from the active conformation, the largest change in Cα interresidue distances is observed between residues 21 and 32 and 106–113, which corresponds to a variation of contacts between the C-terminal half of helix-1 and the following loop and strand-5 and the following loop (which contains Tyr-106 and Lys-109) (Fig. 4, a and b, pink ellipses, and Fig. 4 d). Large chemical shift changes observed by NMR experiments for Val-107 and Lys-109 upon removal of the phosphate are consistent with these data (37,39). In our simulations, large variation in distances is also observed for three regions: 1), residues 65–74; 2), residues 94–98; and 3), residues 115–124, all of which show variation with residues 15–23, the N-terminal half of helix-1 (Fig. 4, a and b, orange ellipses, and Fig. 4 e, orange and red-colored ribbons). Only those simulations starting from the active conformation show these large deviations in interresidue distances between α-carbons. Notably, these sets of residues are located on opposite sides of the phosphate binding pocket (Fig. 4 e), suggesting that the first step in the relaxation of the structure upon removal of the phosphate is a slight “collapsing” of the helices on all sides of the site of phosphorylation. It is interesting to observe that the largest change in this initial step occurs mostly in the helical regions, suggesting that some of the loops are allowing the helices to collapse toward the phosphate binding pocket, but are not moving themselves.
In simulations starting from the inactive structure, large variation is seen with helix-1 (about residues 19–30) (Fig. 4 a, below diagonal; Fig. 4 b, above diagonal, yellow ellipses). Contacts between helix-1 and helix-2 (2cheSt_Ec) and between helix-1 and helix-3 (2cheSt_St and 2cheSt_Ec) are observed to vary (Fig. 4 f; yellow and red-colored ribbons).
It is useful to compare the regions that exhibit deviations based on the starting structure (compare Fig. 4, d and e, to 4, c and f). In all cases, changes in the interresidue distances between residues outside of the strand-4-helix-4 loop are mostly observed between the helices. Helix-1 appears to be a lynchpin, because deviations in all simulations are observed with respect to this structure (red helix in Fig. 4, e and f). In the simulations from the inactive structure, the variations are observed between helix-1 and helices-2 and 3; whereas in simulations from the active structures, variations are observed between helix-1 and helices-3, 4, and 5. Globally, this suggests that the helices are moving with respect to each other, which would imply that the loops are acting as hinges. The observation of increase flexibility in the loop regions (Fig. 3), but larger contact variances between the helical regions (Fig. 4), would support this suggestion. It is interesting to note that the strand-4-helix-4 loop shows deviations with residues in these loops on the front face of the molecule (Fig. 4 c), which implies that, if the loops are acting as hinges, these motions are affected by the conformation of the important strand-4-helix-4 loop, indicating the importance of loop-loop communication in CheY.
Identification of correlated motions specific to sequence or to structure
Analysis of correlated motion can provide information about the regions of the protein that move in a collective manner during a simulation. Several regions of highly positively correlated motion are observed in all simulations (black circles, Fig. 5). As would be expected, many of these correspond to the coupling of motions between the strands forming the β-sheet. The strongest positively correlated motions are between adjacent strands of the sheet: strand 1 with strands 2 and 3, strand 3 with strand 4, and strand 4 with strand 5. Because these correlated motions are common to all simulations they are not specific to the starting structure or sequence. The strong positively correlated motion observed in all simulations between strands 4 and 5 includes Tyr-106 and Thr-87, which is in agreement with the results of NMR spectroscopy (9,14), crystallography of mutant CheY (16,40), and mutant FliM binding experiments (15,41). Motions of these important regions should be strongly correlated because of their key role in CheY function.
FIGURE 5.

Correlated motions of amino acid centroids during the simulations demonstrates the importance of the burial of the Tyr-106 side chain for communicating those motions. Simulations initiated from (a) the E. coli sequence (1fqwEc_Ec, above diagonal; 2cheSt_Ec, below diagonal), and (b) the S. typhimurium sequence (2cheSt_St, above diagonal; 1fqwEc_St, below diagonal), (dark blue ≤ −0.6, red ≥ 0.6). Positively correlated motion between regular secondary structures is indicated by black circles. Positively correlated motion that is structure-dependent is indicated by white circles, whereas correlated motion that is dependent on sequence is marked by red circles. Sequence-dependent correlated motion is more positively correlated in simulations utilizing the S. typhimurium sequence and these regions are indicated by the magenta ribbons in panel c. In panel c, positions of residues that differ between E. coli and S. typhimurium are shown as blue space fill (F51Y, I54V and S76G, S. typhimurium to E. coli). Structure-dependent correlated motion is centered at residue 94 (helix-4) and Tyr-106, as indicated by the red ribbon (helix-4) and yellow van der Waals spheres (Tyr-106) in the active conformation depicted in panel d. In both panels c and d, Thr-87 is a blue side chain. DSSP-derived secondary structure assignment for the reference structure is annotated by the colored blocks above and to the right of the plots (red = α-helix, yellow = extended β-strand, blue = bend).
Sequence-specific correlated motion is observed between helix-2 with the loop between strand-3 and helix-3
There is positively correlated motion present in the two simulations utilizing the S. typhimurium sequence (Fig. 5 b, red circles), which is less correlated in the simulations of the E. coli sequence (Fig. 5 a, red circles). This region corresponds to a coupling of the motions of the N-terminus of helix-2 with the loop between strand-3 and helix-3 (Fig. 5 c, pink ribbons). The strand-3- helix-3 loop contacts the N-terminus of helix-2 in the structure (Figs. 1 a and 5 c). We already noted increased B-factors in the S. typhimurium simulations in two nonadjacent regions in structure: residues 45–52 (helix-2 C-terminus and following loop) and 85–92 (strand-4-to-helix-4 loop) (solid arrows, Fig. 3, a and b, and blue ribbons in Fig. 3, c and d). Analysis of the structure suggests that, if Phe-51 causes increased B-factors in helix-2 in the S. typhimurium simulations, this could cause the correlated motion between helix-2 and the loop between strand-3 and helix-3. Further, these observations suggest a possible path of long-range communication in CheY: from residue 51, to helix 2, to the loop between strand-3 and helix-3 to the active site loop. We can actually observe the path in the S. typhimurium protein because of the increased motion that we attribute to phenylalanine at position 51, rather than the tyrosine found in the E. coli protein. An observation such as this, which appears to be attributable to a sequence difference, suggests mutagenesis as a method for dissecting long-range communications in proteins using MD simulations. Introducing mutations to alter flexibility, without changing the packing, might be used to dissect long-range interactions.
Structure-specific correlated motion, observed between helix-4 and strand-5, is dependent on the Tyr-106 rotamer form
One region of correlated motion is related to the conformation of the starting structure: positively correlated motion between residues 92 and 99 (helix-4) and 105–108 (strand-5) is only observed in the simulations initiated from the active CheY structure (1fqwEc_Ec and 1fqwEc_St) (Fig. 5, a and b, white circles; Fig. 5 d, red ribbon). These two regular secondary structures are adjacent in structure and we would expect that their motions would be positively correlated in all simulations, but they are not. The difference in correlated motion is seen even more clearly in Fig. 6, a and b (arrows). The key functional residue, Tyr-106, is located in the middle of strand-5 (Figs. 1 b and 5 d). The side chain of this residue is buried in the protein in the active structure (compare black and blue side chains, Fig. 1 c, and yellow van der Waals spheres in Fig. 5, c and d). As the Tyr-106 side chain is in the t (trans) χ-1 rotamer and is buried between helix-4 and strand-5 in the simulations initiated from the active structure, the correlated motion in these simulations is apparently the direct result of packing interactions between helix-4 and Tyr-106. In the simulations initiated from the inactive structure, the Tyr-106 χ-1 angle starts from the g+ rotamer, and does not fill the cavity between helix-4 and strand-5. This set of observations strongly suggests that packing and cavities, even small cavities, within protein structures are crucial for facilitating (or not) communication between secondary structures, communication that results in observable correlated motions.
FIGURE 6.

Correlated motion between Tyr-106 with the strand-4-helix-4 loop is only observed in simulations initiated from the active conformation. Correlated motion between Tyr-106 and each residue in the protein during simulations initiated from the active (1fqw) and inactive structures (2che) are shown as black and gray traces, respectively. The significant difference in Tyr-106 correlated motion with the strand-4-helix-4 loop, which depends on starting conformation, is indicated by arrows.
Clustering analysis suggests that helix-4 orientation is linked to burial and χ-1 rotamer state of Tyr-106
Clustering can identify the major conformations sampled in an ensemble. Thus, all-atom clustering was performed on the whole protein (data not shown) and on helix-4 conformations. Clustering on the helix-4 residues was performed because correlated motion analyses indicated that the motions of Tyr-106 and helix-4 were strongly coupled in the simulations initiated from the active structure, but not in the simulations initiated from the inactive structure (Fig. 5, a and b, white circles). Thus, we wanted to observe how the starting structure affected the orientations that helix-4 sampled during each simulation.
The following observations are apparent from overlays of cluster representatives (not shown): 1), helix-4 conformations are more similar to each other in simulations initiated from the active conformation; 2), in simulations starting from the active conformation, the largest difference across the structures occurs at the end of strand-4 and the beginning of the strand-4-to-helix-4 loop (a segment that contains Thr-87); and 3), in both simulations starting from the inactive conformation, helix-4 shows more diverse orientations and its N-terminus exhibits some unraveling. These observations are consistent with the two different behaviors observed for strand-4-helix-4 loop by the other trajectory analyses, for instance in the distance variation observed for Met-85 and Thr-87 for simulations from the active conformation (Fig. 4).
The differences in sampled conformations are especially apparent when the two cluster representatives with the most distinct conformations from each trajectory are overlaid (Fig. 7). Here, again, in simulations initiated from the active conformation, the distinct changes in conformation of the loop around Thr-87 are observed (Fig. 7, a and c). When the loop conformation changes, Thr-87 is associated with a movement of Tyr-106, but in this case, neither side-chain dihedral rotamer changes (Fig. 7 c). In simulations starting from the inactive conformation, the unraveling of the N-terminus of helix-4 and changes in the conformation of the adjacent loop residues are observed (Fig. 7, b and d). Distinct g+ and g− rotamers (discussed subsequently) are observed for Tyr-106 and the helix unraveling is quite distinct in the cluster representatives for the 2cheSt_St simulation (red side chain and cyan helix, Fig. 7 d). These changes, in conjunction with the observation that Tyr-106 remains buried in the simulations initiated from the active structure, suggest that the burial of Tyr-106 in the active conformation of CheY favors a specific subset of helix-4 orientations that improve the correlated motion between strand-5 and helix-4 and increase the rate of CheY binding to the flagella proteins for active relative to inactive CheY.
FIGURE 7.

Ribbon representations of the two cluster representatives that exhibit the largest RMSD in Cα positions from each other show the different conformations sampled during the simulations. Cluster representatives were obtained from all-atom clustering of helix-4 for the simulations of: (a) 1fqwEc_Ec; (b) 2cheSt_Ec; (c) 1fqwEc_St; and (d) 2cheSt_St. Helix-4, Tyr-106, and Thr-87 are indicated by cyan ribbon, red side chain, blue side chain, respectively. Larger differences in the orientation and conformation of helix-4 in simulations initiated from the inactive structure, 2cheSt_St (b) and 2cheSt_Ec, is observed in panel d. The g− rotamer for Tyr-106 is observed for one cluster representative in panel d. The figures were generated in VMD v1.8.3 (20).
Two different mechanisms are observed for rotation of χ-1 dihedral angle of Tyr-106 from g+ and g− in simulations of the inactive conformation
Both the χ-1 dihedral angle and degree of burial of Tyr-106 have been implicated in the activation/deactivation of CheY: the χ-1 dihedral angle exists in the t rotamer and is buried in the active structure (1fqw) and exists in the g+ rotamer and is solvent-exposed in the inactive structure (2che) (Table 1; compare black and blue side chains, Fig. 1 c). To ascertain whether different rotameric forms were sampled during the simulations, we monitored the χ-1 dihedral angles of Tyr-106 during all four trajectories. Tyr-106 remains in the t (+/−180°) rotamer throughout the simulations initiated from the active CheY structure, while it sampled the g− and g+ rotamers in both simulations of the inactive conformation (Fig. 8, a and b). Previous x-ray crystallographic studies of apoCheY (PDB accession code 1jbe) have indicated that the g+ and t Tyr-106 rotamers are associated with the inactive and “meta-active” forms of CheY (42). The role of Thr-87 in the proposed Y-T coupling mechanism involves interactions with the loop between strand-4 and helix-4. The coupling of Tyr-106 to Thr-87 arises from changes in the conformation of the loop between strand-4 and helix-4 (8,14). Our simulations of the inactive conformation indicate that Tyr-106 regularly samples (on a 10-ns timescale) both g+ (60°) and g− (−60°) rotamers as part of the equilibrium ensemble, perhaps facilitating the transition to the active conformation. Similar sampling was observed for Phe-101 in simulations of the FixJ receiver domain (50).
FIGURE 8.

The chi-1 dihedral angles for Tyr-106 (a and b), Thr-87 (c and d), and Ile-95 (e and f) sampled during the course of the simulation show specific side-chain conformational changes. Data for simulations initiated from active (solid symbols) and inactive (shaded symbols) conformations are shown. In the 1fqwEc_Ec and 1fqwEc_St simulations the Tyr-106 χ-1 angle remains in the t rotamer, whereas in the 2cheSt_St and 2cheSt_Ec simulations it samples both the g+ and g− rotamers. The 2cheSt_St simulation shows a rotamer change for Thr-87 and no change in Ile-95 preceding the Tyr-106 rotamer flip, whereas the 2cheSt_Ec simulation shows the converse. The χ-1 dihedral angle is defined by atoms N, Cα, Cβ, and Cγ in each residue. In the static crystal structure of the active conformation, Tyr-106 is in the t (or trans) rotamer, with the dihedral angle at 180°. In the inactive conformation, Tyr-106 is in the g+ (or gauche +) rotamer, with the χ-1 dihedral angle at 60°. To observe a difference in side-chain rotamers, compare blue (active) and black (inactive) Tyr-106 side chains in Fig. 1 c.
If Tyr-106 sampling of the g− rotamer facilitates the process of activation, it is useful to understand what conformational changes are associated with that rotamer change. In both simulations initiated from the inactive conformation, Thr-87 is the focus of the initial changes. Specifically, the backbone containing Thr-87 changes conformation slightly, so that Thr-87 moves toward the Tyr-106 binding pocket. New hydrogen bonds are formed with the backbones of Glu-89 and Ala-88. The Thr-87 hydrogen bond with the Ala-88 backbone subsequently breaks and a new one forms with the side chain of Asn-94. Thereafter, the behavior of the two simulations initiated from the inactive conformations diverges, such that the g− rotamer of Tyr-106 is sampled under one of two conditions: 1), a rotamer change in the Thr-87 side chain (2cheSt_St); or 2), a rotamer change in the Ile-95 side chain (2cheSt_Ec). Both of the observed mechanisms ultimately result in the change from the g+ to the g− rotamer observed for Tyr-106 (Fig. 8).
In the 2cheSt_St simulation, Thr-87 undergoes a chi-1 rotamer flip from t to g− at ∼6 ns (Fig. 8 d) and a hydrogen bond is formed with the Met-85 backbone. Helix-4 moves away from the protein. As a consequence, the Tyr-106 g− rotamer form is accommodated without any change in the Ile-95 χ-1 rotamer (Fig. 8 f). While Tyr-106 is in the g− χ-1 rotamer, the χ-1 rotamer of Thr-87 rotates again, from the g− to the g+ form, resulting in breakage of the hydrogen bond to Met-85 and formation of a new hydrogen bond with the backbone of Val-107. Consequent breakage of Met-85 hydrogen bond is accompanied by Tyr-106 reverting to its g+ rotameric form (Fig. 8 b).
In contrast, the 2cheSt_Ec simulation shows a different mechanism preceding the Tyr-106 rotamer change from g+ to g−. The χ-1 rotamer of Thr-87 does not change; however, there is a change in the rotamer form of Ile-95 (Fig. 8 e), which accommodates the subsequent Tyr-106 rotamer flip. The Tyr-106 g− rotamer is only adopted when a hydrogen bond is formed between the side chain of Thr-87 and the backbone oxygen of Glu-89 (data not shown). Glu-89 stabilizes the t rotamer of Tyr-106 by forming a hydrogen bond between its backbone oxygen and the side chain of Tyr-106 (8).
We can compare these two different mechanisms to static structures observed by crystallography. X-ray crystallographic studies of the only inactive CheY structure in which Tyr-106 is in the g− rotameric form (PDB accession code 1chn) (44) indicate that in order for Tyr-106 to adopt the χ-1 g− rotamer, the conformation of the helix-4 N-terminus and the preceding loop must change significantly from those observed in the 1fqw and 2che crystal structures. One potentially important difference observed in the 1chn structure compared to the 1fqw and 2che structures involves the lack of the Thr-87 side-chain hydrogen bond to the backbone of Glu-89. Thus, a weak hydrogen bond between the side chain of Thr-87 and the backbone of Val-107 is formed in the 1chn crystal structure. This is facilitated in the 1chn structure by a change in the χ-1 rotamer of Thr-87, consistent with the changes in the Thr-87 side-chain rotamer that we observe in our simulations. The consistency of observations of both the simulations and the static crystal structure suggest that the simulations are sampling conformations that are precursors to the conformation observed in the 1chn structure.
Thus, the Tyr-106 side-chain rotation from g+ to g− occurs by different mechanisms in the two simulations of the inactive conformation. The origin of these differences could be related to the S. typhimurium/E. coli sequence differences, although the sequence differences are distant in structure from either Thr-87 or Ile-95 (Fig. 3). Consistent with this explanation is the observation of increased flexibility in the loop containing Phe-51 (which is a Tyr in E. coli) and the active site loop in both simulations starting from the S. typhimurium sequence (Fig. 3). Increased flexibility in the active site loop could accommodate the Thr-87 χ-1 rotamer flip, which precedes the Tyr-106 g+ to g− rotamer flip in 2cheSt_St simulation, without requiring the Ile-95 rotamer change, which is exactly what is observed. What is left unexplained is how alternative residues at position 51 in the helix-2-to-strand-3 loop could affect flexibility at the active site loop, which is on the opposite side of the protein. A second explanation for the observation of two mechanisms is that the sequence differences observed here are a coincidence of sampling and that different conformational changes involving key residues Thr-87 and Ile-95 may provide alternative mechanisms for the Tyr-106 rotamer change. In either case, sampling of the g+ and g− rotamers in both simulations initiated from the inactive conformation suggests that this sampling is part of the equilibrium behavior of inactive CheY and, thus, could be important in overcoming the energy barriers to activation. If so, then the observation of two mechanisms for rotamer sampling suggests specific residues that are important for the phosphorylation.
Ordering of initial events in the relaxation of CheY upon removal of phosphate
MD simulations can provide us with information on the structural ensembles sampled by a given protein and the ordering of events during these initial steps. To observe event ordering following removal of the phosphate mimic, the trajectories were visualized in VMD (20). The conformational changes involving sheet-4, the loop between sheet-4 and helix-4, helix-4, Thr-87 and Tyr-106 were monitored. In the active conformation, the Thr-87 side chain points toward the 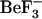 /phosphate binding site; its side-chain conformation starts in the t configuration and remains in that configuration during the simulation (solid symbols, Fig. 8, c and d). This conformation is initially stabilized by a hydrogen bond to the Asp-57 side chain. In the simulations starting from this active conformation, but with
/phosphate binding site; its side-chain conformation starts in the t configuration and remains in that configuration during the simulation (solid symbols, Fig. 8, c and d). This conformation is initially stabilized by a hydrogen bond to the Asp-57 side chain. In the simulations starting from this active conformation, but with 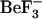 removed (1fqwEc_Ec and 1fqwEc_St), the Asp-57 hydrogen bond to Thr-87 breaks and Asp-57 forms a salt bridge with the side chain of Lys-109. In the absence of the Thr-87–Asp-57 hydrogen bond, the loop backbone containing Thr-87 moves toward the Tyr-106 binding pocket, leading to the formation of new hydrogen bonds to the side chain of Asn-94 and the backbone of Glu-89. Consequently, the hydrogen bond between the side chain of Tyr-106 and the backbone of Glu-89 breaks, and the N-terminus of the strand-4 and helix-4 loop further changes conformation. These steps, breakage of hydrogen bonds and movement of the N-terminus of the strand-4-helix-4 loop, appear to be the first steps in relaxation of the CheY structure.
removed (1fqwEc_Ec and 1fqwEc_St), the Asp-57 hydrogen bond to Thr-87 breaks and Asp-57 forms a salt bridge with the side chain of Lys-109. In the absence of the Thr-87–Asp-57 hydrogen bond, the loop backbone containing Thr-87 moves toward the Tyr-106 binding pocket, leading to the formation of new hydrogen bonds to the side chain of Asn-94 and the backbone of Glu-89. Consequently, the hydrogen bond between the side chain of Tyr-106 and the backbone of Glu-89 breaks, and the N-terminus of the strand-4 and helix-4 loop further changes conformation. These steps, breakage of hydrogen bonds and movement of the N-terminus of the strand-4-helix-4 loop, appear to be the first steps in relaxation of the CheY structure.
DISCUSSION
Activation of CheY, which occurs upon phosphorylation of Asp-57, involves the movement of Thr-87 to form a hydrogen bond with the phosphate group (through the Thr-87 side-chain hydroxyl moiety) and a conformational change of the sheet-4-helix-4 loop, resulting in formation of a cavity into which Tyr-106 can be buried (15). The burial of Tyr-106 results in the formation of a hydrogen bond between the backbone oxygen of Glu-89 and the side-chain hydroxyl of Tyr-106 (40). The direct involvement of Thr-87 and Tyr-106 gives this mechanism the moniker Y-T coupling mechanism (8,14). Despite extensive studies on the static structures and biochemical characteristics of this protein in phosphorylated forms, little is known about the long-range interactions or correlated motions that are exhibited by this protein. As a small, single-domain protein with typical allosteric behavior, analysis of the correlated motions in different states might allow us to understand how the protein structure contributes to the function of the protein and, in particular, how changes in sequence and structure affect the correlated motions within the molecule. To study these questions, we have completed 10-ns simulations on this protein, both in the inactive conformation and the active conformation with the phosphate removed. To distinguish sequence from structure effects, the simulations were performed both with the E. coli and S. typhimurium protein sequences in both states. Analysis of the simulation data indicates that our trajectory ensembles are consistent with experimental data and representative of the structures being analyzed. In addition, correlated motions and fluctuations specific to both sequence and starting structure were observed.
Conformational fluctuations of the strand- 4-helix-4 loop depend on starting structure and on the Tyr-106 rotamer
The conformation and flexibility of the strand-4-helix-4 loop (which is known to be important for CheY activation, but for reasons not fully understood) is dependent on the starting structure. In the inactive conformation, the loop conformation fluctuates in the middle and toward its C-terminal end (Thr-87, Glu-89), which extends to a slight unraveling of the N-terminus of helix-4 (not shown). The helix unraveling is particularly noticeable when Tyr-106 is in the g− conformation (Fig. 7 d). In the active conformation, the flexibility of this loop is located at the N-terminus of the loop and the C-terminus of strand 4 (Met-85 and Thr-87). Crystal structures have identified several conformations of this loop overall and NMR and MD studies show that it is a flexible loop. We now show that the fluctuations within this loop depend upon the initial conformation of the protein.
As mentioned, sampling of the g− χ-1 rotamer of Tyr-106 in the simulations initiated from the inactive structure appears to be linked to the conformation at the N-terminus of helix-4 and the preceding loop (from visual inspection of clusters from this time period of the simulation, Fig. 7 d), both of which are regions associated with FliM binding (40). Based upon the crystal structure of the CheY/CheZ complex (45) and the CheY/CheA complex (46), it has been suggested that the chi-1 rotamer of Tyr-106 has a role in both the phosphorylation of CheY by CheA and the dephosphorylation of CheY by CheZ. The g+ χ-1 rotamer of Tyr-106 forms a hydrogen bond with His-181 in the CheA phosphor-acceptor binding (P2) domain, which cannot be formed by either the g− or t rotamers of Tyr-106. On the other hand, CheZ can bind to conformations of CheY when Tyr-106 is in either the g− or the t χ-1 rotamer without steric clashes between Tyr-106 and CheZ, but this is not the case for the g+ rotamer. From these observations, it is thought that the Tyr-106 χ-1 rotamer might act as a switch, in that CheA binding to CheY, and therefore CheY phosphorylation, is enhanced when Tyr-106 is in the g+ rotamer (i.e., when CheY is in the apo form). Because inactive (unphosphorylated) CheY samples the g− rotamer, one might ask whether CheZ could bind to the apo-form of CheY.
Long-range interaction networks in this small, single-domain protein
We observe several interactions in CheY that are potentially attributable to long-range communication or networks in this protein. In simulations initiated from the inactive structure (with Tyr-106 staring in g+ conformation and solvent exposed), Ala-97 exhibits flexibility that is higher than that in the simulations initiated from the active conformation (Fig. 3). This is easily explained because Ala-97 is adjacent to the Tyr-106 cavity. It is not so easy to explain the increased flexibility observed at residues 76–79, in loop between helix-3 and strand-4, ∼20 Å distant (red regions, Fig. 3, c and d). Simulations of the S. typhimurium sequence show increased flexibility around residues 45–52 and 85–92 (green regions, Fig. 3, c and d). Residue 51 is the site of a sequence difference (Tyr to Phe); however, 85–92 is at the active site loop and is distant from any of the three sites of sequence variation. Again, long-range interactions appear to be involved in transmitting any differences that are local to the sequence differences to the active site loop.
There is an interesting offshoot from these observations. The species-specific differences in the flexibility of the strand-4-helix-4 loop may subtly affect basal CheY activity. For example, FliM binds to helix-4 and the active site loop in CheY. These structures are distant from any sites of sequence different between S. typhimurium and E. coli proteins; thus, one might hypothesize that these are unlikely to affect FliM binding. However, our simulations show that sequence-based effects alter the flexibility of that active site loop, even though these residues are distant from the sites of sequence variation. Because this region of CheY is known to be important for chemotactic signaling, the observation implies that species-specific sequence changes may also affect the basal activity of CheY, including its binding to FliM. Observations such as these, with increased fluctuations in two distinct regions observed from the MD simulations, provide interesting starting points for studying the mechanism of long-range communication in proteins.
Correlated motion and cavities in proteins
The burial of the Tyr-106 side chain (in the t rotamer) results in correlated motion between helix-4 and strand-5 correlations between the motions of helix-4 and Tyr-106 are observed only when the Tyr-106 side chain is buried between helix-4 and strand-5 (Figs. 5 and 6). The origin of correlated motion is not clearly understood. Most often, regular secondary structures that pack against each other in the protein structure exhibit correlated motion, as seen in dihydrofolate reductase and eglin c (47,48). Clearly, helix-4 and strand-5 pack against each other; furthermore, they are adjacent in sequence and connected by a short loop (Fig. 1). One is led to ask why these structures exhibit correlated motion only in simulations initiated from the active conformation. Analysis of the side-chain solvent accessibility (data not shown) and helix-4 clustering indicate that the burial of the Tyr-106 side chain between helix-4 and strand-5 results in a reduction of the number of helix-4 orientations sampled in the simulations. In addition, there is a reduction in calculated Cα B-factors for the residues forming the hydrophobic cavity around buried Tyr-106 (Fig. 3). Thus, when Tyr-106 is buried and the cavity is filled, we see decreased fluctuations in helix-4, decreased flexibility in residues around the Tyr-106, and an increase in correlated motion between helix-4 and strand-5. We conclude that cavities in proteins play a significant role in determining correlated motion. Presence or absence of cavities, even small ones less than the size of a phenyl group, can mean the difference between correlated motion or lack thereof. We propose that analysis of cavities in an ensemble of protein structures (such as that obtained from an MD simulation) might provide insight into residues that will exhibit correlated motion, or into pathways of correlated motion.
The observation that mutation of Tyr-106 to any amino acid other than Trp stops the propagation of chemotactic signals in CheY (40) and the structural and thermodynamic data obtained for helix-4 mutations (49), combined with the results from this study, suggest the possibility of mutant forms of CheY that should result in an increase in the basal activity of apo-CheY. We suggest that stabilization of the helix conformations, through, for example, Ala-98Leu or Ala-98Val mutation, or possibly the Ala-98Leu/Tyr-106Ala and Ala-98Val/Tyr-106Ala double mutations, could demonstrate higher levels of basal activity. This prediction is based upon the assumption that the role of Tyr-106 in the active protein is to pack between helix-4 and strand-5, resulting in the stabilization of helix-4 conformations similar to those in the phosphorylated protein, and introducing correlated motion between helix-4 and strand-5. The Ala-98Leu mutation, in which the Leu-98 side chain is known to partially occupy the hydrophobic cavity between helix-4 and strand-5, should form similar hydrophobic interactions to those of buried Tyr-106. We note that the Ala-98Leu mutation has been accomplished in the context of several other helix mutations and behaves as we would expect: it stabilizes the protein (49). Functional efficiency, which is the effect we are proposing, was not measured in that study. In the Ala-98Leu variant, Tyr-106 was unable to adopt the t rotamer of the χ-1 dihedral angle (49), indicating the importance of the double mutation, Ala-98Leu/Tyr-106Ala, to test our hypothesis.
SUMMARY
We performed 10-ns MD simulations of CheY from two starting conformations and with two sequence variants to identify long-range interactions and correlated motions that were specific to starting sequence or structure. Molecular dynamics simulations were performed on the unphosphorylated, inactive structure from S. typhimurium and the CheY- active mimic structure (with
active mimic structure (with 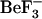 removed) from E. coli. Removal of the
removed) from E. coli. Removal of the 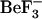 allows us to examine the active conformation and early events in relaxation.
allows us to examine the active conformation and early events in relaxation.
The flexibility of the loop between sheet-4 and helix-4 observed in our simulations agrees with experimental observations. More detailed analysis of the conformations of the loop indicates that its flexibility distinctly depends on the starting structure. The ensemble of inactive conformations shows significant flexibility at Thr-87 and Glu-89, including a slight unwinding of the N-terminus of helix-4. In the trajectory initiated from the active structure, flexibility is located at the N-terminus of the loop and the C-terminus of strand-4, specifically at Met-85 and Thr-87.
The divergence in the events accompanying the Tyr-106 χ-1 rotamer transition from g+ to g− in the simulations initiated from the inactive CheY structure indicates that there may be parallel pathways responsible for this rotameric conversion. Some pathways appear to involve changes in side-chain packing and side-chain conformation of the residues that line the Y106 binding pocket. Another pathway appears to involve larger backbone conformational changes in the loops at the N- and C-terminal of α-helix-4. The changes in the Thr-87 hydrogen bonding pattern that accompany the Tyr-106 χ-1 g+ to g− rotamer transition may have consequences for the dephosphorylation of CheY.
CheY is often used as an example of a typical allosteric protein. Although our simulation results do not allow us to draw conclusions about the mechanism of allostery, or whether this protein is consistent with pathway or ensemble mechanism of allostery, we can gain insights into how small differences in both conformation and sequence may have profound effects on the dynamics, interaction networks, and long-range interactions in this small protein.
Acknowledgments
We thank Stacy Knutson for assistance with figures, Dr. Sarah Knaggs for editorial assistance, and Drs. Ruth Silversmith and Robert Bourret for critical comments before the original submission.
We gratefully acknowledge Wake Forest University start-up funds (JSF) for financial support and use of the DEAC cluster (www.deac.wfu.edu).
References
- 1.Stock, A., D. E. Koshland Jr., and J. Stock. 1985. Homologies between the Salmonella typhimurium CheY protein and proteins involved in the regulation of chemotaxis, membrane protein synthesis, and sporulaton. Proc. Natl. Acad. Sci. USA. 82:7989–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsumura, P., J. J. Rydel, R. Linameir, and D. Vacante. 1984. Overexpression and sequence of the Escherichia coli cheY gene and biochemical activities of the CheY protein. J. Bacteriol. 160:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bren, A., and M. Eisenbach. 2000. How signals are heard during bacterial chemotaxis: protein-protein interactions in sensory signal propagation. J. Bacteriol. 182:6865–6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourret, R. B., and A. M. Stock. 2002. Molecular information processing: lessons from bacterial chemotaxis. J. Biol. Chem. 277:9625–9628. [DOI] [PubMed] [Google Scholar]
- 5.Wadhams, G. H., and J. P. Armitage. 2004. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5:1024–1037. [DOI] [PubMed] [Google Scholar]
- 6.Volz, K., and P. Matsumura. 1991. Crystal structure of Escherichia coli CheY refined at 1.7-Å resolution. J. Biol. Chem. 266:15511–15519. [DOI] [PubMed] [Google Scholar]
- 7.Stock, A. M., E. Martinez-Hackert, B. F. Rasmussen, A. H. West, J. B. Stock, D. Ringe, and G. A. Petsko. 1993. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry. 32:13375–13380. [DOI] [PubMed] [Google Scholar]
- 8.Lee, S.-Y., H. S. Cho, J. G. Pelton, D. Yan, E. A. Berry, and D. E. Wemmer. 2001. Crystal structure of activated CheY: comparison with other activated receiver domains. J. Biol. Chem. 276:16425–16431. [DOI] [PubMed] [Google Scholar]
-
9.Riepl, H., B. Scharf, R. Schmitt, H. R. Kalbitzer, and T. Maurer. 2004. Solution structures of the inactive and
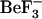 -activated response regulator CheY2. J. Mol. Biol. 338:287–297. [DOI] [PubMed] [Google Scholar]
-activated response regulator CheY2. J. Mol. Biol. 338:287–297. [DOI] [PubMed] [Google Scholar] - 10.Ganguli, S., H. Wang, P. Matsumura, and K. Volz. 1995. Uncoupled phosphorylation and activation in bacterial chemitaxis: the 1.2 Angstrom structure of a threonine to isoleucine mutant at position-87 of CheY. J. Biol. Chem. 270:17386–17393. [PubMed] [Google Scholar]
- 11.Lukat, G. S., A. M. Stock, and J. B. Stock. 1990. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry. 29:5436–5442. [DOI] [PubMed] [Google Scholar]
- 12.Lukat, G. S., B. H. Lee, J. M. Mottonen, A. M. Stock, and J. B. Stock. 1991. Roles of the highly conserved aspartate and lysine residues in the response regulator of bacterial chemotaxis. J. Biol. Chem. 266:8348–8354. [PubMed] [Google Scholar]
- 13.Yan, D., H. S. Cho, C. A. Hastings, M. M. Igo, S.-Y. Lee, J. G. Pelton, V. Stewart, D. E. Wemmer, and S. Kustu. 1999. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. USA. 96:4789–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho, H. S., S.-Y. Lee, D. Yan, X. Pan, J. S. Parkinson, S. Kustu, D. E. Wemmer, and J. G. Pelton. 2000. NMR structure of activated CheY. J. Mol. Biol. 297:543–551. [DOI] [PubMed] [Google Scholar]
- 15.Zhu, X., C. D. Amsler, K. Volz, and P. Matsumura. 1996. Tyrosine 106 of CheY plays an important role in chemotaxis signal transduction in Escherichia Coli. J. Bacteriol. 178:4208–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu, X., J. Rebello, P. Matsumura, and K. Volz. 1997. Crystal structures of CheY mutants Y106W and T87I/Y106W: CheY activation correlates with movement of residue 106. J. Biol. Chem. 272:5000–5006. [DOI] [PubMed] [Google Scholar]
- 17.DeFranco, A. L., J. S. Parkinson, and D. E. Koshland, Jr. 1979. Functional homology of chemotaxis genes in Escherichia coli and Salmonella typhimurium. J. Bacteriol. 139:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4:187–217. [Google Scholar]
- 19.MacKerell, A. D. Jr., D. Bashford, R. L. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 20.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen, W. L., J. Chandreskhar, J. D. Madura, R. W. Imprey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 22.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 23.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular-dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 24.van Gunsteren, W. F., and H. J. C. Berendsen. 1977. Algorithms for macromolecular dynamics and constraint dynamics. Mol. Phys. 34:1311–1327. [Google Scholar]
- 25.Darden, T., T. York, and L. Perdersen. 1993. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 26.Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- 27.Kearsley, S. K. 1989. On the orthogonal transformation used for structural comparisons. Acta Crystallogr. A. 45:208–210. [Google Scholar]
- 28.Aqvist, J., W. F. van Gunsteren, M. Leijonmark, and O. Tapia. 1985. A molecular dynamics study of the C-terminal fragment of the L7/L12 ribosomal protein. Secondary structure motion in a 150 picosecond trajectory. J. Mol. Biol. 183:461–477. [DOI] [PubMed] [Google Scholar]
- 29.Levitt, M., and R. Sharon. 1988. Accurate simulation of protein dynamics in solution. Proc. Natl. Acad. Sci. USA. 85:7557–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCammon, J. A. 1984. Protein dynamics. Rep. Prog. Phys. 47:1–46. [Google Scholar]
- 31.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 32.Hubbard, S. J., and J. M. Thornton. 1993. “NACCESS” Computer Program. Department of Biochemistry and Molecular Biology, University College London, London, UK.
- 33.Lee, B. K., and F. Richards. 1971. The interpretation of protein structures. Estimation of static accessibility. J. Mol. Biol. 55:379–400. [DOI] [PubMed] [Google Scholar]
- 34.Hubbard, S. J., S. F. Campbell, and J. M. Thornton. 1991. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J. Mol. Biol. 220:507–530. [DOI] [PubMed] [Google Scholar]
- 35.Murtagh, F. 1983. A survey of recent advances in hierarchical clustering algorithms. The Computer Journal. 26:354–359. [Google Scholar]
- 36.Kelley, L. A., S. P. Gardner, and M. J. Sutcliffe. 1996. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. 9:1063–1065. [DOI] [PubMed] [Google Scholar]
- 37.Lowery, T. J., M. Doucleff, E. J. Ruiz, S. M. Rubin, A. Pines, and D. E. Wemmer. 2005. Distinguishing multiple chemotaxis Y protein conformations with laser-polarized 129Xe NMR. Protein Sci. 14:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lacroix, E., M. Bruix, E. López-Hernández, L. Serrano, and M. Rico. 1997. Amide hydrogen exchange and internal dynamics the chemotactic protein CheY from Escherichia coli. J. Mol. Biol. 271:472–487. [DOI] [PubMed] [Google Scholar]
- 39.Moy, F. J., D. F. Lowry, P. Matsumura, F. W. Dahlquist, J. E. Krywko, and P. J. Domaille. 1994. Assignments, secondary structure, global fold, and dynamics of chemotaxis Y protein using three- and four-dimensional heteronuclear (13C, 15N) NMR spectroscopy. Biochemistry. 33:10731–10742. [DOI] [PubMed] [Google Scholar]
- 40.Dyer, C. M., M. L. Quillin, A. Campos, J. Lu, M. M. McEvoy, A. C. Hausrath, E. M. Westbrook, P. Matsumura, B. W. Matthews, and F. W. Dahlquist. 2004. Structure of the constitutively active double mutant CheYD13K Y106W alone and in complex with a FliM peptide. J. Mol. Biol. 342:1325–1335. [DOI] [PubMed] [Google Scholar]
- 41.Scharf, B. E., K. A. Fahrner, L. Turner, and H. C. Berg. 1998. Control of the direction of flagellar rotation in bacterial chemotaxis. Proc. Natl. Acad. Sci. USA. 95:202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simonovic, M., and K. Volz. 2001. A distinct meta-active conformation in the 1.1-Å resolution structure of wild-type apoCheY. J. Biol. Chem. 276:28637–28640. [DOI] [PubMed] [Google Scholar]
- 43.Roche, P., L. Mouawad, D. Perahia, J.-P. Samama, and D. Kahn. 2002. Molecular dynamics of the FixJ receiver domain: movement of the β4-α4 loop correlates with the in and out flip of Phe101. Protein Sci. 11:2622–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bellsolell, L., T. Prieto, L. Serrano, and M. Coll. 1994. Magnesium binding to the bacterial chemotaxis protein CheY results in large conformational changes involving its functional surface. J. Mol. Biol. 238:489–495. [DOI] [PubMed] [Google Scholar]
- 45.Zhao, R., E. J. Collins, R. B. Bourret, and R. E. Silversmith. 2002. Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat. Struct. Biol. 9:570–575. [DOI] [PubMed] [Google Scholar]
- 46.McEvoy, M. M., A. C. Hausrath, G. B. Randolph, S. J. Remington, and F. W. Dahlquist. 1998. Two binding modes reveal flexibility in kinase/response regulator interactions in the bacterial chemotaxis pathway. Proc. Natl. Acad. Sci. USA. 95:7333–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rod, T. H., J. L. Radkiewicz, and C. L. Brooks III. 2003. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA. 100:6980–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fetrow, J. S., S. T. Knutson, and M. H. Edgell. 2006. Mutations in α-helical solvent exposed sites of eglin c have long-range effects: evidence from molecular dynamics simulations. Proteins. 63:356–72. [DOI] [PubMed] [Google Scholar]
- 49.Sola, M., E. Lopez-Hernandez, P. Cronet, E. Lacroix, L. Serrano, M. Coll, and A. Parraga. 2000. Towards understanding a molecular switch mechanism: thermodynamics and crystallographic studies of the signal transduction protein CheY. J. Mol. Biol. 303:213–225. [DOI] [PubMed] [Google Scholar]