

Abstract
1,2,3,4-Tetrahydrobenz[h]isoquinoline (THBQ, 11) is a potent, inhibitor of phenylethanolamine N-methyltransferase (PNMT). Docking studies indicated that the enhanced PNMT inhibitory potency of 11 (hPNMT Ki = 0.49 μM) versus 1,2,3,4-tetrahydroisoquinoline (5, hPNMT Ki = 5.8 μM) was likely due to hydrophobic interactions with Val53, Met258, Val272, and Val269 in the PNMT active site. These studies also suggested that the addition of substituents to the 7-position of 11 that are capable of forming hydrogen bonds to the enzyme could lead to compounds (14–18) having enhanced PNMT inhibitory potency. However, these compounds are in fact less potent at PNMT than 11. Furthermore, 7-bromo-THBQ (19, hPNMT Ki = 0.22 μM), which has a lipophilic 7-substituent that cannot hydrogen bond to the enzyme, is twice as potent at PNMT than 11. This once again illustrates the limitations of docking studies for lead optimization.
1. Introduction
The role of epinephrine as a hormone in the sympathetic nervous system was first described by Walter Cannon in 1914.2 An acute stress response triggers the release of epinephrine from the adrenal gland, which initiates the physiological changes associated with Fight–Flight Syndrome (FFS). In contrast to the vast understanding of the functions of epinephrine relating to FFS, its role in the mammalian central nervous system (CNS), where it accounts for approximately 5% of the total CNS catecholamine content,3-5 remains unclear.6 Primarily on the basis of CNS localization, epinephrine neurons are thought to be involved in the regulation of blood pressure, respiration, and body temperature,7 the secretion of hormones from the pituitary gland,8 the regulation of α2-adrenoceptors in the hypothalamus,9 and some of the neurodegeneration seen in Alzheimer's disease.10-12 As one approach to elucidate the role(s) of epinephrine in the CNS, our laboratory has targeted phenylethanolamine N-methyltransferase (PNMT; EC 2.1.1.28). This enzyme catalyzes the terminal step in the biosynthesis of epinephrine (Figure 1), in which a methyl group is transferred from S-adenosyl-l-methionine (AdoMet; 3) to the primary amine of norepinephrine (1) to form epinephrine (2) and the cofactor product S-adenosyl-l-homocysteine (AdoHcy; 4).
Figure 1.

The terminal step in epinephrine (2) biosynthesis is the transfer of a methyl group from AdoMet (3) to norepinephrine (1) to form epinephrine (2) and the cofactor product AdoHcy (4).
Compounds based on the 1,2,3,4-tetrahydroisoquinoline (THIQ, 5, Table 1) nucleus have been found to be some of the most potent inhibitors of PNMT reported, particularly those compounds having electron-withdrawing substituents in the 7-position (6–10).13 SK&F 6413914 (6) and SK&F 2966115 (7) are two of the most well-studied inhibitors of PNMT, but the former is nonselective due to significant affinity for the α2-adrenoceptor,16 while the latter is selective for PNMT, but does not cross the blood-brain barrier (BBB),15 most likely due to the high polarity of the 7-aminosulfonyl substituent. According to an in vitro BBB model,17, 18 the apparent minimum lipophilicity (ClogP) for partial permeability through the BBB is 0.13–0.57 for THIQs.19 A potent inhibitor of PNMT, which exhibits minimal affinity for the α2-adrenoceptor and is able to cross the BBB, would be a useful pharmacological tool for defining the role(s) of epinephrine in the CNS.
Table 1.
In Vitro Human PNMT (hPNMT) and α2-Adrenoceptor Affinity of Some PNMT Inhibitors.
 |
| Ki (μM) ± SEMa | ||||||
|---|---|---|---|---|---|---|
| Compd | R7 | R8 | hPNMT | α2b | Selectivity α2/hPNMT | ClogPc |
| 5d | H | H | 5.8 ± 0.5 | 0.35 ± 0.11 | 0.060 | 1.60 |
| 6e | Cl | Cl | 0.0031 ± 0.0006f | 0.021 ± 0.005 | 6.8 | 2.90 |
| 7g | SO2NH2 | H | 0.28 ± 0.02f | 100 ± 10 | 360 | −0.24 |
| 8h | NO2 | H | 0.12 ± 0.01 | 4.3 ± 0.3 | 36 | 1.34 |
| 9h | CN | H | 1.5 ± 0.1 | 7.3 ± 0.2 | 4.9 | 1.03 |
| 10h | Br | H | 0.056 ± 0.003 | 0.23 ± 0.13 | 77 | 2.46 |
| 11i,j | H | H | 0.49 ± 0.05 | 0.016 ± 0.002 | 0.033 | 2.77 |
| 12i,k | H | OH | 1.4 ± 0.1 | 0.078 ± 0.002 | 0.056 | 2.10 |
| 13i,k | H | OMe | 2.3 ± 0.3 | 0.84 ± 0.03 | 0.37 | 2.69 |
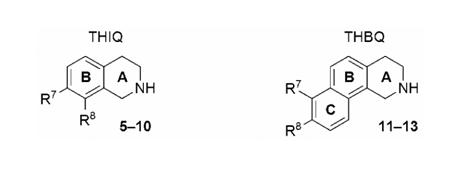
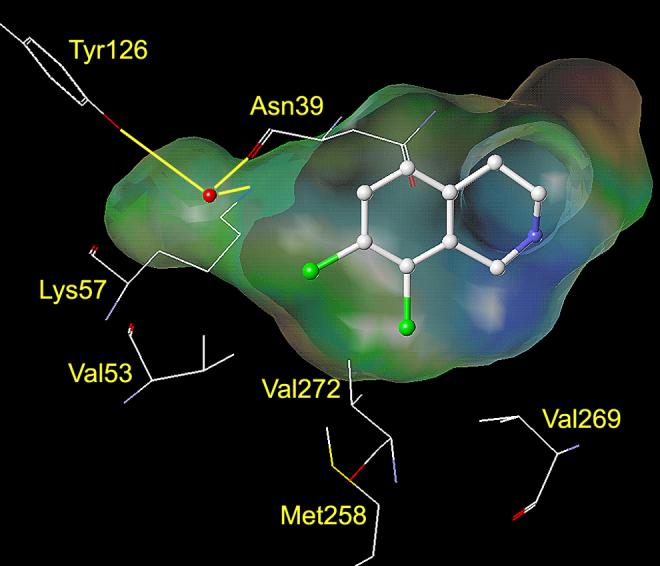
Analysis of the crystal structure of human PNMT (hPNMT) co-crystallized with inhibitors20-23 and substrates,24 coupled with site directed mutagenesis studies,22 has helped to identify the key amino acids within the hPNMT active site, some of which are shown in Figures 2 and 3. Examination of the crystal structure of hPNMT in complex with 6 and 4 (hPNMT·6·4, Figure 2) indicated that the increased inhibitory potency of 6 versus 5 was due to hydrophobic interactions between the 7-chloro and Val53, the 8-chloro and Val269 and Val272, and both the 7- and 8-chloro with Met258,22 while a comparable study of hPNMT in complex with 7 and 4 (hPNMT·7·4, Figure 3) indicated that the increased inhibitory potency of 7 versus 5 was due to hydrogen bonds between both sulfonamide oxygens of 7 and the side chain of Lys57.20 Also, water-mediated hydrogen bonds are indicated between one of the sulfonamide oxygens of 7 and the main chain carbonyl oxygen of Arg44 and the side chains of Tyr126 and Lys57.20 Additional crystallography studies have also indicated the presence of a hydrogen bond acceptor, the main chain carbonyl oxygen of Asn39, adjacent to the 7-position of THIQ.23
Figure 2.
This figure shows the amino acids (carbon is white, nitrogen is blue, oxygen is red, chlorine is green, and sulfur is yellow) interacting with SK&F 64139 (6) at the active site of hPNMT.22, 48 A Connolly (solvent accessible) surface exposing 6 is shown and a lipophilic potential is mapped on the Connolly surface whereby the areas shown in green are neutral, blue are hydrophilic, and brown are lipophilic. The brown lipophilic region of the Connolly surface adjacent to Val53, Met258, Val272, and Val269 is not easily observed, as it is perpendicular to the plane of the page. Hydrogens are not shown for clarity.
Figure 3.
This figure shows the amino acids (carbon is white, nitrogen is blue, oxygen is red, and sulfur is yellow) interacting with SK&F 29661 (7) at the active site of hPNMT.20, 48 A Connolly (solvent accessible) surface exposing 7 is shown and indicates the presence of a binding pocket adjacent to the sulfonamide group of 7. A lipophilic potential is mapped on the Connolly surface whereby the areas shown in green are neutral, blue are hydrophilic, and brown are lipophilic. The brown lipophilic region of the Connolly surface adjacent to Val53, Met258, Val272, and Val269 is not easily observed, as it is perpendicular to the plane of the page. Hydrogen bonds between the sulfonamide oxygens of 7 and Lys57 are shown in magenta. Water mediated hydrogen bonds between one of the sulfonamide oxygens, the side chain of Tyr126, the main chain carbonyl oxygen of Arg44, and the side chain of Lys57 are shown in yellow. Hydrogens are not shown for clarity.
Prior to obtaining the crystal structure of hPNMT, the 10-fold increase in PNMT inhibitory potency of 1,2,3,4-tetrahydrobenz[h]isoquinoline (THBQ, 11, Table 1) versus 5 was attributed to interactions of the additional aromatic ring (ring C, Table 1), which likely bound in the same area in the active site as the 7- and 8-chloro groups of 6.25 On the basis of the PNMT inhibitory potency of lead compound 11, 8-substituted-THBQs 12 and 13 were prepared and evaluated in order to mimic some of the apparent interactions of the sulfonamide of 7 with PNMT.26 Compounds 12 and 13 were found to be slightly less potent than parent compound 11 suggesting that they did not form additional favorable interactions with PNMT.
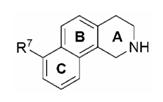
When the X-ray crystal structures of hPNMT·6·4 and hPNMT·7·4 became available, docking studies with 11–13 (see section 4.2) indicated that the 8-position was likely not the ideal site to add substituents that could form hydrogen bonds to the enzyme and that substituents on the 7-position of 11 would be more likely to form hydrogen bonding interactions with the enzyme (see section 4.3). Thus, a series of 7-substituted-THBQs (14–18, section 4.1, Table 2) having functional groups in the 7-position that could act as either hydrogen bond donors or acceptors were prepared and evaluated at hPNMT and the α2-adrenoceptor. In addition, 19, which has a lipophilic electron-withdrawing substituent, was included in this series.
Table 2.
In Vitro Human PNMT (hPNMT) and α2-Adrenoceptor Affinity of Some PNMT Inhibitors.
 |
| Ki (μM) ± SEMa | |||||
|---|---|---|---|---|---|
| Compd | R7 | hPNMT | α2b | Selectivity α2/hPNMT | ClogPc |
| 14 | NO2 | 0.90 ± 0.10 | 0.41 ± 0.04 | 0.46 | 2.51 |
| 15 | SO2NH2 | 33 ± 3 | 2.9 ± 0.3 | 0.088 | 0.93 |
| 16 | OMe | 2.1 ± 0.2 | 0.16 ± 0.02 | 0.76 | 2.69 |
| 17 | OH | 0.91 ± 0.06 | 0.011 ± 0.001 | 0.012 | 2.10 |
| 18 | CN | 2.3 ± 0.2 | 0.59 ± 0.06 | 0.26 | 2.20 |
| 19 | Br | 0.22 ± 0.03 | 0.10 ± 0.01 | 0.45 | 3.63 |
Standard error of the mean.
In vitro activities for the inhibition of [3H]clonidine binding to the α2-adrenoceptor.
Calculated log P.
Compound 11 is a nonselective inhibitor of PNMT due to significant affinity for the α2-adrenoceptor. THIQs having hydrophilic electron-withdrawing 7-substituents (e.g., 7–9) have reduced affinity for the α2-adrenoceptor.13 If 7-substituted-THBQs bind to the α2-adrenoceptor in a similar manner as 7-substituted-THIQs, the proposed THBQs having hydrophilic electron-withdrawing 7-substituents (14, 15, and 18) would likely show reduced affinity for the α2-adrenoceptor in comparison to 11, thus increasing their overall selectivity. In addition, some THIQs having hydrophilic 7-substituents (e.g., 7) are not able to penetrate the BBB.19 Since the THBQ scaffold (ClogP = 2.77) is more lipophilic than the THIQ scaffold (ClogP = 1.60), the proposed THBQs are more likely to cross the BBB than similarly substituted THIQs.
2. Chemistry
The synthesis of THBQs 14 and 15 (Table 2) is shown in Scheme 1. 2-(Naphthylen-2-yl)ethanamine (20) was converted to carbamate 21 with methylchloroformate and pyridine. Cyclization of 21 was accomplished under dehydrating conditions with polyphosphoric acid to form 22.27 Treatment of 22 with KNO3 in H2SO4 yielded a mixture of mononitro- and dinitro-3,4-dihydrobenz[h]isoquinolin-1(2H)-ones. The major monosubstituted products, 23 and 24, were isolated via flash chromatography. The structures of these two regioisomers were confirmed by X-ray crystallography. Treatment of 22 with chlorosulfonic acid yielded a mixture of monosubstituted regioisomers, from which 25 and 26 were isolated as the major products. The structures of these two regioisomers were also confirmed by X-ray crystallography.28 Compound 25 was converted to aminosulfonyl 27 with ammonium hydroxide in acetonitrile. Compounds 23 and 27 were reduced with diborane to yield THBQs 14 and 15, respectively.
Scheme 1a.
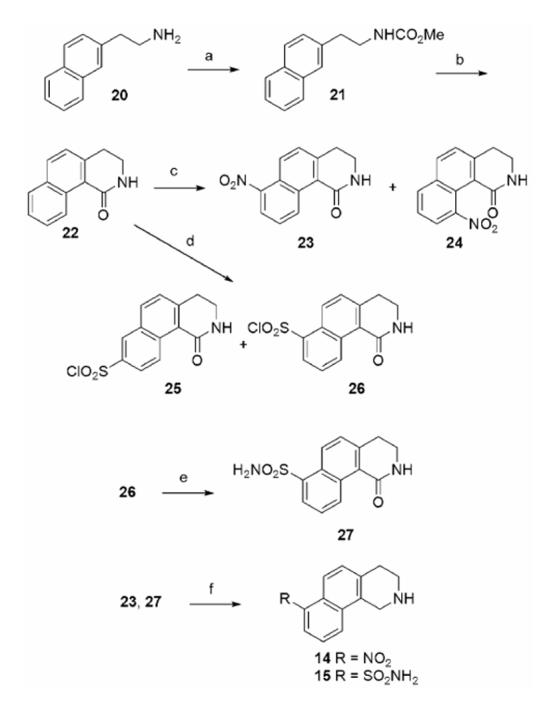
a Reagents and conditions: (a) ClCO2Me, pyridine, CH2Cl2; (b) polyphosphoric acid; (c) H2SO4, KNO3; (d) chlorosulfonic acid; (e) NH4OH, CH3CN; (f) BH3·THF.
The synthesis of THBQs 16 and 17 (Table 2) is shown in Scheme 2. 1-Chloro-5-methoxy-3,4-dihydronaphthalene-2-carboxaldehyde (28) was prepared from 5-methoxytetralone according to a known procedure.29 Treatment of 28 with methyl thioglycolate, TEA, and pyridine, followed by KOH (aq.) formed cyclized product 29.30 Oxidation of 29 with DDQ yielded 30.30 Desulfurization with Raney Nickel31 afforded ester 31, which was hydrolyzed with LiOH to form acid 32. The intermediate isocyanate formed by a Curtius rearrangement of the acid chloride of 32 was cyclized with AlCl3 to yield 33.32 Cleavage of the methyl ether of 33 with HBr/AcOH afforded 34. Lactams 33 and 34 were reduced with diborane to yield THBQs 16 and 17, respectively.
Scheme 2a.
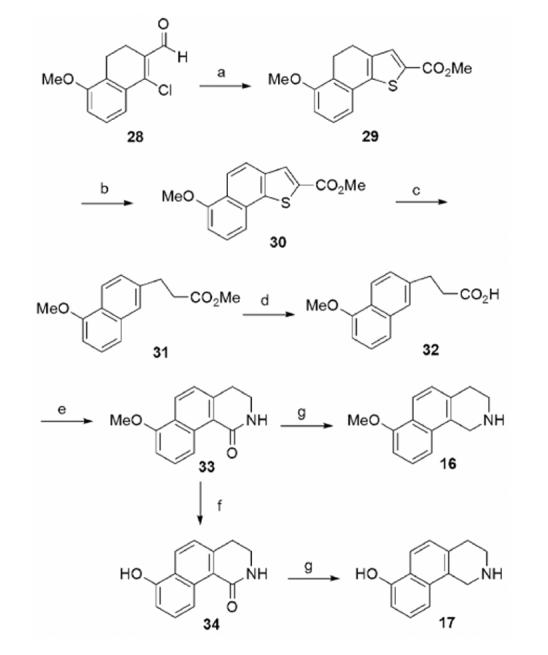
a Reagents and conditions: (a) methyl thioglycolate, TEA, pyridine; then KOH; (b) DDQ; (c) Raney Nickel; (d) LiOH; (e) (COCl)2; then NaN3; then toluene (reflux); then AlCl3; (f) HBr, AcOH; (g) BH3·THF.
The synthesis of THBQs 18 and 19 (Table 2) is shown in Scheme 3. Compound 34 was reacted with PhNTf2 and TEA to afford 35. Aryl triflate 35 was reduced to 36 with diborane. Workup with acetic acid was required because standard diborane workup conditions (methanol/HCl) led to cleavage of the triflate group.33 Boc protection of 36 followed by microwave-promoted palladium catalyzed cyanation afforded 38,34 which was deprotected with trifluoroacetic acid to afford 18. Attempts to convert 34 directly to aryl bromide 39 using Ph3PBr235, 36 were unsuccessful, thus an alternative route was employed. Aryl triflate 35 was converted to the trimethyltin intermediate with hexamethylditin37 and then reacted with NBS38 to yield 39, which was subsequently reduced with diborane to afford 19.
Scheme 3a.
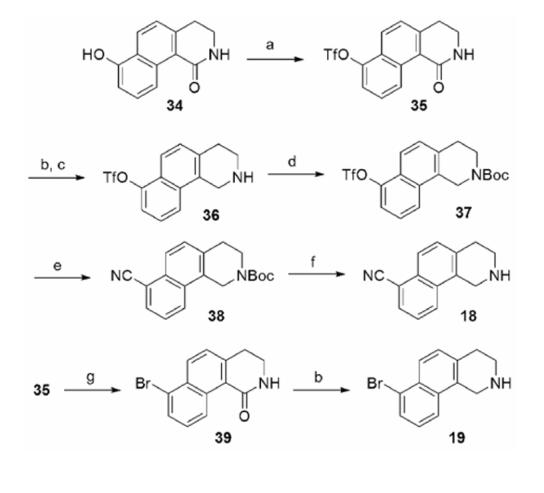
a Reagents and conditions: (a) PhNTf2, TEA; (b) BH3·THF; (c) AcOH, H2O; (d) Boc2O, TEA; (e) Zn(CN)2, Pd(Ph3P)4; (f) trifluoroacetic acid; (g) Sn2Me6, Pd(Ph3P)4, LiCl; then NBS.
3. Biochemistry
The PNMT inhibitory potency for compounds 11–13 was originally determined using bovine PNMT. The hPNMT inhibitory potency for these compounds is now reported for comparison purposes. In the current study, human PNMT (hPNMT) with a C-terminal hexahistidine tag was expressed in E. coli.39, 40 The radiochemical assay conditions, previously reported for the bovine enzyme,41 were modified to account for the high binding affinity of some inhibitors.39, 42 Inhibition constants were determined using four concentrations of phenylethanolamine as the variable substrate, and three concentrations of inhibitor.
α2-Adrenergic receptor binding assays were performed using cortex obtained from male Sprague Dawley rats.43 [3H]Clonidine was used as the radioligand to define the specific binding and phentolamine was used to define the nonspecific binding. Clonidine was used as the ligand to define α2-adrenergic binding affinity to simplify the comparison with previous results.44
4. Results and Discussion
4.1 Biochemical evaluation of 7-substituted-THBQs
The biochemical data for 7-substituted-THBQs 14–19 are shown in Table 2. Contrary to the predictions from docking studies, the addition of a 7-substituent (NO2, SO2NH2, OMe, OH, or CN, 14–18) to THBQ (11) that could potentially form hydrogen bonds with the enzyme did not lead to an increase in PNMT inhibitory potency. In comparison to parent compound 11, THBQs 14 (NO2) and 17 (OH) are two-fold less potent at hPNMT, THBQs 16 (OMe) and 18 (CN) are four-fold less potent, and THBQ 15 (SO2NH2) is almost seventy-fold less potent. Unexpectedly, 7-bromo-THBQ, which has a lipophilic 7-substituent, is 2-fold more potent as an inhibitor of PNMT than is 11. As anticipated, THBQs having hydrophilic electron-withdrawing 7-substituents (14, 15, and 18) show a significant reduction in affinity for the α2-adrenoceptor (25-, 180-, and 37-fold, respectively) versus 11. In the following sections, docking studies using the X-ray crystal structures of hPNMT·6·4 and hPNMT·7·4 and SAR analysis of the α2-adrenoceptor are used to explain these data.
4.2 hPNMT docking studies with 8-substituted-THBQs
Docking studies (AutoDock 3.0)45 with THBQ 11 (Table 1) indicate that, similarly to 6, its enhanced inhibitory potency as compared to that of 5 is likely due to favorable hydrophobic interactions with Val53, Met258, Val272, and Val269 (Figure 4A). Interactions between the amine group of THIQ inhibitors and Glu219 are known to play a key role in their binding to hPNMT.20-22 The amine group of 11 is predicted to interact with Glu219 in the same manner as that of 6. According to the docking studies with 11, it is apparent that, in order for 8-substituted-THBQs to bind in the active site of hPNMT, a significant shift of Val53 away from the 8-substituent or a shift of the inhibitor in the active site would be required. Docking studies with 8-hydroxy-THBQ (12, Figure 4B) illustrate this point. Compound 12 is predicted to twist in the active site relative to 11, which shifts the C ring of 12 away from Val53, Met258, Val272, and Val269. The reduced ability of the C ring of 12 to participate in hydrophobic interactions with Val53, Met258, Val272, or Val269 is consistent with the observed reduction in potency of 12 (hPNMT Ki = 1.4 μM) versus 11 (hPNMT Ki = 0.49 μM). Similar results were obtained from docking studies with 8-methoxy-THBQ 13 (results not shown, hPNMT Ki = 2.3 μM). Docking studies with 12 (or 13) indicate that the phenol (or methoxy) group could interact with Lys57 or Asn39 through hydrogen bonds. However, if these interactions are taking place, their benefit is apparently counteracted by the loss of the hydrophobic interactions with Val53, Met258, Val272, or Val269.
Figure 4.
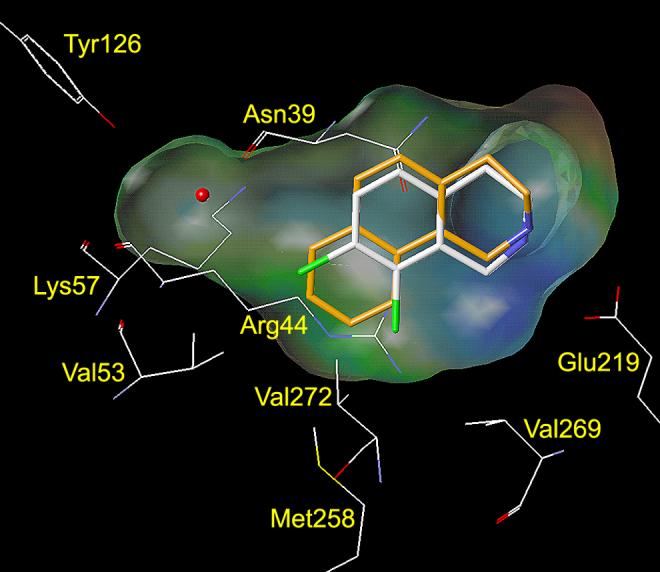
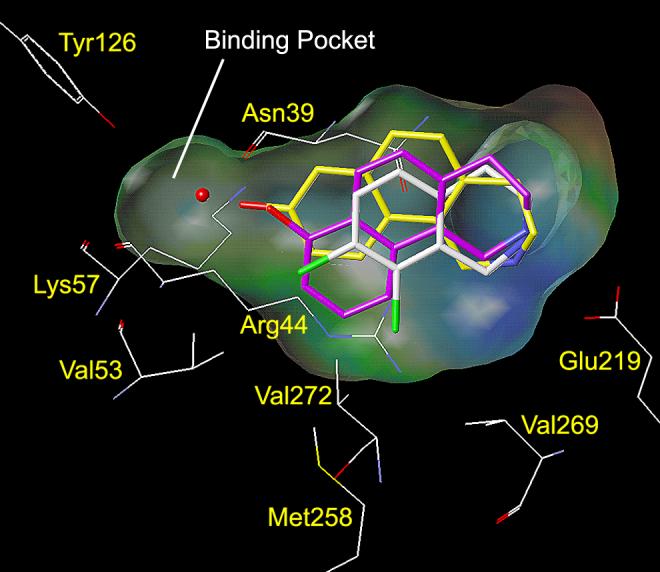
This figure shows the amino acids (carbon is white, nitrogen is blue, oxygen is red, chlorine is green, and sulfur is yellow) interacting with SK&F 64139 (6, carbons are shown in white) at the active site of hPNMT.22, 48 A Connolly (solvent accessible) surface exposing 6 is shown and a lipophilic potential is mapped on the Connolly surface whereby the areas shown in green are neutral, blue are hydrophilic, and brown are lipophilic. The brown lipophilic region of the Connolly surface adjacent to Val53, Met258, Val272, and Val269 is not easily observed, as it is perpendicular to the plane of the page. Hydrogens are not shown for clarity. Figure 4A (top) shows 11 (carbons are shown in orange) docked into the active site of hPNMT. The C ring of 11 is predicted to occupy the same area in the active site as the 7- and 8-chloro groups of 6, thus forming favorable hydrophobic interactions with Val53, Met258, Val272, and Val269. Figure 4B (bottom) shows 12 (carbons are shown in yellow) and 17 (carbons are shown in purple) docked into the active site of hPNMT. Compounds 12 and 17 are predicted to twist in the active site relative to 11, which shifts the C rings of 12 and 17 away from Val53, Met258, Val272, and Val269. Additional docking studies showed that the THBQ nucleus of 13 overlays with that of 12 and the THBQ nuclei of 14–16, 18, and 19 overlay with that of 17. Thus, the 8-methoxy group of 13 is predicted to occupy the same area in the active site as the 8-hydroxy group of 12, and the 7-substituents of 14–16, 18, and 19 are predicted to occupy the same area in the active site as the 7-hydroxy group of 17.
4.3 hPNMT docking studies with 7-substituted-THBQs
Docking studies using both hPNMT·6·4 (Figure 4B) and hPNMT·7·4 (not shown) with 7-substituted-THBQs 14–19 (Table 2) showed that the 7-substituents of these compounds could occupy an auxiliary binding pocket. It is predicted that 7-substituted-THBQs would need to twist in the active site relative to 11, which would shift the C ring of THBQ away from Val53, Met258, Val272, and Val269, but to a lesser degree than the 8-substituted-THBQs (e.g., 12, Figure 4B). These docking studies also suggested that 7-substituted-THBQs could maintain these hydrophobic interactions while simultaneously forming hydrogen bonding interactions with Lys57 or Asn39, resulting in compounds having enhanced PNMT inhibitory potency versus 11. This does not appear to be the case, however, as the THBQs having 7-substituents that could act as either hydrogen bond acceptors (14, 16, and 18) or as both hydrogen bond donors and acceptors (15 and 17) were less potent at PNMT than 11. Similarly to 12 and 13, if hydrogen bonding interactions are taking place, their benefit is apparently offset by the loss of the hydrophobic interactions of the THBQ C ring with Val53, Met258, Val272, or Val269. 7-Bromo-THBQ (19), which is the only analogue in this series having a lipophilic 7-substituent, is twice as potent at PNMT than 11. This is possibly due to hydrophobic interactions between the bromine of 19 and the aliphatic portion of the side chain of Arg44. Such favorable hydrophobic interactions would not be expected to occur between Arg44 and the hydrophilic 7-substituents of 14–18.
4.4 α2-Adrenoceptor binding studies
THIQs having hydrophilic electron-withdrawing 7-substituents, such as 7–9, have considerably less affinity for the α2-adrenoceptor than unsubstituted THIQ 5 (Table 3).13 Similarly, THBQs having hydrophilic electron-withdrawing 7-substituents (14, 15, and 18) have less affinity for the α2-adrenoceptor than 11. For the nitro, cyano, and aminosulfonyl THIQ and THBQ derivatives, the relative α2-adrenoceptor affinity versus the parent compounds is very similar. Also, the rank order of affinity (nitro > cyano > aminosulfonyl) is the same. These observations suggest that the 7-substituents of these THBQs are occupying the same area of the α2-adrenoceptor binding site as 7-substituents of THIQ. THIQs having lipophilic 7-substituents, such as 6 and 7-bromo-THIQ (10), have more affinity for the α2-adrenoceptor than 5. It was thus expected that 19 would have more affinity for the α2-adrenoceptor than 11. However, 19 has 6-fold less affinity for the α2-adrenoceptor than 11, indicating that lipophilic 7-substituents of THBQ are likely not occupying the same area of the α2-adrenoceptor binding site as lipophilic 7-substituents of THIQ. Although 14, 15, 18, and 19 have less affinity for the α2-adrenoceptor than 11, they nonetheless maintain sub-micromolar to low micromolar affinity for this target and are thus nonselective for PNMT.
Table 3.
Effects of 7-Substituents on THIQ (5) and THBQ (11) α2-Adrenoceptor Affinity.
 |
| Compd | R7 | α2aKi (μM) ± SEMb | Ratio |
|---|---|---|---|
| 8 | NO2 | 4.3 ± 0.3 | 12c |
| 9 | CN | 7.3 ± 0.2 | 21c |
| 7 | SO2NH2 | 100 ± 10 | 290c |
| 10 | Br | 0.23 ± 0.13 | 0.66c |
| 14 | NO2 | 0.41 ± 0.04 | 26d |
| 18 | CN | 0.59 ± 0.06 | 37d |
| 15 | SO2NH2 | 2.9 ± 0.3 | 180d |
| 19 | Br | 0.10 ± 0.01 | 6.3d |
In vitro activities for the inhibition of [3H]clonidine binding to the α2-adrenoceptor.
Standard error of the mean.
Ratio of α2-adrenoceptor affinity versus the unsubstituted (R7 = H) parent compound (5, α2 Ki = 0.35).
Ratio of α2-adrenoceptor affinity versus the unsubstituted (R7 = H) parent compound (11, α2 Ki = 0.016).
5. Conclusions
Although docking studies suggested that the addition of substituents to the 7-position of 11 that are capable of forming hydrogen bonds to the enzyme could lead to compounds having enhanced PNMT inhibitory potency, these compounds (14–18) were in fact less potent than 11 at PNMT, illustrating the limitations of docking studies for the optimization of lead compounds.46 Compound 19, which has a lipophilic 7-substituent, is twice as potent as 11 at PNMT, but is nonselective due to significant affinity for the α2-adrenoceptor. The co-crystallization of one or more of these THBQs with hPNMT would be required to determine the exact nature of their binding interactions.
6. Experimental
6.1. General methods
All reagents and solvents were of reagent grade or were purified by standard methods before use. Melting points were determined in open capillary tubes on a Thomas-Hoover melting point apparatus calibrated with known compounds. Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were taken on a Bruker DRX-400, Bruker AM-500, or Bruker AV-800 spectrophotometer. High resolution mass spectra (HRMS) were obtained on a Ribermag R 10-10 mass spectrophotometer. Thin-layer chromatography (TLC) was performed on K6F silica gel 60 Å (Whatman) glass backed plates. Flash chromatography was performed using silica gel 60 (230–400 mesh) supplied by Universal Adsorbents, Atlanta, Georgia.
Anhydrous methanol and ethanol were used unless stated otherwise, and were prepared by distillation over magnesium. Other solvents were routinely distilled prior to use. Anhydrous tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled from sodium-benzophenone ketyl. Hexanes refers to the mixture of hexane isomers (bp 40–70 °C) and brine refers to a saturated solution of NaCl. All reactions that required anhydrous conditions were performed under a positive nitrogen or argon flow, and all glassware was either oven-dried or flame-dried before use. [methyl-3H]AdoMet and [3H]clonidine were obtained from PerkinElmer (Boston, MA).
6.2. Radiochemical Assay of PNMT Inhibitors
A typical assay mixture consisted of 25 μL of 0.5 M phosphate buffer (pH 8.0), 25 μL of 50 μM unlabeled AdoMet, 5 μL of [methyl-3H]AdoMet, containing approximately 3 × 105 dpm (specific activity approximately 15 Ci/mmol), 25 μL of substrate solution (phenylethanolamine), 25 μL of inhibitor solution, 25 μL of enzyme preparation (containing 30 ng hPNMT and 25 μg of bovine serum albumin), and sufficient water to achieve a final volume of 250 μL. After incubation for 30 min at 37 °C, the reaction mixture was quenched by addition of 250 μL of 0.5 M borate buffer (pH 10.0) and was extracted with 2 mL of toluene/isoamyl alcohol (7:3). A 1 mL portion of the organic layer was removed, transferred to a scintillation vial and diluted with cocktail for counting. The mode of inhibition was ascertained to be competitive in all cases reported in Tables 1 and 2 by examination of the correlation coefficients (r2) for the fit routines as calculated in the Enzyme Kinetics module (version 1.1) in SigmaPlot (version 7.0).32 While all Ki values reported were calculated using competitive kinetics, it should be noted that there was not always a great difference between the r2 values for the competitive model versus the non-competitive model. All assays were run in duplicate with 3 inhibitor concentrations over a 5-fold range. Ki values were determined by a hyperbolic fit of the data using the Single Substrate – Single Inhibitor routine in the Enzyme Kinetics module (version 1.1) in SigmaPlot (version 7.0). For inhibitors with apparent IC50 values less than 0.1 μM (as determined by a preliminary screen of the compounds to be assayed), the Enzyme Kinetics Tight Binding Inhibition routine was used to calculate the Ki values.
6.3. α2-Adrenoceptor Radioligand Binding Assay
The radioligand receptor binding assay was performed according to the method of U'Prichard et al.29 Male Sprague-Dawley rats were decapitated, and the cortexes were dissected out and homogenized in 20 volumes (w/v) of ice-cold 50 mM Tris/HCl buffer (pH 7.7 at 25 °C). Homogenates were centrifuged thrice for 10 min at 50,000 × g with resuspension of the pellet in fresh buffer between spins. The final pellet was homogenized in 200 volumes (w/v) of ice-cold 50 mM Tris/HCl buffer (pH 7.7 at 25 °C). Incubation tubes containing [3H]clonidine (specific activity approximately 55 Ci/mmol, final concentration 2.0 nM), various concentrations of drugs and an aliquot of freshly resuspended tissue (800 μL) in a final volume of 1 mL were used. Tubes were incubated at 25 °C for 30 min and the incubation was terminated by rapid filtration under vacuum through GF/B glass fiber filters. The filters were rinsed with three 5 mL washes of ice-cold 50 mM Tris buffer (pH 7.7 at 25 °C). The filters were counted in vials containing premixed scintillation cocktail. Non-specific binding was defined as the concentration of bound ligand in the presence of 2 μM of phentolamine. All assays were run in quadruplicate with 5 inhibitor concentrations over a 16-fold range. IC50 values were determined by a log-probit analysis of the data and Ki values were determined by the equation Ki = IC50 / (1 + [Clonidine] / KD), as all Hill coefficients were approximately equal to 1.
6.4. Molecular Modeling
Connolly surfaces were generated in SYBYL® on a Silicon Graphics Octane workstation.47 Docking of the various inhibitors into the PNMT active site was performed using AutoDock 3.0.45 The default settings for AutoDock 3.0 were used. The compound to be docked was initially overlayed with the co-crystallized ligand and minimized with the Tripos force field.
6.5 Synthesis
6.5.1. Methyl 2-Naphthylethylcarbamate (21)
2-Naphthyleneethylamine (20, 1.60 g, 9.34 mmol) was dissolved in dry CH2Cl2 (50 mL) and pyridine (5 mL) and the solution was stirred at 0 °C. Methyl chloroformate (0.87 mL, 11 mmol) was added dropwise and the solution was stirred overnight at ambient temperature. Ice-cold water (40 mL) was added and the mixture was stirred for 30 min. The organic phase was removed, washed with 3N HCl (3 × 30 mL) and brine (30 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield the crude product as a yellow oil, which was purified by flash chromatography eluting with hexanes/EtOAc (4:1) to yield carbamate 21 as a white solid (1.55 g, 6.76 mmol, 72%): mp 95–97 °C; 1H NMR (400 MHz, CDCl3) δ 7.85–7.81 (m, 2H), 7.66 (s, 1H), 7.52–7.45 (m, 2H), 7.37–7.34 (m, 1H), 4.72 (b, 1H), 4.26 (b, 1H), 3.68 (s, 3H), 3.59–3.54 (m, 2H), 3.00 (m, J = 6.8 Hz, 2H); 13C NMR (400 MHz, CDCl3) δ 157.0, 136.2, 133.5, 132.3, 128.4, 127.7, 127.5, 127.2, 127.1, 126.2, 125.6, 52.1, 42.1, 36.3; HRMS (ESI+) m/z calcd for C14H16NO2 (MH+) 230.1181, obsd 230.1179.
6.5.2. 3,4-Dihydrobenz[h]isoquinolin-1(2H)-one (22)
Polyphosphoric acid (75 g) was heated to 120 °C and carbamate 21 (3.00 g, 15.2 mmol) was added. After stirring for 45 min, the reaction was poured into ice water (100 mL) and stirred vigorously for 5 min. This aqueous mixture was extracted with CH2Cl2 (3 × 75 mL). The combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield an off-white solid, which was purified by flash chromatography eluting with EtOAc/hexanes (1:1) to yield lactam 22 as a white solid (1.80 g, 9.13 mmol, 60%): mp 158–160 °C; 1H NMR (500 MHz, CDCl3) δ 9.42 (d, J = 8.7 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.65–7.62 (m, 1H), 7.53–7.50 (m, 1H), 7.33 (d, J = 8.3 Hz, 1H), 7.17 (b, 1H), 3.59 (t, J = 6.6 Hz, 2H), 3.13 (t, J = 6.6 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 167.4, 140.3, 133.6, 133.2, 132.1, 128.6, 128.3, 127.1, 126.1, 125.8, 124.3, 39.8, 30.9; HRMS (ESI+) m/z calcd for C13H12NO (MH+) 198.0919, obsd 198.1924.
6.5.3. 7-Nitro-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (23) and 10-Nitro-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (24)
Lactam 22 (300 mg, 1.52 mmol) was dissolved in concentrated H2SO4 (5 mL) and stirred at 0 °C. KNO3 (115 mg, 1.14 mmol) was added in small portions over the course of 15 min. The reaction continued for 1 h at 0 °C and then was poured slowly onto ice (50 g). The aqueous mixture was extracted with CH2Cl2 (4 × 40 mL) and the combined organic extracts were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield an off-white solid, which, according to HNMR (crude), consisted of unreacted starting material and a mixture of numerous mono- and di-substituted nitration products. Purification by flash chromatography eluting with hexanes/2-propanol (4:1) yielded unreacted starting material (70 mg, 0.35 mmol, 23%, Rf = 0.45 in 4:1 hexanes/2-propanol), the desired product, lactam 23 (Rf = 0.30 in 4:1 hexanes/2-propanol) and the undesired regioisomer, lactam 24 (Rf = 0.25 in 4:1 hexanes/2-propanol) as the major products. Compound 23 was recrystallized from CHCl3/hexanes to yield yellow needles (45 mg, 0.19 mmol, 12%): mp 184–186 °C; 1H NMR (800 MHz, CDCl3) δ 9.82 (d, J = 8.7 Hz, 1H), 8.61 (d, J = 8.8 Hz, 1H), 8.19 (d, J = 7.5 Hz, 1H), 7.70–7.68 (m, 1H), 7.59 (d, J = 8.9 Hz, 1H), 6.30 (b, 1H), 3.64–3.62 (m, 2H), 3.20 (t, J = 6.5 Hz, 2H); 13C NMR (800 MHz, CDCl3) δ 165.9, 147.1, 141.1, 133.2, 132.5, 128.7, 126.0, 124.8, 124.4, 123.3, 39.2, 30.3; HRMS (ESI+) m/z calcd for C13H11N2O3 (MH+) 243.0770, obsd 243.0773. Compound 24 was recrystallized from CH2Cl2/hexanes to yield yellow needles (38 mg, 0.16 mmol, 10%): mp dec 297–299 °C; 1H NMR (500 MHz, CDCl3) δ 8.14–8.12 (m, 1H), 8.07–8.05 (m, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.58–7.55 (m, 1H), 7.48 (d, J = 8.4 Hz, 1H), 6.23 (b, 1H), 3.71–3.67 (m, 2H), 3.15 (t, J = 6.2 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 164.7, 148.4, 142.4, 134.2, 133.2, 131.6, 126.8, 125.0, 124.9, 124.7, 122.1, 39.0, 30.8; HRMS (ESI+) m/z calcd for C13H11N2O3 (MH+) 243.0770, obsd 243.0759. The structures of 23 and 24 were confirmed by X-ray crystallography. See section 7.
6.5.4. 8-Chlorosulfonyl-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (25) and 7-Chlorosulfonyl-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (26)
Lactam 22 (500 mg, 2.54 mmol) was treated with chlorosulfonic acid (10 mL) with stirring at 0 °C. The reaction was permitted to warm to ambient temperature and was continued for 30 min. with stirring. The reaction mixture was cautiously added via pipet onto ice (200 mL). A white precipitate formed. The mixture was extracted with CH2Cl2 (4 × 50 mL). The combined organic extracts were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield a white solid, which consisted of a mixture of monosubstituted sulfonyl chloride regioisomers according to HNMR (crude). Hot filtration with CHCl3 yielded a white solid, which was further purified by recrystallization from CH2Cl2/hexanes to yield 25 (95 mg, 0.321 mmol, 13%) as white needles: mp 219–221 °C; 1H NMR (500 MHz, CDCl3) δ 9.64 (d, J = 9.4 Hz, 1H), 8.49 (d, J = 2.0 Hz, 1H), 8.04–8.01 (m, 2H), 7.48 (d, J = 8.3 Hz, 1H), 7.48–7.43 (m, 2H), 6.35 (b, 1H), 3.57–3.54 (m, 2H), 3.14 (t, J = 6.5 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 165.7, 144.3, 140.9, 134.5, 134.2, 131.7, 129.6, 128.7, 128.1, 124.4, 123.1, 39.2, 30.6; HRMS (ESI+) m/z calcd for C13H11ClNO3S (MH+) 296.0148, obsd 296.0152. The filtrate from the hot filtration was purified by flash chromatography eluting with hexanes/2-propanol (2:1) to yield the desired regioisomer as a white solid, which was further purified by recrystallization from CH2Cl2/hexanes to yield 26 (120 mg, 0.406 mmol, 16%) as white needles: mp dec 215–217 °C; 1H NMR (500 MHz, CDCl3) δ 9.94 (d, J = 8.8 Hz, 1H), 8.97 (d, J = 8.8 Hz, 1H), 8.42–8.41 (m, 1H), 7.76–7.73 (m, 1H), 7.69 (d, J = 8.8 Hz, 1H), 6.25 (b, 1H), 3.67–3.63 (m, 2H), 3.24 (t, J = 6.5 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 165.6, 141.4, 139.7, 135.9, 132.9, 129.2, 128.8, 128.2, 127.3, 125.7, 124.9, 39.2, 30.2; HRMS (ESI+) m/z calcd for C13H11ClNO3S (MH+) 296.0148, obsd 296.0152. The structures of 25 and 2628 were confirmed by X-ray crystallography. See section 7.
6.5.5. 7-Aminosulfonyl-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (27)
Conc. NH4OH (8 mL) was added to a stirred solution of sulfonyl chloride 26 (50 mg, 0.169 mmol) in acetonitrile (4 mL). The reaction was stirred 16 h at ambient temperature. The solvent was removed under reduced pressure and the resulting residue was purified by flash chromatography eluting with acetone/hexanes (2:1) to yield lactam 27 as a white solid (37 mg, 0.13 mmol, 79%): mp 298–300 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.37 (d, J = 8.8 Hz, 1H), 8.55 (d, J = 8.8 Hz, 1H), 8.03 (br, 1H), 7.91–7.89 (m, 1H), 7.49 (br, 2H), 7.48– 7.43 (m, 2H), 3.18–3.14 (m, 2H), 2.86 (t, J = 6.5 Hz, 2H); 13C NMR (500 MHz, DMSO-d6) δ 164.9, 141.0, 139.6, 131.8, 131.1, 128.6, 127.4, 127.1, 125.7, 125.5, 124.6, 38.1, 29.5; HRMS (ESI+) m/z calcd for C13H13N2O3S (MH+) 277.0647, obsd 277.0648.
6.5.6. Methyl 6-methoxy-4,5-dihydronaphtho[1,2-b]thiophene-2-carboxylate (29)
To a stirred solution of 28 (11.5g, 51.8 mmol) and methyl thioglycolate (5.09 mL, 57.0 mmol) in pyridine (60 mL), was added triethylamine (10.5 mL, 75.1 mmol) dropwise. The reaction was continued for 1.5 h at 50 °C and then cooled to ambient temperature. An aqueous solution of 50% KOH (15 mL) was added and the reaction was stirred for an additional 20 min. The reaction was poured over crushed ice (100 g) and then partitioned between CH2Cl2 (300 mL) and 2M HCl (200 mL). The organic layer was removed and washed with 2M HCl (2 × 100 mL), 10% NaOH (100 mL) and brine (100 mL). The CH2Cl2 solution was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to yield a yellow oil. This crude product was purified by flash chromatography eluting with hexanes/ether (5:1) to yield a white solid. Recrystallization from ether/hexanes gave 29 as white needles (12.8 g, 46.7 mmol, 90%): mp 107–109 °C; 1H NMR (500 MHz, CDCl3) δ 7.59 (s, 1H), 7.21–7.18 (m, 1H), 7.07 (d, J = 7.7 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 2.96 (t, J = 7.9 Hz, 2H), 2.80 (t, J = 7.9 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 163.0, 156.8, 143.2, 137.8, 133.8, 131.6, 130.0, 127.5, 123.7, 116.3, 110.5, 55.6, 52.1, 23.2, 20.9; HRMS (ESI+) m/z calcd for C15H15O3S (MH+) 275.0742, obsd 275.0729.
6.5.7. Methyl 6-methoxynaphtho[1,2-b]thiophene-2-carboxylate (30)
Compound 29 (12.4g, 45.2 mmol) and DDQ (11.3g, 49.7 mmol) were dissolved in dry benzene (150 mL) and heated at reflux for 3.5 h. After cooling to ambient temperature, the resulting solution was purified by flash chromatography eluting with CHCl3/hexanes/EtOAc (5:5:1) to yield 30 as a white solid. (11.8 g, 43.4 mmol, 96%): mp 119–121 °C; 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 9.2 Hz, 1H), 7.59 (s, 1H), 7.72 (d, J = 9.1 Hz, 1H), 7.64 (d, J = 8.2 Hz, 1H), 7.46–7.43 (m, 1H), 6.87 (d, J = 7.8 Hz, 1H), 3.96 (s, 3H), 3.90 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 163.2, 156.3, 140.9, 137.0, 132.3, 131.6, 129.6, 127.5, 123.4, 121.8, 120.1, 116.1, 105.7, 55.7, 52.5; HRMS (ESI+) m/z calcd for C15H13O3S (MH+) 273.0585, obsd 273.0582.
6.5.8. Methyl 3-(5-methoxynaphthalen-2-yl)propanoate (31)
Compound 30 (5.80g, 21.3 mmol) and Raney nickel were refluxed in MeOH (75 mL) for 18 h. The reaction was filtered through Celite, which was washed with MeOH (2 × 75 mL). The solvent was removed under reduced pressure and the resulting oil was redissolved in EtOAc (150 mL). This organic solution was washed with saturated NaHCO3 (75 mL) and brine (50 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (8:1) to yield 31 as a clear oil (4.10g, 16.8 mmol, 79%): 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.6 Hz, 1H), 7.62 (d, J = 0.84 Hz, 1H), 7.39–7.34 (m, 3H), 6.82–6.78 (m, 1H), 4.02 (s, 3H), 3.70 (s, 3H), 3.14 (t, J = 7.8 Hz, 2H), 2.75 (t, J = 7.8 Hz, 2H); 13C NMR (400 MHz, CDCl3) δ 173.4, 155.5, 138.6, 134.7, 126.2, 126.1, 126.1, 124.2, 122.3, 119.9, 103.4, 55.5, 51.7, 35.6, 31.1; HRMS (ESI+) m/z calcd for C15H17O3 (MH+) 245.1178, obsd 245.1160.
6.5.9. 3-(5-Methoxynaphthalen-2-yl)propanoic acid (32)
Compound 31 (3.50g, 14.3 mmol) was dissolved in THF/H2O (5:1, 75 mL) and cooled to 0 °C. LiOH·H2O (3.61g, 86.0 mmol) was added and the reaction was stirred for 6 h at ambient temperature. The reaction was partitioned between CH2Cl2 (250 mL) and brine (150 mL) and was acidified (pH ≈ 1) with 1M HCl. The organic layer was removed and the aqueous layer was extracted with CH2Cl2 (2 × 75 mL). The combined organic extracts were washed with brine (25 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CHCl3/EtOAc (2:1) to yield 32 as a white solid (3.15g, 13.7 mmol, 96%): mp 125–127 °C; 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 8.6 Hz, 1H), 7.54 (d, J = 0.75 Hz, 1H), 7.30–7.29 (m, 1H), 7.27–7.25 (m, 1H), 6.72–6.69 (m, 1H), 3.92 (s, 3H), 3.05 (t, J = 7.8 Hz, 2H), 2.71 (t, J = 7.8 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 177.1, 155.5, 138.2, 134.7, 126.2, 126.2, 126.1, 124.3, 122.4, 119.9, 103.5, 55.5, 35.2, 30.7; HRMS (ESI+) m/z calcd for C14H15O3 (MH+) 231.1021, obsd 231.1039.
6.5.10. 7-Methoxy-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (33)
Compound 32 (400 mg, 1.74 mmol) was treated with oxalyl chloride (2.0 mL) and stirred for 2 h at ambient temperature. The reaction mixture was evaporated to dryness, redissolved in dry benzene (1 mL) and again evaporated to dryness to remove trace amounts of oxalyl chloride. The crude acid chloride was dissolved in acetone (5 mL) and cooled to 0 °C. Sodium azide (250 mg, 3.84 mmol) was dissolved in H2O (750 μL) and added to this solution. The reaction was stirred at ambient temperature for 1 h and quenched with ice water (25 mL). The resulting solution was made basic (pH ≈ 10) with Na2CO3 and extracted with EtOAc (3 × 20 mL). The organic layers were pooled and dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The resulting crude acyl azide (390 mg, 1.53 mmol, 88%) was dissolved in dry toluene (10 mL) and heated at reflux for 3 h. The toluene was removed under reduced pressure and the crude isocyanate was dissolved in DCE (15 mL). AlCl3 (600 mg, 4.50 mmol) was added and the reaction was stirred for 16 h at 60 °C. The reaction was cooled to ambient temperature, quenched with 1M HCl (40 mL), and extracted with CH2Cl2 (3 × 50 mL). The organic layers were pooled, washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CHCl3/EtOAc (3:1) to yield 33 as a white solid (190 mg, 0.836 mmol, 48%): mp 182–184 °C; 1H NMR (500 MHz, CDCl3) δ 8.87 (d, J = 8.9 Hz, 1H), 8.35 (d, J = 8.6 Hz, 1H), 7.45–7.42 (m, 1H), 7.23 (d, J = 8.6 Hz, 1H), 6.78 (d, J = 7.7 Hz, 1H), 6.14 (b, 1H), 3.93 (s, 3H), 3.50–3.47 (m, 2H), 3.05 (t, J = 6.5 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 167.0, 155.3, 140.4, 132.8, 128.1, 126.8, 125.4, 124.6, 123.3, 118.9, 103.9, 55.6, 39.5, 30.5; HRMS (ESI+) m/z calcd for C14H14NO2 (MH+) 228.1024, obsd 228.1014.
6.5.11. 7-Hydroxy-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (34)
Compound 33 (1.25g, 5.50 mmol) was dissolved in 48% HBr (45 mL) and glacial acetic acid (35 mL) and was heated at reflux (120 °C) for 2 h. The reaction was poured into ice–cold H2O (300 mL), the pH was adjusted to neutral with NaHCO3, and extracted with EtOAc (3 × 100 mL). The organic layers were pooled and extracted with 10% (w/v) NaOH (2 × 100 mL). The basic aqueous extracts were combined, the pH was adjusted to neutral with 3M HCl, and extracted with EtOAc (3 × 100 mL). The combined organic extracts were pooled, washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield compound 34 (1.06 g, 4.97 mmol, 90%) as a crystalline off-white solid: mp dec 246–248 °C; 1H NMR (500 MHz, DMSO-d6 with 5% D2O) δ 10.1 (s, 1H), 8.77 (d, J = 8.7 Hz, 1H), 8.32 (d, J = 8.5 Hz, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.39–7.37 (m, 1H), 6.91 (d, J = 7.4 Hz, 1H), 3.40 (t, J = 6.5 Hz, 2H), 3.06 (t, J = 6.5 Hz, 2H); 13C NMR (800 MHz, DMSO-d6) δ 137.9, 137.6, 130.3, 129.5, 127.3, 126.8 (q, J = 276 Hz), 126.4, 81.8 (d, J = 172 Hz), 53.6, 53.1 (d, J = 18.6 Hz), 31.3 (q, J = 29 Hz), 44.4, 26.2 (d, J = 5.9 Hz), 15.8 (q, J = 3.5 Hz); HRMS (ESI+) m/z calcd for C13H12NO2 (MH+) 214.0868, obsd 214.0862.
6.5.12. 7-Trifluoromethanesulfonate-3,4-dihydrobenz[h]isoquinolin-1(2H)-one. (35)
Compound 34 (400 mg, 1.88 mmol) and PhNTf2 (804 mg, 2.25 mmol) were dissolved in dry CH2Cl2 (50 mL) and stirred at 0 °C. Triethylamine (523 μL, 3.75 mmol) was added dropwise and reaction was permitted to warm to ambient temperature and stirred for 15 h. The solvent was removed under reduced pressure and the resulting residue was purified by flash chromatography eluting with EtOAc/hexanes (1:1) to yield a white solid. Recrystallization from CH2Cl2/hexanes yielded 35 as white needles (590 mg, 1.71 mmol, 91%): mp 144–146 °C; 1H NMR (500 MHz, CDCl3) δ 9.42 (d, J = 8.8 Hz, 1H), 8.13 (d, J = 8.6 Hz, 1H), 7.56–7.53 (m, 1H), 7.45–7.40 (m, 2H), 6.09 (b, 1H), 3.54–3.51 (m, 2H), 3.10 (t, J = 6.6 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 165.9, 145.5, 141.2, 133.2, 127.6, 127.5, 127.1, 126.3, 125.1, 124.2, 118.7 (q, J = 318.3), 117.7, 39.3, 30.4; HRMS (FAB+) m/z calcd for C14H11F3NO4S (MH+) 346.0361, obsd 346.0358.
6.5.13. 7-Trifluoromethanesulfonate-1,2,3,4-tetrahydrobenz[h]isoquinoline (36)
Compound 35 (120 mg, 0.348 mmol) was dissolved in THF (10 mL). 1M BH3·THF (1.4 mL, 1.4 mmol) was added dropwise to the solution, which was heated at reflux for 3 h. The reaction was cooled to 0 °C and AcOH (1 mL) was added dropwise. The reaction was permitted to warm to ambient temperature and was stirred for 10 min. H2O (2 mL) was added and the THF was removed under reduced pressure. MeOH (10 mL) was added and the resulting mixture was heated at reflux for 30 min. The MeOH was removed under reduced pressure and the resulting solution was added to brine (50 mL), made basic (pH ≈ 12) with 1M NaOH, and extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were pooled and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CH2Cl2/MeOH (6:1) to yield 36 (108 mg, 0.326 mmol, 94%) as a clear oil that solidified on standing: 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.7 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.54–7.50 (m, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 8.7 Hz, 1H), 4.46 (s, 2H), 3.26 (t, J = 5.7 Hz, 2H), 3.00 (t, J = 5.5 Hz, 2H); 13C NMR (400 MHz, CDCl3) δ 146.6, 134.5, 132.7, 131.0, 130.4, 125.9, 125.4, 122.9, 119.3, 119.1 (q, J = 320.3), 117.4, 45.8, 43.4, 30.1; HRMS (ESI+) m/z calcd for C14H13F3NO3S (MH+) 332.0568, obsd 332.0560.
6.5.14. N-Boc-7-trifluoromethanesulfonate-1,2,3,4-tetrahydrobenz[h]isoquinoline (37)
To a solution of 36 (95.0 mg, 0.287 mmol) and Boc2O (81 mg, 0.373 mmol) in CH2Cl2 (10 mL), was added triethylamine (60 μL, 0.43 mmol). The reaction was stirred for 5 h at ambient temperature and the solvent was removed under reduced pressure. The resulting oil was purified by flash chromatography eluting with CH2Cl2/hexanes (2:1) to yield 37 (123 mg, 0.285 mmol, 99%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.95–7.93 (m, 2H), 7.59–7.55 (m, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H), 5.02 (s, 2H), 3.79 (t, J = 5.6 Hz, 2H), 3.03 (t, J = 5.5 Hz, 2H), 1.54 (s, 9H); HRMS (ESI+) m/z calcd for C19H24F3N2O5S (MNH4+) 449.1358, obsd 449.1358.
6.5.15. N-Boc-7-cyano-1,2,3,4-tetrahydrobenz[h]isoquinoline (38)
DMF (7.0 mL), 37 (70 mg, 0.162 mmol), Zn(CN)2 (48 mg, 0.41 mmol) and 15 mol% tetrakis(triphenylphosphine)palladium (28 mg, 0.024 mmol) were added to a 10 mL pressurized vial containing a magnetic stirrer. The vial was sealed and heated (using an Emrys Creator from Personal Chemistry AB, Uppsala, Sweden) at 80 °C for 45 min. The reaction was partitioned between a saturated solution of Na2CO3 (50 mL) and CH2Cl2 (50 mL). The organic layer was removed and the aqueous phase was extracted with CH2Cl2 (2 × 25 mL). The combined organic extracts were pooled, washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CH2Cl2/EtOAc (30:1) to yield 38 (38.5 mg, 0.125 mmol, 77%) as a white solid: mp 154–156 °C; 1H NMR (500 MHz, CDCl3, 45 °C) δ 8.14−8.10 (m, 2H), 7.91 (d, J = 7.4 Hz, 1H), 7.61−7.58 (m, 1H), 7.47 (d, J = 8.7 Hz, 1H), 5.03 (s, 2H), 3.79 (t, J = 5.9 Hz, 2H), 3.04 (t, J = 5.8 Hz, 2H), 1.56 (s, 9H); 13C NMR (500 MHz, CDCl3, 45 °C) δ 154.8, 133.9, 131.6, 131.3, 130.1, 129.1, 126.9, 125.3, 123.6, 117.7, 110.9, 80.1, 43.4, 40.9, 29.5, 28.4; HRMS (FAB+) m/z calcd for C19H21N2O2 (MH+) 309.1603, obsd 309.1592.
6.5.16. 7-Cyano-1,2,3,4-tetrahydrobenz[h]isoquinoline hydrochloride (18·HCl)
To a stirred solution of 38 (50 mg, 0.16 mmol) in CH2Cl2 (4 mL) was added TFA (1 mL). The reaction was stirred at ambient temperature for 2 h and then partitioned between basic brine (50 mL) and CH2Cl2 (50 mL). The organic layer was removed and the aqueous phase was extracted with CH2Cl2 (25 mL). The combined organic extracts were pooled and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CH2Cl2/MeOH (6:1) to yield 18 as a white solid. This amine was dissolved in CH2Cl2 and dry HCl(g) was bubbled through the solution to form the hydrochloride salt, which was recrystallized from EtOH/hexanes to yield 18·HCl (37 mg, 0.15 mmol, 93%) as white crystals: mp 255–257 °C; 1H NMR (500 MHz, CD3OD) δ 8.25 (d, J = 8.8 Hz, 1H), 7.92−7.90 (m, 2H), 7.54−7.51 (m, 2H), 4.82 (s, 2H), 3.63 (t, J = 6.3 Hz, 2H), 3.34 (d, J = 6.3 Hz, 1H); 13C NMR (800 MHz, CD3OD) δ 132.5, 131.6, 131.3, 129.8, 129.6, 127.0, 126.3, 124.5, 124.2, 116.9, 110.6, 42.2, 40.7, 25.3; HRMS (ESI+) m/z calcd for C14H13N2 (MH+) 209.1079, obsd 209.1077; Anal. Calcd for C14H12N2·⅛H2O: C, 68.08; H, 5.41; N, 11.34. Found: C, 68.10; H, 5.16; N, 11.22.
6.5.17. 7-Bromo-3,4-dihydrobenz[h]isoquinolin-1(2H)-one (39)
To a stirred solution of 35 (100 mg, 0.290 mmol) in dioxane (3 mL) was added lithium chloride (38.6 mg, 0.869 mmol), 5 mol% tetrakis(triphenylphosphine)palladium (16.7 mg, 0.0145 mmol), and hexamethylditin (60 μL, 0.29 mmol). The resulting mixture was stirred at 105 °C for 4 h. The reaction was then cooled to 0 °C and N-bromosuccinamide (62 mg, 0.35 mmol) was added. The reaction was stirred for 30 min at 0 °C and then permitted to warm to ambient temperature and stirred for an additional 30 min. The reaction was poured into a saturated solution of Na2S2O4/NaCl (1:1, 50 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were pooled, washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CH2Cl2/acetonitrile (8:1) to yield 39 (64.0 mg, 0.232 mmol, 80%) as a white solid: mp 199−201 °C; 1H NMR (500 MHz, CDCl3) δ 9.19 (d, J = 8.8 Hz, 1H), 8.19 (d, J = 8.6 Hz, 1H), 7.58−7.57 (m, 1H), 7.23−7.21 (m, 2H), 5.89 (b, 1H), 3.37−3.34 (m, 2H), 2.93 (t, J = 6.5 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 166.3, 140.6, 133.2, 131.8, 131.6, 130.1, 128.1, 126.7, 126.7, 122.9, 39.4, 30.4; HRMS (ESI+) m/z calcd for C13H11BrNO (MH+) 276.0024, obsd 276.0017.
6.6. General Procedure for Lactam Reduction. Synthesis of 14–17 and 19
The appropriate lactam (23, 27, 33, 34, or 39) (0.1–0.4 mmol) was dissolved in THF (10 mL) and 1M BH3·THF (10 equivalents) was added. The solution was heated at reflux for 4 h, cooled to ambient temperature, and MeOH (10 mL) was added dropwise. The solvent was removed under reduced pressure and a solution of MeOH (10 mL) and 6N HCl (10 mL) was added to the residue. The mixture was heated at reflux for 3 h and the MeOH was removed under reduced pressure. Water (25 mL) was added to the mixture, which was then made basic (pH ≈ 10) with 10% NaOH. The basic solution was extracted with CH2Cl2 (4 × 30 mL) and the combined organic extracts were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield the free amine, which was purified by flash chromatography eluting with EtOAc. The free amine was dissolved in CHCl3 and dry HCl(g) was bubbled through the solution to form the hydrochloride salt, which was recrystallized from EtOH/hexanes or MeOH/Et2O to yield THBQs 14–17 or 19.
6.6.1. 7-Nitro-1,2,3,4-tetrahydrobenz[h]isoquinoline hydrochloride (14·HCl)
Compound 23 (45 mg, 0.186 mmol) was reduced to THBQ 14 according to the General Procedure for Lactam Reduction (section 6.6). The hydrochloride salt was recrystallized from MeOH/ether to yield 14·HCl (37.0 mg, 0.140 mmol, 75%) as white crystals: mp 236−238 °C; 1H NMR (800 MHz, CD3OD) δ 8.34 (d, J = 9.0 Hz, 1H), 8.26−8.24 (m, 2H), 7.80−7.78 (m, 1H), 7.64 (d, J = 9.0 Hz, 1H), 4.87 (s, 2H), 3.64 (t, J = 6.3 Hz, 2H), 3.35 (t, J = 6.3 Hz, 2H),); 13C NMR (800 MHz, CD3OD) δ 148.0, 131.4, 131.0, 130.0, 127.3, 125.6, 123.8, 123.7, 122.8, 122.3, 42.5, 40.7, 25.3; HRMS (ESI+) m/z calcd for C13H13N2O2 (MH+) 229.0977, obsd 229.0970; Anal. Calcd for C13H12N2O2·¼H2O: C, 58.00; H, 5.05; N, 10.41. Found: C, 58.28; H, 4.76; N, 10.24.
6.6.2. 7-Aminosulfonyl-1,2,3,4-tetrahydrobenz[h]isoquinoline hydrochloride (15·HCl)
Compound 27 (30.0 mg, 0.109 mmol) was reduced to THBQ 15 according to the General Procedure for Lactam Reduction (section 6.6). The hydrochloride salt was recrystallized from MeOH/ether to yield 15·HCl (24.0 mg, 0.0803 mmol, 74%) as white crystals: mp 231–233 °C; 1H NMR (800 MHz, CD3OD) δ 8.72 (d, J = 9.0 Hz, 1H), 8.31 (d, J = 7.4 Hz, 1H), 8.14 (d, J = 8.6 Hz, 1H), 7.75−7.71 (m, 1H), 7.57 (d, J = 9.0 Hz, 1H), 4.86 (s, 2H), 3.64 (t, J = 6.3 Hz, 2H), 3.35 (t, J = 6.3 Hz, 2H); 13C NMR (800 MHz, CD3OD) δ 165.6, 141.4, 139.7, 135.9, 132.9, 129.2, 128.8, 128.2, 127.3, 125.7, 124.9, 39.2, 30.2; HRMS (ESI+) m/z calcd for C13H15N2O2S (MH+) 263.0854, obsd 263.0852; Anal. Calcd for C13H14N2O2S·¼H2O: C, 51.48; H, 5.15; N, 9.24. Found: C, 51.64; H, 5.16; N, 8.99. The structure of 15 was confirmed by X-ray crystallography. See section 7.
6.6.3. 7-Methoxy-1,2,3,4-tetrahydrobenz[h]isoquinoline hydrochloride (16·HCl)
Compound 33 (90.0 mg, 0.396 mmol) was reduced to THBQ 16 according to the General Procedure for Lactam Reduction (section 6.6). The hydrochloride salt was recrystallized from MeOH/ether to yield 16·HCl (78 mg, 0.31 mmol, 79%) as white crystals: mp 241–243 °C; 1H NMR (500 MHz, CD3OD) δ 8.23 (d, J = 8.8 Hz, 1H), 7.57−7.54 (m, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.34 (d, J = 8.8 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 4.75 (s, 2H), 4.03 (s, 3H), 3.60 (t, J = 6.3 Hz, 2H), 3.28 (t, J = 6.2 Hz, 2H); 13C NMR (500 MHz, CD3OD) δ 156.0, 131.0, 129.7, 127.4, 125.6, 124.2, 122.1, 121.9, 113.2, 104.2, 54.7, 42.6, 40.9, 25.3; HRMS (ESI+) m/z calcd for C14H14NO2 (MH+) 228.1024, obsd 228.1014.HRMS (ESI+) m/z calcd for C14H16NO (MH+) 214.1232, obsd 214.1227; Anal. Calcd for C14H15NO·HCl: C, 67.33; H, 6.46; N, 5.61. Found: C, 67.16; H, 6.26; N, 5.45.
6.6.4. 7-Hydroxy-1,2,3,4-tetrahydrobenz[h]isoquinoline hydrochloride (17·HCl)
Compound 34 (75.0 mg, 0.352 mmol) was reduced to THBQ 17 according to the General Procedure for Lactam Reduction (section 6.6). The hydrochloride salt was recrystallized from MeOH/ether to yield 17·HCl (61.8 mg, 0.262 mmol, 74%) as white crystals: mp 241–243 °C; 1H NMR (500 MHz, CD3OD) δ 8.22 (d, J = 8.7 Hz, 1H), 7.44−7.40 (m, 1H), 7.29 (d, J = 8.7 Hz, 1H), 7.28 (d, J = 8.5 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 4.72 (s, 2H), 3.60 (t, J = 6.3 Hz, 2H), 3.27 (t, J = 6.2 Hz, 2H); 13C NMR (500 MHz, CD3OD) δ 154.1, 131.3, 129.3, 127.5, 124.9, 123.6, 122.4, 121.9, 111.9, 108.1, 42.7, 41.0, 25.3; HRMS (ESI+) m/z calcd for C13H13NO (MH+) 200.1075, obsd 200.1057; Anal. Calcd for C13H14ClNO: C, 66.24; H, 5.99; N, 5.94. Found: C, 68.10; H, 5.90; N, 5.91.
6.6.5. 7-Bromo-1,2,3,4-tetrahydrobenz[h]isoquinoline hydrochloride (19·HCl)
Compound 39 (50.0 mg, 0.181 mmol) was reduced to THBQ 19 according to the General Procedure for Lactam Reduction (section 6.6). The hydrochloride salt was recrystallized from EtOH/hexanes to yield 19·HCl (39 mg, 0.13 mmol, 72%) as white crystals: mp dec 280–282 °C; 1H NMR (500 MHz, CD3OD) δ 8.25 (d, J = 8.8 Hz, 1H), 7.92−7.90 (m, 2H), 7.54−7.51 (m, 2H), 4.82 (s, 2H), 3.63 (t, J = 6.3 Hz, 2H), 3.33 (t, J = 6.3 Hz, 2H), 13C NMR (500 MHz, CD3OD) δ 131.3, 130.7, 130.6, 130.1, 128.2, 127.3, 126.9, 123.3, 123.1, 121.5, 42.4, 40.8, 25.3; HRMS (ESI+) m/z calcd for C13H13NBr (MH+) 262.0231, obsd 262.0227; Anal. Calcd for C13H12BrClN: C, 52.29; H, 4.39; N, 4.69. Found: C, 52.13; H, 4.23; N, 4.65.
7. X-ray Crystallography
The structures of compounds 23–25 and 15 have been deposited in the Cambridge Crystallographic Data Centre, reference numbers 619681–619684, respectively.
Acknowledgements
This research was supported by National Institutes of Health (NIH) Grant HL 34193. The funding for M.R.S. was also supported by NIH Predoctoral Training Grant GM 07775 and the American Foundation for Pharmaceutical Education. The funding for R.C.R. was also supported by the Madison and Lila Self Graduate Fellowship and the Pfizer Summer Undergraduate Research Fellowship. We thank David VanderVelde and Sarah Neuenswander of the University of Kansas Nuclear Magnetic Resonance Laboratory for their assistance. The 500 MHz NMR spectrometer was partially funded by National Science Foundation Grant CHE-9977422. We thank Victor Day of the University of Kansas X-ray Crystallography Laboratory, Gerald Lushington of the University of Kansas Molecular Graphics and Modeling Laboratory, and Todd Williams of the University of Kansas Mass Spectrometry Laboratory for their assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1. The contents of this paper were taken in large part from the Ph.D. dissertation (University of Kansas, 2006) of Mitchell R. Seim.
- 2.Cannon WB. The emergency function of the adrenal medulla in pain and the major emotions. Am. J. Physiol. 1914;33:356–372. [Google Scholar]
- 3.Vogt M. The Concentration of Sympathin in Different Parts of the Central Nervous System Under Normal Conditions and After the Administration of Drugs. J. Physiol. (London) 1954;123:451–81. doi: 10.1113/jphysiol.1954.sp005064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunne LM. Relative Adrenaline Content in Brain Tissue. Acta Biochim. Pol. 1962;56:324–333. doi: 10.1111/j.1748-1716.1962.tb02509.x. [DOI] [PubMed] [Google Scholar]
- 5.Axelrod J. Purification and Properties of Phenylethanolamine-N-Methyltransferase. J. Biol. Chem. 1962;237:1657–1660. [PubMed] [Google Scholar]
- 6.Fuller RW. Epinephrine in the Central Nervous System. Oxford University Press; New York: 1988. pp. 366–369. [Google Scholar]
- 7.Ruggiero DA, Cravo SL, Golanov E, Gomez R, Anwar M, Reis DJ. Adrenergic and Non-Adrenergic Spinal Projections of a Cardiovascular-Active Pressor Area of Medulla Oblongata: Quantitative Topographic Analysis. Brain Res. 1994;663:107–120. doi: 10.1016/0006-8993(94)90468-5. [DOI] [PubMed] [Google Scholar]
- 8.Crowley WR, Terry LC. Effects of an Epinephrine Synthesis Inhibitor, SKF64139, on the Secretion of Luteinizing Hormone in Ovariectomized Female Rats. Brain Res. 1981;204:231–235. doi: 10.1016/0006-8993(81)90670-3. [DOI] [PubMed] [Google Scholar]
- 9.Masaharu K, Atobe M, Nakagawara M, Kariya T. Effect of a Phenylethanolamine N-Methyltransferase Inhibitor, 2,3-Dichloro-methylbenzylamine, on the Alpha-2-adrenoceptor Function in the Hypothalamus in Rats. Neuropsychobiology. 1996;33:132–137. doi: 10.1159/000119263. [DOI] [PubMed] [Google Scholar]
- 10.Burke WJ, Galvin NJ, Chung HD, Stoff SA, Gillespie KN, Cataldo AM, Nixon RA. Degenerative Changes in Epinephrine Tonic Vasomotor Neurons in Alzheimer's Disease. Brain Res. 1994;661:35–42. doi: 10.1016/0006-8993(94)91177-0. [DOI] [PubMed] [Google Scholar]
- 11.Burke WJ, Chung HD, Marshall GL, Gillespie KN, Joh TH. Evidence for Decreased Transport of PNMT Protein in Advanced Alzheimer's Disease. J. Am. Geriatr. Soc. 1990;38:1275–1282. doi: 10.1111/j.1532-5415.1990.tb03448.x. [DOI] [PubMed] [Google Scholar]
- 12.Burke WJ, Chung HD, Strong R, Mattammal MB, Marshall GL, Nakra R, Grossberg GT, Haring JH, Joh TH. Mechanism of Degeneration of Epinephrine Neurons in Alzheimer's Disease. In: Strong R, editor. Central Nervous System Disorders of Aging: Clinical Intervention and Research. Raven Press; New York: 1988. pp. 41–70. [Google Scholar]
- 13.Grunewald GL, Dahanukar VH, Jalluri RK, Criscione KR. Synthesis, Biochemical Evaluation, and Classical and Three-Dimensional Quantitative Structure-Activity Relationship Studies of 7-Substituted-1,2,3,4-tetrahydroisoquinolines and Their Relative Affinities Toward Phenylethanolamine N-Methyltransferase and the α2-Adrenoceptor. J. Med. Chem. 1999;42:118–134. doi: 10.1021/jm980429p. [DOI] [PubMed] [Google Scholar]
- 14.Pendleton RG, Kaiser C, Gessner G. Studies on Adrenal Phenylethanolamine N-Methyltransferase (PNMT) with SK&F 64139, a Selective Inhibitor. J. Pharmacol. Exp. Ther. 1976;197:623–632. [PubMed] [Google Scholar]
- 15.Pendleton RG, Gessner G, Weiner G, Jenkins B, Sawyer J, Bondinell W, Intoccia A. Studies on SK&F 29661, an Organ-Specific Inhibitor of Phenylethanolamine N-Methyltransferase. J. Pharmacol. Exp. Ther. 1979;208:24–30. [PubMed] [Google Scholar]
- 16.Toomey RE, Horng JS, Hemrick-Luecke SK, Fuller RW. α2-Adrenoreceptor Affinity of Some Inhibitors of Norepinephrine N-Methyltransferase. Life Sci. 1981;29:2467–2472. doi: 10.1016/0024-3205(81)90701-3. [DOI] [PubMed] [Google Scholar]
- 17.Takakura Y, Audus KL, Borchardt RT. Blood-Brain Barrier: Transport Studies in Isolated Brain Capillaries and in Cultured Brain Endothelial Cells. Adv. Pharmacol. 1991;22:137–165. doi: 10.1016/s1054-3589(08)60034-4. [DOI] [PubMed] [Google Scholar]
- 18.Audus KL, Borchardt RT. Characterization of an In Vitro Blood-Brain Barrier Model System for Studying Drug Transport and Metabolism. Pharm. Res. 1986;3:81–87. doi: 10.1023/A:1016337202335. [DOI] [PubMed] [Google Scholar]
- 19.Grunewald GL, Caldwell TM, Li QF, Slavica M, Criscione KR, Borchardt RT, Wang W. Synthesis and Biochemical Evaluation of 3-Fluoromethyl-1,2,3,4-tetrahydroisoquinolines as Selective Inhibitors of Phenylethanolamine N-Methyltransferase Versus the α2-Adrenoceptor. J. Med. Chem. 1999;42:3588–3601. doi: 10.1021/jm990045e. [DOI] [PubMed] [Google Scholar]
- 20.Martin JL, Begun J, McLeish MJ, Caine JM, Grunewald GL. Getting the Adrenaline Going: Crystal Structure of the Adrenaline-Synthesizing Enzyme PNMT. Structure. 2001;9:977–985. doi: 10.1016/s0969-2126(01)00662-1. [DOI] [PubMed] [Google Scholar]
- 21.McMillan FM, Archbold J, McLeish MJ, Caine JM, Criscione KR, Grunewald GL, Martin JL. Molecular Recognition of Sub-Micromolar Inhibitors by the Epinephrine-Synthesizing Enzyme Phenylethanolamine N-Methyltransferase. J. Med. Chem. 2004;47:37–44. doi: 10.1021/jm0205752. [DOI] [PubMed] [Google Scholar]
- 22.Wu Q, Gee CL, Lin F, Tyndall JD, Martin JL, Grunewald GL, McLeish MJ. Structural, Mutagenic, and Kinetic Analysis of the Binding of Substrates and Inhibitors of Human Phenylethanolamine-N-Methyltransferase. J. Med. Chem. 2005;48:7243–7252. doi: 10.1021/jm050568o. [DOI] [PubMed] [Google Scholar]
- 23.Grunewald GL, Seim MR, Regier RC, Martin JL, Gee CL, Drinkwater N, Criscione KR. Comparison of the Binding of 3-Fluoromethyl-7-sulfonyl-1,2,3,4-tetrahydroisoquinolines with Their Isosteric Sulfonamides to the Active Site of Phenylethanolamine N-Methyltransferase. J. Med. Chem. 2006;49:5424–5433. doi: 10.1021/jm060466d. [DOI] [PubMed] [Google Scholar]
- 24.Gee CL, Tyndall JD, Grunewald GL, Wu Q, McLeish MJ, Martin JL. Binding mode of methyl acceptor substrates to the adrenaline-synthesizing enzyme phenylethanolamine N-methyltransferase: implications for catalysis. Biochemistry. 2005;44:16875–16885. doi: 10.1021/bi051636b. [DOI] [PubMed] [Google Scholar]
- 25.Girard GR, Bondinell WE, Hillegass LM, Holden KG, Pendleton RG, Uzinskas I. Tetrahydro thiadiazolo isoquinolines: synthesis and inhibition of phenylethanolamine-Nmethyltransferase. J. Med. Chem. 1989;32:1566–1571. doi: 10.1021/jm00127a027. [DOI] [PubMed] [Google Scholar]
- 26.Grunewald GL, Dahanukar VH, Caldwell TM, Criscione KR. Examination of the Role of the Acidic Hydrogen in Imparting Selectivity of 7-(Aminosulfonyl)-1,2,3,4-tetrahydroisoquinoline (SK&F 29661) Toward Inhibition of Phenylethanolamine-NMethyltransferase vs the α2-adrenoceptor. J. Med. Chem. 1997;40:3997–4005. doi: 10.1021/jm960235e. [DOI] [PubMed] [Google Scholar]
- 27.Beaumont D, Waigh RD. Bischler-Napieralski cyclization of N-[2-(2-naphthyl)ethyl] amides. J. Chem. Res., Synop. 1979:332. [Google Scholar]
- 28. The structure of 26 was confirmed by the X-ray crystal structure of THBQ 15.
- 29.Donkor IO, Devraj R, Queener SF, Barrows LR, Gangjee A. Synthesis of a series of diaminobenzo[f]- and diaminobenzo[h]pyrimido[4,5-b]quinolines as 5-deaza tetracyclic nonclassical antifolates. J. Heterocycl. Chem. 1996;33:1653–1661. [Google Scholar]
- 30.Kar GK, Karmakar AC, Ray JK. Studies on polycyclic thiaarenes. Part II. An improved synthesis of phenanthro[1,2-b]thiophene and a synthesis of acenaphtho[1,2-b]naphtho[2,3-d]thiophene, a novel polynuclear thiaarene. J. Heterocycl. Chem. 1991;28:999–1002. [Google Scholar]
- 31.Pavlic AA, Adkins H. Preparation of a Raney nickel catalyst. J. Am. Chem. Soc. 1946;68:1471. [Google Scholar]
- 32.Smith AB, III, Cantin L-D, Pasternak A, Guise-Zawacki L, Yao W, Charnley AK, Barbosa J, Sprengeler PA, Hirschmann R, Munshi S, Olsen DB, Schleif WA, Kuo LC. Design, Synthesis, and Biological Evaluation of Monopyrrolinone-Based HIV-1 Protease Inhibitors. J. Med. Chem. 2003;46:1831–1844. doi: 10.1021/jm0204587. [DOI] [PubMed] [Google Scholar]
- 33.Lumma WC., Jr. Modification of the Passerini Reaction: Facile Synthesis of Analogs of Isoproterenol and (Aryloxy)propanolamine β-Adrenergic Blocking Agents. J. Org. Chem. 1981;46:3668–3671. [Google Scholar]
- 34.Zhang A, Neumeyer JL. Microwave-Promoted Pd-Catalyzed Cyanation of Aryl Triflates: A Fast and Versatile Access to 3-Cyano-3-desoxy-10-ketomorphinans. Org. Lett. 2003;5:201–203. doi: 10.1021/ol027256p. [DOI] [PubMed] [Google Scholar]
- 35.Wiley GA, Hershkowitz RL, Rein BM, Chung BC. Organophosphorus chemistry. I. Conversion of alcohol and phenols to halides by tertiary phosphine dihalides. J. Am. Chem. Soc. 1964;86:964–965. [Google Scholar]
- 36.Wiley GA, Rein BM, Herschkowitz RL. Organophosphorus chemistry. II. Mechanism of reaction of tertiary phosphine dihalides with alcohols. Tetrahedron Lett. 1964:2509–13. [Google Scholar]
- 37.Wulff WD, Peterson GA, Bauta WE, Chan K-S, Faron KL, Gilbertson SR, Kaesler RW, Yang DC, Murray CK. A regioselective entry to vinyl lithiums from unsymmetrical ketones via enol triflates. J. Org. Chem. 1986;51:277–279. [Google Scholar]
- 38.Eaborn C, Najam AA, Walton DRM. 1,2-Dihydrobenzocyclobutene, (‘benzocyclobutene’). Lithiation, and the preparation of 3-substituted derivatives. J. Chem. Soc., Perkin Trans. 1972;1:2481–2484. [Google Scholar]
- 39.Romero FA, Vodonick SM, Criscione KR, McLeish MJ, Grunewald GL. Inhibitors of Phenylethanolamine N-Methyltransferase that are Predicted to Penetrate the Blood-Brain Barrier: Design, Synthesis, and Evaluation of 3-Fluoromethyl-7-(N-substituted aminosulfonyl)-1,2,3,4-tetrahydroisoquinolines that Possess Low Affinity Toward the α2-Adrenoceptor. J. Med. Chem. 2004;47:4483–4493. doi: 10.1021/jm0400653. [DOI] [PubMed] [Google Scholar]
- 40.Caine JM, Macreadie IG, Grunewald GL, McLeish MJ. Recombinant Human Phenylethanolamine N-Methyltransferase: Overproduction in Escheria coli, Purification and Characterization. Protein Expression Purif. 1996;8:160–166. doi: 10.1006/prep.1996.0088. [DOI] [PubMed] [Google Scholar]
- 41.Grunewald GL, Borchardt RT, Rafferty MF, Krass P. Conformational Preferences of Amphetamine Analogues for Inhibition of Phenylethanolamine N-Methyltransferase: Conformationally Defined Adrenergic Agents. 5. Mol. Pharmacol. 1981;20:377–381. [PubMed] [Google Scholar]
- 42.Wu Q, Criscione KR, Grunewald GL, McLeish MJ. Phenylethanolamine N-Methyltransferase Inhibition: Re-evaluation of Kinetic Data. Bioorg. Med. Chem. Lett. 2004;14:4217–4220. doi: 10.1016/j.bmcl.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 43.U'Prichard DC, Greenberg DA, Snyder SH. Binding Characteristics of a Radiolabeled Agonist and Antagonist at Central Nervous System Alpha Noradrenergic Receptors. Mol. Pharmacol. 1977;13:454–473. [PubMed] [Google Scholar]
- 44.Grunewald GL, Romero FA, Seim MR, Criscione KR, Deupree JD, Spackman CC, Bylund DB. Exploring the Active Site of Phenylethanolamine N-Methyltransferase with 3-Hydroxyethyl- and 3-Hydroxypropyl-7-Substituted-1,2,3,4-tetrahydroisoquinolines. Bioorg. Med. Chem. Lett. 2005;15:1143–1147. doi: 10.1016/j.bmcl.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 45.Morris GM, Goodsell DS, Halliday RS, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 46.Davis AM, Teague SJ, Kleywegt GJ. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew. Chem. 2003;42:2718–2736. doi: 10.1002/anie.200200539. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 47. SYBYL® 7.0 Tripos Inc., 1699 South Hanley Rd., St. Louis, MO 63144, USA.
- 48. All of the compounds in this study show competitive kinetics with phenylethanolamine. This figure shows only the norepinephrine binding site, and not the AdoHcy binding site.
- 49.Sall DJ, Grunewald GL. Inhibition of Phenylethanolamine N-Methyltransferase (PNMT) by Aromatic Hydroxy-substituted 1,2,3,4-Tetrahydroisoquinolines: Further Studies on the Hydrophilic Pocket of the Aromatic Ring Binding Region of the Active Site. J. Med. Chem. 1987;30:2208–2216. doi: 10.1021/jm00395a006. [DOI] [PubMed] [Google Scholar]