

Summary
Postmortem brain loss of reelin is noted in schizophrenia patients. Accordingly, heterozygous reeler mutant mice have been proposed as a putative model of this disorder. Little is known, however, about the involvement of the two receptors for reelin, Very-Low-Density Lipoprotein Receptor (VLDLR) and Apolipoprotein E Receptor 2 (ApoER2), on pre-cognitive processes of relevance to deficits seen in schizophrenia. Thus, we evaluated sensorimotor gating in mutant mice heterozygous or homozygous for the two reelin receptors. Mutant mice lacking one of these reelin receptors were tested for prepulse inhibition (PPI) of the acoustic startle reflex prior to and following puberty, and on a crossmodal PPI task, involving the presentation of acoustic and tactile stimuli. Furthermore, because schizophrenia patients show increased sensitivity to N-methyl-D-aspartate (NMDA) receptor blockade, we assessed the sensitivity of these mice to the PPI-disruptive effects of the NMDA receptor antagonist phencyclidine. The results demonstrated that acoustic PPI did not differ between mutant and wildtype mice. However, VLDLR homozygous mice displayed significant deficits in crossmodal PPI, while ApoER2 heterozygous and homozygous mice displayed significantly increased crossmodal PPI. Both ApoER2 and VLDLR heterozygous and homozygous mice exhibited greater sensitivity to the PPI-disruptive effects of phencyclidine than wildtype mice. These results indicate that partial or complete loss of either one of the reelin receptors results in a complex pattern of alterations in PPI function that include alterations in crossmodal, but not acoustic, PPI and increased sensitivity to NMDA receptor blockade. Thus, reelin receptor function appears to be critically involved in crossmodal PPI and the modulation of the PPI response by NMDA receptors. These findings have relevance to a range of neuropsychiatric disorders that involve sensorimotor gating deficits, including schizophrenia..
Keywords: Crossmodal, Knockout mouse, Prepulse inhibition, Phencyclidine, Reelin, Schizophrenia
Introduction
The large extracellular matrix protein reelin is expressed by Cajal-Retzius cells during cortical development, and plays an essential role in the organization of the mammalian brain. In mice, loss of the reelin gene product results in dramatic disturbances in brain architecture, as exemplified by the reeler mouse, which displays severe neuronal migratory aberrations in regions such as the cortex, hippocampus and cerebellum (Rice and Curran, 2001). The two known receptors for reelin, ApoER2 and VLDLR, transduce the reelin signal to intracellular signaling molecules. In early cortical development, reelin-induced signal transduction regulates neuronal positioning along radial glia, as well as dendrite development (D'Arcangelo, 2006). In adulthood, reelin mediated signaling regulates synaptic plasticity (Beffert et al., 2005). The loss of both reelin receptors results in neuroarchitectural changes that appear to be identical to those found in the reeler mouse (Trommsdorff et al., 1999).
Postmortem studies have reported consistently decreased levels of reelin and its message, by approximately 30–50%, in the frontal cortex, hippocampus, caudate and cerebellum of schizophrenia and bipolar patients, with less consistent findings for unipolar depression (Fatemi et al., 2000; Fatemi et al., 2005; Guidotti et al., 2000; Impagnatiello et al., 1998; Torrey et al., 2005). Levels of ApoER2 and VLDLR in the brains of patients remain undetermined. The finding of reduced reelin levels in these psychiatric disorders has prompted interest in the reelin signaling pathway as a neurobiological substrate that may contribute to the etiology of these disorders. Consequently, there has been substantial interest in the heterozygous reeler mouse, that is reelin haploinsufficient, as an animal model of psychotic disorders, and schizophrenia in particular (Fatemi, 2001; Pappas et al., 2003; Tueting et al., 1999). Unlike the striking neuroanatomical aberrations of the homozygous reeler mouse, the heterozygous reeler mouse exhibits more constrained neuropathology resembling that of schizophrenia, including decreased neuropil and dendritic spine density (Ballmaier et al., 2002; Costa et al., 2002).
Behavioral phenotyping of the heterozygous reeler mouse has uncovered deficits homologous to those noted in schizophrenia. Specifically, heterozygous reeler mice exhibit increased sensitivity to the disruptive effects of the non-competitive NMDA receptor antagonist dizocilpine on cognition (Carboni et al., 2004). This finding is relevant, as NMDA receptor blockade mimics aspects of schizophrenia in healthy human volunteers and exacerbates negative and positive symptoms in schizophrenia patients (Krystal et al., 2003; Steinpresis, 1996). An initial behavioral study by Tueting and colleagues also reported that post-pubertal heterozygous reeler mice had reduced prepulse inhibition (PPI) of the acoustic startle reflex. Prepulse inhibition is a pre-attentional sensorimotor gating phenomenon frequently decreased in psychotic disorders, such as schizophrenia (Braff et al., 2001), and can be modeled in homologous behavioral paradigms with rodents (Barr et al., 2004a; Barr et al., 2006). More recently, however, two separate research groups have been unable to confirm the presence of PPI deficits in heterozygous reeler mice (Podhorna and Didriksen, 2004; Salinger et al., 2003). The discrepancy in findings between research groups remains unresolved. Differences in behavior are unlikely to result from strain differences, as mice were obtained from the same supplier (Jackson labs). One possible explanation may be differences in the PPI protocol, as this paradigm is sensitive to experimental adjustments (Geyer et al., 2002), including the age at which animals are tested.
To evaluate further the hypothesis that reelin signaling is involved in sensorimotor gating deficits, in the present study we assessed PPI in mutant mice lacking either of the two receptors (ApoER2 or VLDLR) for reelin under a variety of experimental conditions, including a pharmacological challenge. As far as we are aware, there are no previous reports of PPI measures in mice lacking either of the reelin receptors. Thus, these mice provide the opportunity to assess further the potential role of reelin signaling in sensorimotor gating deficits using an alternative animal procedure than the reelin mutant mice that have provided inconsistent findings.
PPI can consist of either unimodal or crossmodal protocols. In the former, both prepulse and pulse are of the same sensory modality (typically acoustic); in the crossmodal task, prepulse and pulse are of different modalities, such as acoustic and tactile. Although reduced unimodal and crossmodal PPI have both been reported in schizophrenia (Braff et al., 1992), mouse studies indicate that performance on unimodal PPI may be unrelated to crossmodal PPI (Bullock et al., 1997). Further, there is evidence suggesting that different brain regions are involved in unimodal versus crossmodal PPI (Swerdlow et al., 2001a). Therefore, to provide a comprehensive analysis of PPI in these animals, mice were tested for unimodal acoustic PPI throughout development, starting before completion of puberty and continuing into adulthood. To test whether mutant mice may also display differential response to unimodal versus crossmodal stimuli, animals were additionally tested in adulthood with a crossmodal PPI paradigm that combined an acoustic prepulse with a tactile pulse stimulus. Finally, as both schizophrenia patients and heterozygous reeler mice are more sensitive to the disruptive effects of NMDA receptor antagonists on cognition, we determined whether mutant mice were more sensitive to the effects of a pharmacological challenge with the NMDA receptor antagonist phencyclidine (PCP) on unimodal acoustic PPI behavior.
Materials and Methods
Animals
ApoER2 homozygous knockout (−/−) and VLDLR homozygous knockout (−/−) mutant mice on a C57/B6;129S6 background were obtained from The Jackson Laboratory (JAX; Bar Harbor, ME) and back-crossed onto a pure C57/Bl6 background for 5 generations. For all studies, ApoER2 and VLDLR knockout (−/−), heterozygous (−/+) and wildtype (+/+) male and female mice were obtained from heterozygous breeding pairs of mice. Mice were group-housed and given ad libitum access to food and water; all animals were maintained on a 12 h light-dark cycle, with lights on at 07:00 am. Behavioral testing occurred during the light cycle. Mice were weighed with an electronic scale before each test session. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Genotyping was performed by PCR using protocols provided by JAX.
Drugs
Phencyclidine (NIDA, Bethesda MD) was dissolved in vehicle (isotonic saline). Three different doses of PCP (10, 15 and 20 mg/kg) were used in Experiment 3. All drug doses were dissolved in a volume of 5 ml/kg, and injected intraperitoneally (i.p.) with a 27-gauge syringe needle. Doses are expressed as the salt.
Prepulse inhibition apparatus and procedure
Four startle chambers were used to measure the startle response (SR-LAB, San Diego Instruments, San Diego, CA), as described previously (Barr et al., 2004a). Each chamber consisted of a non-restrictive Plexiglas™ cylinder mounted on a frame inside a lit ventilated box (39 × 38 × 58 cm). Movements within the cylinder were detected by a piezoelectric accelerometer that was attached beneath the cylinder. Vibrations detected by the accelerometer were transduced into analog electrical signals that were subsequently digitized and stored by the computer. Sixty-five readings were recorded at 1 msec intervals, commencing at stimulus onset, and the average amplitude was used to describe the acoustic startle response. A high frequency loudspeaker inside the box, mounted 24 cm above the chamber, generated the broadband background noise and acoustic stimuli, which were controlled by the SR-LAB software system and interface. Tactile stimuli (airpuff) were governed by computer-controlled activity of compressed air, at a pressure of 30 p.s.i. leaving the regulator. Sound levels [dB(A) scale] and accelerometer sensitivities were monitored routinely to ensure consistent sensitivities across test sessions.
Mice were assessed on different PPI tasks commencing at postnatal day 40 (Table 1), beginning with unimodal acoustic PPI prior to puberty, followed by postpubertal testing at postnatal day 80 using the same protocol (Experiment 1). Subjects were then tested for crossmodal PPI using a task with acoustic prepulses and tactile pulse stimuli (Experiment 2). Finally, mice were assessed for their sensitivity to the disruptive effects of PCP on the same unimodal acoustic PPI session as before (Experiment 3). PCP studies were conducted last to prevent any possible “carryover” effects of the drug on PPI (Spielewoy and Markou, 2003). Analysis of data did not indicate statistically significant difference between sexes on PPI measures, therefore data from males and females were pooled. For Experiments 1 and 2, male and female ApoER2 (+/+) (10 ♂, 12♀), (+/−) (17 ♂, 14 ♀) and (−/−) (13 ♂, 13 ♀) mice were used; male and female VLDLR (+/+) (9 ♂, 13 ♀), (+/−) (29 ♂, 23♀) and (−/−) (18 ♂, 21 ♀) mice were used. For Experiment 3, for both ApoER2 and VLDLR mice, the same (+/+) and (−/−) mice were used as for Experiments 1 and 2. However, a number of VLDLR mice, of both sexes, were randomly selected to be removed for breeding mutant mice for future work. Due to their breeding experience, these mice were not included in the PCP studies of Experiment 3 (see Table 1). The numbers of animals represent the random numbers of offspring generated by equal numbers of breeding pairs.
Table 1.
Timeline of testing prepulse inhibition (PPI) behavior in ApoER2 and VLDLR wildtype, heterozygous and homozygous mutant mice. Animals were tested at different stages of development and using different PPI protocols.
| Postnatal Day | PPI Protocol | # ApoER2 mice | # VLDLR mice |
|---|---|---|---|
| 40 | Unimodal acoustic PPI | 22 WT, 31 HT, 26 KO | 22 WT, 56 HT, 39 KO |
| 80 | Unimodal acoustic PPI | 22 WT, 31 HT, 26 KO | 22 WT, 55 HT, 39 KO |
| 90 | Crossmodal PPI (acoustic and tactile stimuli) | 22 WT, 31 HT, 26 KO | 22 WT, 55 HT, 39 KO |
| 120–150 | PCP challenge (10, 15, 20 mg/kg or vehicle) | 22 WT, 31 HT, 26 KO | 19 WT, 26 HT, 27 KO |
PCP = phencyclidine; WT = wildtype; HT = heterozygous; KO = knockout.
Experiment 1
For the acoustic startle experiments at postnatal days 40 and 80 (see Table 1), mice were tested with observers blind to genotype. Postnatal days 40 and 80 were chosen as they represent time points in mice that span from early adolescence (before completion of puberty) to postpubertal early adulthood. These time points in humans represent stages of development that are typically prior to and after the first episode in schizophrenia (Larsen et al., 1996). For this reason, PND 40 was used in the original study of PPI deficits in heterozygous reeler mice (Tueting et al., 1999); we have chosen to be consistent with this study. PND 80 occurs well after puberty is completed in mice. The experimental session consisted of a 5 min acclimatization period in which only broadband background noise (65 dB) was presented. The acclimatization was followed by a PPI session that consisted of five different trial types: no stimulus trials (NOSTIM); a startle pulse alone trial of 40 msec at 120 dB (P120); and three prepulse + pulse trials of a 20 msec noise prepulse at either 2 (PP2), 4 (PP4) or 8 (PP8) dB above 65 dB background noise, resulting in 67, 69 and 73 dB prepulses followed by a 80 msec delay, then a 40 msec 120 dB startle pulse. The NOSTIM trial consisted of only background broadband noise (65 dB background). All test sessions commenced and concluded with five presentations of the P120 trial, while the remainder of the session consisted of 10 presentations of each trial type in a pseudorandom order, with varying intertrial intervals (mean of 15 sec, range of 12–30 sec). Chambers were cleaned between every session, and each animal was always tested in the same startle chamber.
Experiments 2–3
At postnatal day 90, the same mice from Experiment 1 were used for the crossmodal experiments in Experiment 2 (see Table 1). For crossmodal studies, testing sessions were used that contained both acoustic prepulse/pulse trials as above (P120 and PP8) as well as airpuff alone trials (PUF) and a prepulse 73 db (8 dB above background) followed 80 msec later by an airpuff (PREPUF). These sessions started and ended with five presentations of the P120 trial while the remainder of the session consisted of ten presentations of each trial type in a pseudorandom order, with varying intertrial intervals (mean of 15 sec, range of 12–30 sec). To assess the effects of PCP, in Experiment 3, the same mice used previously in Experiments 1 and 2 were used, with testing initiated on postnatal day 120. Animals were randomly assigned to one of four counterbalanced groups (vehicle, 10, 15 and 20 mg/kg PCP) and tested with all doses on separate days, in a within-subjects design, separated by at least one week between each treatment. Doses of PCP were based on those described in the literature to have behavioral effects in mice of a similar C57/Bl6 background (Dulawa and Geyer, 1996; Yee et al., 2004). Drug was injected 30 min before the animals were placed into the startle chambers. A standard acoustic session, identical to the one used in Experiment 1, was used to test the effects of PCP on PPI after PCP/vehicle administration.
Data and statistical analyses
Prepulse inhibition was calculated as a percentage score for each prepulse intensity by the equation: % prepulse inhibition = 100 − {(startle response for prepulse + pulse trials [PP2, PP4 or PP8]) / (startle response for pulse alone [P120]) × 100} (Barr et al., 2006). The startle magnitude was calculated as the average of all pulse alone trials, excluding the first and last five such trials in each session. As preliminary analysis of data indicated that there was no significant main effect or interactive effect of Sex (data not shown), data from both sexes were combined for all indices. Startle magnitude and habituation data for Experiments 1 and 2 were analyzed by one factor within-group design ANOVAs, with Genotype as the between group factor. To analyze the PPI data from Experiments 1 and 2, two-way mixed-design ANOVAs were used, with Genotype as the between-subjects factor and Prepulse Intensity as the within-subject factor. The PPI and habituation data from Experiment 3, involving the assessment of the effects of PCP, were analyzed using a three-factor mixed-design ANOVA, with Drug and Prepulse Intensity as the within-subjects factors and Genotype as the between-subjects factor. Fisher’s LSD tests were conducted for post-hoc analysis when applicable. The level of significance was set at .05. Data were analyzed using SYSTAT (SPSS, Inc., Chicago, IL).
Results
Consistent with previous reports, both ApoER2 and VLDLR (+/−) and (−/−) mice appeared indistinguishable from wildtypes and did not display any obvious behavioral abnormalities with casual inspection. There were no significant differences in bodyweight between genotypes for either ApoER2 or VLDLR mutant mice prior to or following puberty (data not shown).
Experiment 1
Testing of ApoER2 mice on postnatal day 40 with a unimodal acoustic PPI session did not reveal any significant effect of Genotype on startle magnitude, but there was a significant effect on magnitude of startle habituation [F(2,76) = 4.45, p < 0.05] (Table 2). Posthoc analysis indicated that this was due to significantly lower startle habituation in ApoER2 (−/−) mice compared to (+/−) animals; although there was a trend for habituation levels of (−/−) mice to be lower than (+/+) mice, this effect was not statistically significant. The ANOVA on PPI data (Figure 1) indicated a significant main effect of Prepulse Intensity [F(2,152) = 188.17, p < 0.001], but no main effect of Genotype, and no interaction effect. Analysis of the VLDLR mice on postnatal day 40 indicated no significant main effect of Genotype on startle magnitude or startle habituation. The ANOVA indicated a significant main effect of Prepulse Intensity [F(2,230) = 318.47, p < 0.001] on PPI, but no effect of Genotype, even though PPI levels tended to be lower across all three prepulse intensities for VLDLR (−/−) mice compared to VLDLR (+/−) and VLDLR (+/+) mice (Figure 1). There was no significant interaction effect of Genotype × Prepulse Intensity.
Table 2.
Startle habituation in ApoER2 and VLDLR mice. Values represent group mean (± SEM) percentage habituation to an acoustic startle stimulus.
| ApoER2 | Postnatal Day 40 | Postnatal Day 80 | PCP study Vehicle | PCP study 10 mg/kg | PCP study 15 mg/kg | PCP study 20 mg/kg# |
|---|---|---|---|---|---|---|
| WT | 24.2 ± 4.1 | 30.6 ± 6.0 | 21.1 ± 6.5 | 25.4 ± 6.2 | 15.9 ± 8.5 | 5.6 ± 7.5 |
| HT | 31.0 ± 3.3 | 18.2 ± 5.3 | 10.5 ± 10.2 | 5.4 ± 8.8§ | 14.4 ± 5.5 | −17.4 ± 16.3 |
| KO | 17.1 ± 5.3* | 14.4 ± 9.8 | 16.2 ± 6.3 | 21.5 ± 4.9 | 17.7 ± 5.1 | 10.8 ± 6.1 |
| VLDLR | ||||||
| WT | 13.8 ± 4.7 | 27.1 ± 5.0 | 26.5 ± 4.8 | 25.5 ± 5.8 | 10.9 ± 8.5 | 5.3 ± 13.4 |
| HT | 22.1 ± 3.0 | 23.5 ± 3.1 | 30.5 ± 3.5 | 3.9 ± 12.7 | −2.0 ± 9.7 | −5.3 ± 8.8 |
| KO | 18.0 ± 3.4 | 23.2 ± 3.8 | 22.3 ± 4.4 | 26.7 ± 4.5 | 15.3 ± 5.0 | 8.8 ± 6.7 |
= significantly different compared to ApoER2 heterozygous (HT) mice, p < 0.05;
= significantly different compared to ApoER2 wildtype (WT) mice, p < 0.05;
= significantly different compared to same genotype vehicle-treated mice, p < 0.01. KO = homozygous knockout.
Figure 1.
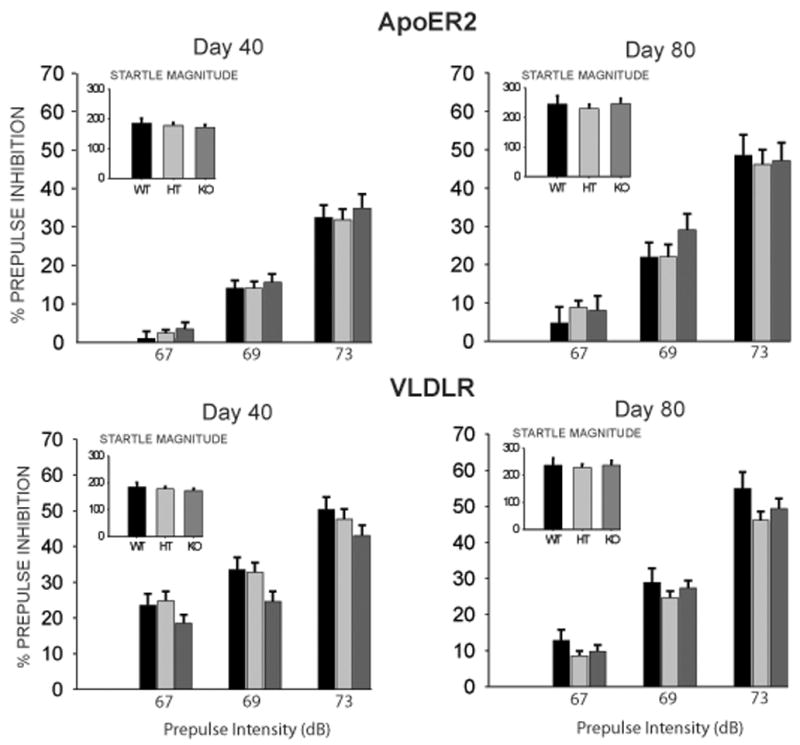
(A) ApoER2 and (B) VLDLR wildtype, heterozygous and homozygous knockout mice were tested for prepulse inhibition to an acoustic startle stimulus prior to puberty (postnatal day 40) and after puberty (postnatal day 80). Test sessions consisted of individual trials with prepulse intensitites 2, 4 and 8 dB above 65 dB background noise (i.e., 67, 69 and 73 dB prepulses). Values represent group means (± SEM). Startle magnitudes (arbitrary units) are inset in graph. No significant differences between genotypes were evident at either point in development (see text for details). WT = Wildtype (ApoER2, n = 22; VLDLR, n = 22); HT = Heterozygote (ApoER2, n = 31; VLDLR, n = 52); KO = Homozygous knockout (ApoER2, n = 26; VLDLR, n = 39).
When mice were tested again with the identical acoustic PPI session following puberty on postnatal day 80, similar results were obtained as on postnatal day 40. For ApoER2 mice, there was no effect of Genotype on acoustic startle, and though levels of startle habituation in (−/−) mice were only half those of (+/+) mice, this difference was not statistically significant as there was no main effect of Genotype. There was also no significant main effect of Genotype on PPI (Figure 1), nor a significant interaction between Genotype and Prepulse Intensity. For the VLDLR mice on postnatal day 80, the ANOVA indicated no significant effect of Genotype on acoustic startle, startle habituation or PPI, and no interaction effect of Genotype and Prepulse intensity (Figure 1).
Experiment 2
In Experiment 2, mice were assessed for PPI in a crossmodal test session that combined both acoustic PPI trials as well as crossmodal trials that consisted of an acoustic prepulse with a tactile (airpuff) startle stimulus (Figure 2). Analysis indicated that there was no significant main effect of Genotype of ApoER2 mice on acoustic or tactile startle magnitude, nor was there an effect on acoustic startle habituation (data not shown). Further, there was no significant main effect of Genotype on unimodal acoustic PPI, consistent with the results of Experiment 1 at postnatal day 80. However, there was a striking effect of Genotype on crossmodal PPI [F(2,75) = 5.73, p < 0.005]. Further, posthoc analysis indicated that this effect was due to a large and significant increase in the levels of crossmodal PPI in ApoER2 (−/−) and (+/−) mice compared to wildtype (+/+) animals. Results of VLDLR mutant mice revealed a contrasting effect of Genotype on crossmodal PPI. That is, while the ANOVA indicated no significant main effect of Genotype on acoustic startle, tactile startle, acoustic startle habituation or acoustic PPI, there was a significant main effect of Genotype on crossmodal PPI [F(2,114) = 4.68, p < 0.05]. Further, posthoc analysis indicated that unlike ApoER2 mice, the VLDLR (−/−) mice exhibited significantly reduced levels of cross-modal PPI compared to both (+/−) and (+/+) animals.
Figure 2.
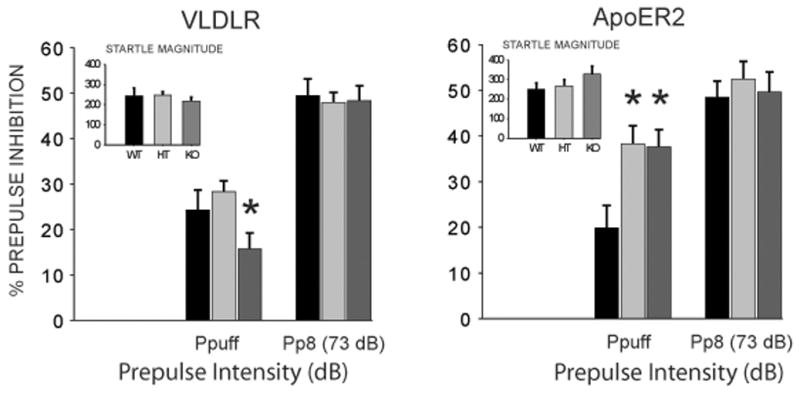
On postnatal day 90, ApoER2 and VLDLR mutant mice were tested for deficits in acoustic (Pp8, 73 dB) and multimodal (Ppuff) trials of prepulse inhibition. The latter task consisted of a combination of acoustic prepulses with a tactile (airpuff) startle stimulus. Values represent group means (± SEM). Startle magnitudes (arbitrary units) are inset in graph. * indicates significantly different compared to wildtype and heterozygous mice, p < 0.05. WT = Wildtype (ApoER2, n = 22; VLDLR, n = 22); HT = Heterozygote (ApoER2, n = 31; VLDLR, n = 52); KO = Homozygous knockout (ApoER2, n = 26; VLDLR, n = 39).
Experiment 3
In Experiment 3, all animals were treated in a randomized manner with either vehicle or 10, 15 or 20 mg/kg of PCP, and tested with the same unimodal acoustic PPI session that was used in Experiment 1. Analysis of startle magnitude in ApoER2 mice indicated that while there was no effect of Genotype on startle, nor a significant interaction of Genotype × Drug dose, there was a highly significant effect of Drug dose on startle levels [F(3,228) = 17.03, p < 0.001], with the 15 and 20 mg/kg doses significantly reducing startle magnitude. Analysis of startle habituation revealed a significant main effect of Genotype [F(2,76) = 3.91, p < 0.05] and Drug dose [F(3,228) = 2.93, p < 0.05] as levels of habituation decreased with the 20 mg/kg dose, although there was no Genotype × Dose interaction. The Genotype effect was due to lower levels of startle habituation in the (+/−) mice compared to the (+/+) and (−/−) animals (see Table 2).
Analysis of PPI data by ANOVA demonstrated that there were highly significant main effects of Drug dose [F(3,228) = 37.37, p < 0.0001] and Prepulse Intensity [F(2,152) = 293.84, p < 0.0001]. There was also a significant interaction of Drug × Prepulse Intensity [F(6,456) = 8.44, p < 0.001] and a significant three-way interaction between Genotype × Drug × Prepulse Intensity [F(12,456) =1.76, p < 0.05]. Posthoc tests revealed that levels of PPI were significantly reduced across all prepulse intensities with increasing PCP doses for all genotypes (Figure 3). In addition, both (+/−) and (−/−) mutant mice displayed significantly greater reductions in PPI than (+/+) mice across most prepulse intensities, with significantly greater reductions at the 69 dB prepulse intensity in the 15 mg/kg dose, and the 73 dB prepulse intensity in both the 10 and 15 mg/kg doses. Lack of genotype effect at the 20 mg/kg dose may represent a “floor effect”, as levels of PPI were already suppressed strongly in (+/+) mice, decreasing the power to detect group differences. The latter findings demonstrate a greater sensitivity to the sensorimotor gating disruptive effects of PCP in ApoER2 (+/−) and (−/−) mutant mice.
Figure 3.
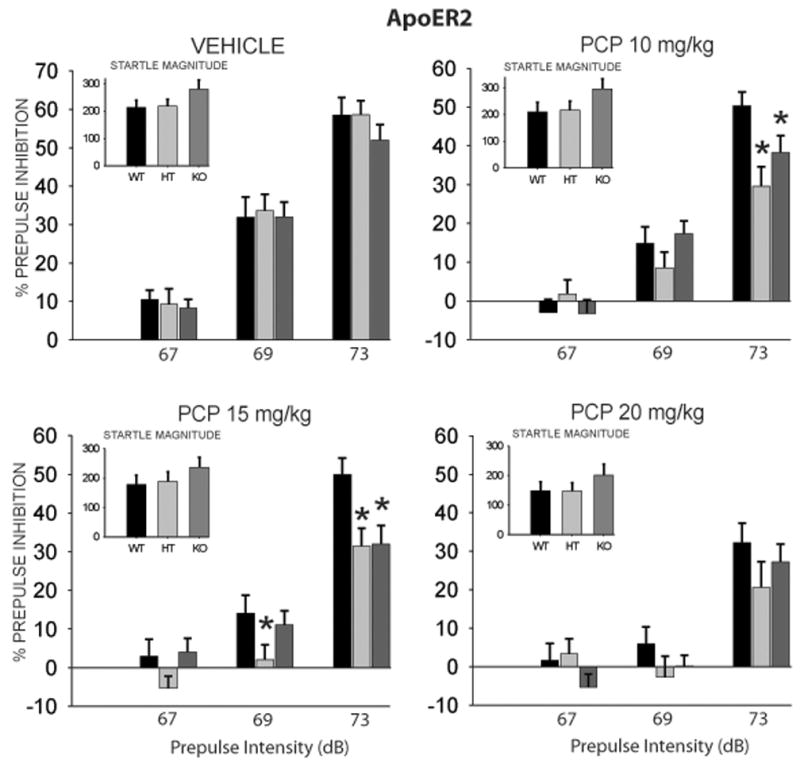
ApoER2 mice on postnatal day 120 were randomly assigned to treatment with vehicle, 10 mg/kg, 15 mg/kg, or 20 mg/kg of the NMDA-receptor antagonist phencyclidine (PCP). All mice received all doses, in a counterbalanced order. Animals were tested for prepulse inhibition to an acoustic startle stimulus. Test sessions consisted of individual trials that presented prepulse acoustic stimuli 2, 4 and 8 dB above 65 dB background noise (i.e., 67, 69 and 73 dB). Values represent group means (± SEM). Startle magnitudes (arbitrary units) at each dose are inset in graph. * indicates significantly different compared to wildtype mice, p < 0.05. WT = Wildtype (n = 22); HT = Heterozygote (n = 31); KO = Homozygous knockout (n = 26).
For the VLDLR mice, the analysis of startle magnitude indicated that there was no effect of Genotype on startle, but a significant effect of Drug dose on startle levels [F(3,207) = 15.10, p < 0.001], with all three PCP doses significantly decreasing startle magnitude. There was no interaction of Genotype × Drug dose. The analysis of startle habituation in VLDLR mice confirmed a strong trend for a main effect of Genotype [F(2,69) = 2.85, p = 0.06] and a significant effect of Drug dose [F(3,297) = 5.57, p < 0.001] (see Table 2), although no significant interaction of Genotype × Drug dose. Posthoc analysis of startle habituation values revealed that the marginal Genotype effect was due to lowered habitation in the (+/−) mice for all PCP doses, while the Drug effect was due to significantly greater loss of habituation with increasing dose of PCP for all genotypes.
Analysis of PPI data in VLDLR mice revealed a similar pattern of results as for the ApoER2 mice. There was a significant main effect of Drug dose [F(3,207) = 30.51, p < 0.0001] and Prepulse Intensity [F(2,138) = 272.33, p < 0.0001]. Unlike with the ApoER2 mice, there was also a significant main effect of Genotype [F(2,69) = 4.20, p < 0.05]. Results of the analysis also indicated a significant interaction of Drug dose × Prepulse Intensity [F(6,414) = 10.65, p < 0.0001], as well as a strong trend for a three-way interaction of Genotype × Drug dose × Prepulse Intensity [F(12,414) = 1.72, p = 0.06]. Posthoc tests revealed that PPI was significantly reduced across all Prepulse intensities with increasing doses of PCP, for all Genotypes (Figure 4). Furthermore, and similarly to the ApoER2 animals, both the VLDLR (+/−) and (−/−) mice displayed significantly greater reductions in PPI than (+/+) mice, confirming greater sensitivity to the sensorimotor gating disruptive effects of PCP. This effect was significant at the 69 dB prepulse intensity for the 20 mg/kg dose, and at the 73 dB prepulse intensity for the 10 and 15 mg/kg doses.
Figure 4.
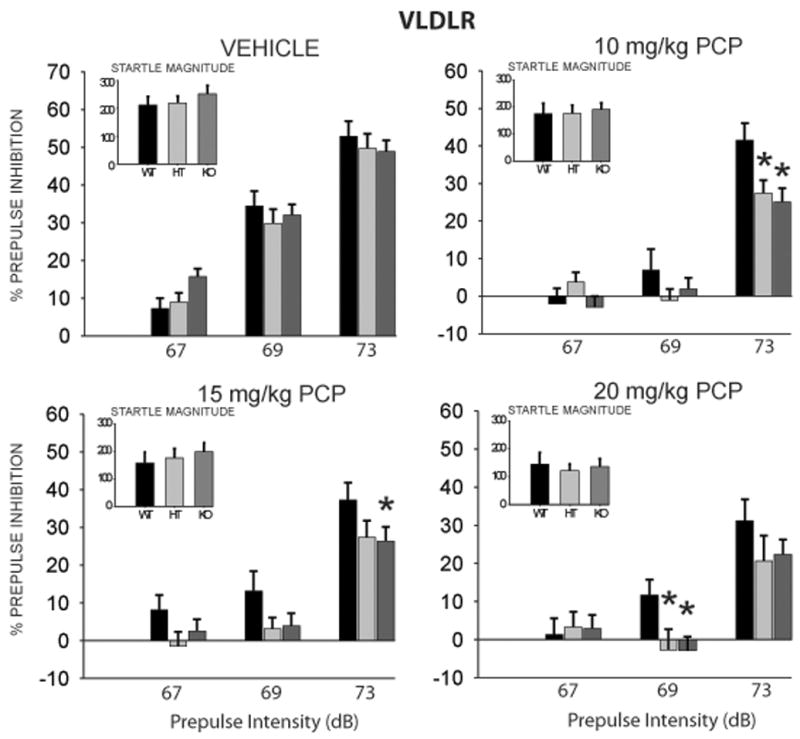
VLDLR mice on postnatal day 120 were randomly assigned to treatment with vehicle, 10 mg/kg, 15 mg/kg, or 20 mg/kg of the NMDA-receptor antagonist phencyclidine (PCP). All mice received all doses, in counterbalanced order. Animals were tested for prepulse inhibition to an acoustic startle stimulus. Test sessions consisted of individual trials that presented prepulse acoustic stimuli 2, 4 and 8 dB above 65 dB background noise (i.e., 67, 69 and 73 dB). Values represent group means (± SEM). Startle magnitudes (arbitrary units) at each dose are inset in graph. * indicates significantly different compared to wildtype mice, p < 0.05. WT = Wildtype (n = 19); HT = Heterozygote (n = 26); KO = Homozygous knockout (n = 27).
Discussion
The present study provides a detailed evaluation of sensorimotor gating in ApoER2 and VLDLR mutant mice in a number of different PPI paradigms, and at different stages of development. When mice were tested for unimodal acoustic PPI prior to completion of puberty on postnatal day 40, ApoER2 mutant mice displayed similar PPI as wildtype mice, although levels of startle habituation were significantly reduced in ApoER2 (−/−) mice. Following puberty on postnatal day 80, the habituation deficits in ApoER2 (−/−) mice were no longer significantly different. VLDLR mutant mice did not exhibit significantly altered unimodal acoustic PPI or startle habituation on postnatal day 40 or postnatal day 80. Consistent with previous studies demonstrating that ApoER2 and VLDLR mutant mice’s motor and sensory functions did not differ from that of wildtype control mice (Trommsdorff et al., 1999; Weeber et al., 2002), neither strain displayed altered levels of startle magnitude in response to an acoustic startle stimulus.
Subsequent testing of ApoER2 and VLDLR mice in a crossmodal PPI session that combined acoustic prepulses with tactile startle stimuli revealed significant differences between the genotypes, and between the two different strains. ApoER2 (+/−) and (−/−) mice displayed a significant increase of crossmodal PPI compared to ApoER2 (+/+) mice, whereas VLDLR (−/−) mice displayed a significant decrease in crossmodal PPI compared to VLDLR (+/+) and (+/−) mice. These differences in crossmodal PPI could not be ascribed to differences in the magnitude of tactile startle, as all genotypes of each mutant strain exhibited equivalent levels of startle.
In the third series of experiments, mice were tested after administration of vehicle or 10, 15 or 20 mg/kg of the NMDA-receptor antagonist PCP, representing doses that are commonly used with mice in the literature (Dulawa and Geyer, 1996). Similar to the findings from prior studies in wildtype mice, increasing doses of this drug decreased levels of unimodal acoustic PPI (Dulawa and Geyer, 1996; Wiley, 1998; Yee et al., 2004). Most relevant to the hypotheses assessed with the present studies, both ApoER2 and VLDLR (+/−) and (−/−) mice displayed significantly greater loss of PPI after administration of PCP than wildtype animals. Interestingly, there were no differences between male and female mice in any of the three different experimental conditions.
The present results represent the first comprehensive report of sensorimotor gating behavior in reelin receptor mutant mice. The general perspective provided by our findings is that the partial or complete loss of either of the reelin receptors results in behavioral phenotypes that are distinct of those of the heterozygous reeler mouse (Tueting et al., 1999), (Podhorna and Didriksen, 2004; Salinger et al., 2003). In our studies, both VLDLR and ApoER2 (+/−) and (−/−) mice did not display significant deficits in unimodal acoustic PPI either before or after completion of puberty. Our findings of normal levels of unimodal acoustic PPI in ApoER2 and VLDLR mutant mice were replicated twice here, once in the acoustic-only trials of the crossmodal experiments and the second time in vehicle condition in the PCP challenge studies. Thus, the current findings do not support the hypothesis that reelin receptors are necessary for unimodal acoustic PPI, although reelin receptors appear to play a role in crossmodal PPI, and sensitivity to the disruptive effects of NMDA blockade on PPI in adulthood.
In the current study, we did not measure crossmodal PPI or the PPI-disruptive effects of PCP prior to completion of puberty. This was due to the relatively short developmental period of the mouse, as well as concerns about potential long-term effects of PCP exposure during adolescence on adult behavior (Jentsch and Roth, 1999). Also, PCP was only used in the unimodal PPI test, and not the crossmodal task, for a number of reasons. Firstly, the use of PCP with crossmodal procedures is not well characterized, and would have required substantial additional experiments for validation beyond the scope of this study (Swerdlow et al., 2001a). Secondly, PCP can have long-term effects that could confound subsequent tests with PCP. Thirdly, the absence of genotype differences in the unimodal PPI sessions allowed us to observe a differential response to the drug, and thus test our hypothesis that different genotypes could be more sensitive to PCP. Use of the crossmodal paradigm, where there were significant differences in the absence of the drug, would have confounded this approach, and complicated the study with possible floor / ceiling effects. It should be noted, however, that future studies are required to determine if crossmodal PPI deficits follow a developmental trajectory, and if crossmodal PPI is differentially sensitive to PCP, in reelin receptor mutant mice.
The precise bases of the PPI alterations in ApoER2 or VLDLR mutant mice remain unknown. Both reelin receptors transduce reelin through the intracellular adaptor protein disabled-1 (Dab-1) and modify the activity of similar signaling molecules (Trommsdorff et al., 1999). Thus, it is unlikely that physiological differences between the two receptors could account for crossmodal PPI differences, and levels of reelin are not significantly different between the two lines (Trommsdorff et al., 1999). While neuroanatomical aberrations occur in both ApoER2 and VLDLR (−/−) mice, there are no obvious neurodevelopmental changes in (+/−) mice of either strain (Trommsdorff et al., 1999) unlike schizophrenia, which is a disorder of altered connectivity (Barr et al., 2004b; Sawada et al., 2005; Sawada et al., 2002). Thus, macroarchitectural changes may not account for PPI alterations in ApoER2 mutant mice, as both (+/−) and (−/−) mice displayed similar and parallel behavioral alterations. For VLDLR mutant mice, only the (−/−) animals exhibited decreased crossmodal PPI, although – like ApoER2 mice – both (+/−) and (−/−) mice were more sensitive to the disruptive effects of PCP on PPI.
The neural circuitry underlying unimodal acoustic PPI has been well characterized in rodents (Swerdlow et al., 2001a). Multiple forebrain sites can modify PPI, with lesions to these regions typically decreasing PPI. The one major exception is lesions to the medial prefrontal cortex, which induce increases in unimodal acoustic PPI (Lacroix et al., 2000; Schwabe et al., 2004). By contrast, the neural circuitry underlying crossmodal PPI is not well understood, and clearly the integration of input from different sensory modalities may be expected to recruit different or additional neural circuits to those regulating unimodal acoustic PPI. Studies with different strains of wildtype mice showed that levels of unimodal acoustic and crossmodal PPI do not correlate with each other (Bullock et al., 1997; Ralph et al., 2001), and are therefore suggestive of regulation by different genes (Bullock et al., 1997), possibly mediated through different brain regions. The physiological basis of the increased sensitivity of ApoER2 (+/− and −/−) and VLDLR (+/− and −/−) mice to the disruptive effects of PCP on unimodal acoustic PPI also remains unknown, although the circuitry underlying this form of sensorimotor gating is relatively well understood. At a cellular level, GABA turnover rate appears decreased in heterozygous reeler mice (Carboni et al., 2004). Thus, reduced GABA turnover rate could account for the greater sensitivity of heterozygous reeler mice to the disruptive effects of the NMDA-receptor antagonist dizocilpine on locomotor activity and cognitive performance on a maze task. Decreased function of GABAergic interneurons has been hypothesized to contribute to the cognitive and affective symptoms of psychotic disorders (Benes and Berretta, 2001; Eyles et al., 2002). The above observations considered together with the parallel findings of behavioral sensitivity to the effects of NMDA-receptor antagonists in both heterozygous reeler mice and receptor mutant mice suggests that decreased GABA turnover may also mediate the increased sensitivity to NMDA receptor blockade in the receptor mutants. Alternatively, it has been proposed that deficits in learning and long-term potentiation in ApoER2 (−/−) (Beffert et al., 2005; Weeber et al., 2002) and VLDLR (−/−) (Weeber et al., 2002) mice were due to loss of reelin-receptor mediated activation of tyrosine and serine/threonine kinases. In addition, primary culture studies showed that stimulation of ApoER2 or VLDLR receptors results in phosphorylation of Dab-1, leading to activation of kinases, including Src and Fyn (Rice and Curran, 2001), that upregulate the activity of the NMDA receptor through phosphorylation of tyrosine residues (Chen et al., 2005). Hypothetically, a reduction in this signaling pathway by complete or partial loss of receptor signaling would lead to an exaggerated behavioral response to further loss of NMDA receptor function following treatment with PCP. Further research will be required to evaluate these possible mechanisms of dysfunctional crossmodal PPI and increased sensitivity to PCP in the reelin receptor mutant mice.
PPI deficits have been reported in a number of neuropsychiatric disorders, such as schizophrenia (Braff et al., 2001; Braff et al., 1992), obsessive-compulsive disorder (Hoenig et al., 2005), Tourette's syndrome (Swerdlow et al., 2001b), Huntington’s Disease (Swerdlow et al., 1995) and Parkinson’s Disease (Valls-Sole et al., 2004). We are not aware of any example of a disorder that is characterized by alterations in crossmodal PPI but not unimodal acoustic PPI, except in response to a pharmacological challenge. Disorders such as schizophrenia are usually associated with decreased levels of both unimodal acoustic and crossmodal PPI under non-pharmacological challenge conditions (Braff et al., 2001; Braff et al., 1992). Thus, the present findings indicate that loss of reelin receptors leads to phenotypes that model aspects of PPI deficits in psychotic disorders, but not in their entirety. Since the study of mice with genetic alterations of the reelin signaling pathway has provided essential insights into the nature of early neurodevelopment (D'Arcangelo, 2006), we expect that the further evaluation of the mechanisms mediating PPI deficits in reelin receptor mutant mice will elucidate how anatomical and cellular changes in these mutants are related to behavioral deficits.
Acknowledgments
This research was supported by NIMH grant R01 MH62527 to AM, a NARSAD Young Investigator Award and NIMH grant K01 MH064372 to KNF, and CIHR grant MOP77606 to AMB. AMB is a MSFHR Scholar. We would like to thank Dr. Mark Geyer for the provision of multimodal PPI test sessions and intellectual input to this work, and Mr. Mike Arends for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ballmaier M, Zoli M, Leo G, Agnati LF, Spano P. Preferential alterations in the mesolimbic dopamine pathway of heterozygous reeler mice: an emerging animal-based model of schizophrenia. Eur J Neurosci. 2002;15(7):1197–205. doi: 10.1046/j.1460-9568.2002.01952.x. [DOI] [PubMed] [Google Scholar]
- Barr AM, et al. The selective serotonin-2A receptor antagonist M100907 reverses behavioral deficits in dopamine transporter knockout mice. Neuropsychopharmacology. 2004a;29(2):221–8. doi: 10.1038/sj.npp.1300343. [DOI] [PubMed] [Google Scholar]
- Barr AM, Powell SB, Markou A, Geyer MA. Iloperidone reduces sensorimotor gating deficits in pharmacological models, but not a developmental model, of disrupted prepulse inhibition in rats. Neuropharmacology. 2006;51(3):457–65. doi: 10.1016/j.neuropharm.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Barr AM, et al. Abnormalities of presynaptic protein CDCrel-1 in striatum of rats reared in social isolation: relevance to neural connectivity in schizophrenia. Eur J Neurosci. 2004b;20(1):303–7. doi: 10.1111/j.0953-816X.2004.03457.x. [DOI] [PubMed] [Google Scholar]
- Beffert U, et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47(4):567–79. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25(1):1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156(2–3):234–58. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49(3):206–15. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Bullock AE, Slobe BS, Vazquez V, Collins AC. Inbred mouse strains differ in the regulation of startle and prepulse inhibition of the startle response. Behav Neurosci. 1997;111(6):1353–60. doi: 10.1037//0735-7044.111.6.1353. [DOI] [PubMed] [Google Scholar]
- Carboni G, et al. Enhanced dizocilpine efficacy in heterozygous reeler mice relates to GABA turnover downregulation. Neuropharmacology. 2004;46(8):1070–81. doi: 10.1016/j.neuropharm.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Chen Y, et al. Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci. 2005;25(36):8209–16. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E, Davis J, Pesold C, Tueting P, Guidotti A. The heterozygote reeler mouse as a model for the development of a new generation of antipsychotics. Curr Opin Pharmacol. 2002;2(1):56–62. doi: 10.1016/s1471-4892(01)00121-7. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G. Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 2006;8(1):81–90. doi: 10.1016/j.yebeh.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Dulawa SC, Geyer MA. Psychopharmacology of prepulse inhibition in mice. Chin J Physiol. 1996;39(3):139–46. [PubMed] [Google Scholar]
- Eyles DW, McGrath JJ, Reynolds GP. Neuronal calcium-binding proteins and schizophrenia. Schizophr Res. 2002;57(1):27–34. doi: 10.1016/s0920-9964(01)00299-7. [DOI] [PubMed] [Google Scholar]
- Fatemi SH. Reelin mutations in mouse and man: from reeler mouse to schizophrenia, mood disorders, autism and lissencephaly. Mol Psychiatry. 2001;6(2):129–33. doi: 10.1038/sj.mp.4000129. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle JA, McMenomy T. Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry. 2000;5(6):654–63. 571. doi: 10.1038/sj.mp.4000783. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72(2–3):109–22. doi: 10.1016/j.schres.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry. 2002;7(10):1039–53. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- Guidotti A, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57(11):1061–9. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Hoenig K, Hochrein A, Quednow BB, Maier W, Wagner M. Impaired prepulse inhibition of acoustic startle in obsessive-compulsive disorder. Biol Psychiatry. 2005;57(10):1153–8. doi: 10.1016/j.biopsych.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Impagnatiello F, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A. 1998;95(26):15718–23. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20(3):201–25. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Krystal JH, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169(3–4):215–33. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Lacroix L, Spinelli S, White W, Feldon J. The effects of ibotenic acid lesions of the medial and lateral prefrontal cortex on latent inhibition, prepulse inhibition and amphetamine-induced hyperlocomotion. Neuroscience. 2000;97(3):459–68. doi: 10.1016/s0306-4522(00)00013-0. [DOI] [PubMed] [Google Scholar]
- Larsen TK, McGlashan TH, Moe LC. First-episode schizophrenia: I. Early course parameters. Schizophr Bull. 1996;22(2):241–56. doi: 10.1093/schbul/22.2.241. [DOI] [PubMed] [Google Scholar]
- Pappas GD, et al. Immunocytochemical localization of reelin in the olfactory bulb of the heterozygous reeler mouse: an animal model for schizophrenia. Neurol Res. 2003;25(8):819–30. doi: 10.1179/016164103771953916. [DOI] [PubMed] [Google Scholar]
- Podhorna J, Didriksen M. The heterozygous reeler mouse: behavioural phenotype. Behav Brain Res. 2004;153(1):43–54. doi: 10.1016/j.bbr.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Geyer MA. Strain-specific effects of amphetamine on prepulse inhibition and patterns of locomotor behavior in mice. J Pharmacol Exp Ther. 2001;298(1):148–55. [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–39. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Salinger WL, Ladrow P, Wheeler C. Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav Neurosci. 2003;117(6):1257–75. doi: 10.1037/0735-7044.117.6.1257. [DOI] [PubMed] [Google Scholar]
- Sawada K, et al. Hippocampal complexin proteins and cognitive dysfunction in schizophrenia. Arch Gen Psychiatry. 2005;62(3):263–72. doi: 10.1001/archpsyc.62.3.263. [DOI] [PubMed] [Google Scholar]
- Sawada K, et al. Altered immunoreactivity of complexin protein in prefrontal cortex in severe mental illness. Mol Psychiatry. 2002;7(5):484–92. doi: 10.1038/sj.mp.4000978. [DOI] [PubMed] [Google Scholar]
- Schwabe K, Enkel T, Klein S, Schutte M, Koch M. Effects of neonatal lesions of the medial prefrontal cortex on adult rat behaviour. Behav Brain Res. 2004;153(1):21–34. doi: 10.1016/j.bbr.2003.10.030. [DOI] [PubMed] [Google Scholar]
- Spielewoy C, Markou A. Withdrawal from chronic phencyclidine treatment induces long-lasting depression in brain reward function. Neuropsychopharmacology. 2003;28(6):1106–16. doi: 10.1038/sj.npp.1300124. [DOI] [PubMed] [Google Scholar]
- Steinpresis RE. The behavioral and neurochemical effects of phenyclidine in humans and animals: some implications for modeling psychosis. Behav Brain Res. 1996;74(1–2):45–55. doi: 10.1016/0166-4328(95)00162-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 2001a;156(2–3):194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Tactile prepuff inhibition of startle in children with Tourette's syndrome: in search of an "fMRI-friendly" startle paradigm. Biol Psychiatry. 2001b;50(8):578–85. doi: 10.1016/s0006-3223(01)01164-7. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington's disease. J Neurol Neurosurg Psychiatry. 1995;58(2):192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, et al. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 2005;57(3):252–60. doi: 10.1016/j.biopsych.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97(6):689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Tueting P, et al. The phenotypic characteristics of heterozygous reeler mouse. Neuroreport. 1999;10(6):1329–34. doi: 10.1097/00001756-199904260-00032. [DOI] [PubMed] [Google Scholar]
- Valls-Sole J, Munoz JE, Valldeoriola F. Abnormalities of prepulse inhibition do not depend on blink reflex excitability: a study in Parkinson's disease and Huntington's disease. Clin Neurophysiol. 2004;115(7):1527–36. doi: 10.1016/j.clinph.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, et al. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277(42):39944–52. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- Wiley JL. Nitric oxide synthase inhibitors attenuate phencyclidine-induced disruption of prepulse inhibition. Neuropsychopharmacology. 1998;19(1):86–94. doi: 10.1016/S0893-133X(98)00008-6. [DOI] [PubMed] [Google Scholar]
- Yee BK, Chang DL, Feldon J. The Effects of dizocilpine and phencyclidine on prepulse inhibition of the acoustic startle reflex and on prepulse-elicited reactivity in C57BL6 mice. Neuropsychopharmacology. 2004;29(10):1865–77. doi: 10.1038/sj.npp.1300480. [DOI] [PubMed] [Google Scholar]