

Abstract
Studies of the nuclear transcriptional regulatory activities of nonphysiological estrogens have not explained their actions in mediating endocrine disruption in animals and humans at the low concentrations widespread in the environment. However, xenoestrogens have rarely been tested for their ability to participate in the plethora of nongenomic steroid signaling pathways elucidated over the last several years. Here we review what is known about such responses in comparison to our recent evidence that xenoestrogens can rapidly and potently elicit signaling through nongenomic pathways culminating in functional endpoints. Both estradiol (E2) and compounds representing various classes of xenoestrogens (diethylstilbestrol, coumestrol, bisphenol A, DDE, nonylphenol, endosulfan, and dieldrin) act via a membrane version of the estrogen receptor-α on pituitary cells, and can provoke Ca++ influx via L-type channels, leading to prolactin (PRL) secretion. These hormones and mimetics can also cause the oscillating activation of extracellular regulated kinases (ERKs). However, individual estrogen mimetics differ in their potency and temporal phasing of these activations compared to each other and to E2. It is perhaps in these ways that they disrupt some endocrine functions when acting in combination with physiological estrogens. Our quantitative assays allow comparison of these outcomes for each mimetic, and let us build a detailed picture of alternative signaling pathway usage. Such an understanding should allow us to determine the estrogenic or antiestrogenic potential of different types of xenoestrogens, and help us to develop strategies for preventing xenoestrogenic disruption of estrogen action in many tissues.
Keywords: membrane, environmental estrogen, kinases, calcium, prolactin, low concentrations
INTRODUCTION
Xenoestrogens and their known modes of action
Xenoestrogens are compounds other than physiological estrogens that can nonetheless evoke estrogenic responses. Xenoestrogens are known to contaminate our environment and alter the reproductive health of wildlife, and probably humans [1]. Such estrogen mimetics were noted for their effects on wildlife in the 1960’s when naturalists such as Rachel Carson drew attention to the endocrine-disrupting effects of some pesticides (notably DDT, [2]). These compounds may act as inappropriate estrogens, and/or could interfere with the actions of endogenous estrogens. For many years the mechanisms via which many xenoestrogens act remained a mystery. This lack of a mechanistic explanation existed because while these compounds can affect animal functions and development at relatively low concentrations, experimental systems for testing the classical nuclear transcriptional activities of xenoestrogens showed weak or no activity [3–10]. Therefore, the question remained, via what cellular mechanisms do xenoestrogens act? Actions mediated through nongenomic pathways and plasma membrane receptors for steroids [11–13] were largely unstudied until very recently.
Compounds known as xenoestrogens have wide structural diversity, but all have in common lipophillic phenolic rings and other hydrophobic components, a characteristic they share with steroid hormones and related nuclear receptor-activating compounds (see Fig. 1). It has been suggested that the “promiscuity” of estrogen receptors in accepting many diverse ligands may be due to their status as the most evolutionarily primitive versions of ligand-activatable regulatory proteins [14]; as such they probably initially evolved to respond to a diverse set of molecules in the cell's environment. Therefore, many compounds that are byproducts of our modern industrialized life-style (pesticides, herbicides, plastics manufacturing byproducts, fungicides, cosmetics additives, and pharmaceuticals) can serve as estrogenic ligands in an inappropriate way.
Fig. 1. Structurally diverse xenoestrogens examined in our studies represent various common classes of these compounds.
17ß estradiol is the predominate physiological estrogen. DES is a pharmaceutical estrogen. Coumestrol is a plant estrogen. Bisphenol A (a plastics monomer) and nonylphenol (a detergent) are byproducts of plastics manufacturing. DDE, endosulfan and dieldrin are organochlorine pesticides or their metabolites.
We and others have very recently studied compounds representing different functional and structural xenoestrogen classes for actions initiated at the plasma membrane. Our studies, summarized in this review, examined the following diverse xenoestrogenic compound classes displayed in Fig. 1: Dieldrin, endosulfan, and the DDT metabolite o,p'-dichlorodiphenylethylene (DDE) are organochlorine pesticides; because of widespread past usage they still contaminate many agricultural and runoff sites. Detergents used in plastics manufacturing (eg. p-nonylphenol) and a common precursor monomer that leaches from polycarbonate plastics (bisphenol A) are widespread contaminants in food and water via packaging, and as manufacturing byproducts in the environment [15]. Naturally occurring estrogens from plants and molds can also be abundant; we studied the phytoestrogen coumestrol, which is present in alfalfa sprouts and red clover (entering the food cycle via animals grazing in pastures containing this plant) [16]. Finally, some estrogen mimetics (such as diethylstilbesterol, DES) were designed as pharmaceuticals, but later found to have health-threatening side effects such as vaginal cancer in the neonatally exposed [17]. The potencies of these compounds in nuclear transcription reporter assays range from very weak (dieldrin, DDE, endosulfan), to somewhat weak (bisphenol A and nonylphenol), to quite strong (DES and coumestrol). There is a paucity of data on the ability of environmental estrogens to mediate nongenomic effects at low concentrations [18–24]. Most published studies examine only very high (μM-mM) concentrations (for example, [25]) in the range required to see any effects on nuclear transcription responses, but which are rarely reached at contamination sites.
The debate about the identity of steroid receptor proteins that mediate nongenomic effects, including those for xenoestrogens
Representative examples of the proposed membrane steroid receptor types have recently been reviewed [12]. Such an abundance of credible reports indicates that nongenomic steroid and mimetic actions are likely to result from a very complex sequence of events which can assemble a repertoire of proteins likely to function together. These proteins are probably differentially represented in different cell types and circumstances, and at different response stages. The existence of multiple kinds of steroid-binding proteins (receptors, enzymes, transporters, and blood and cellular binding globulins and their receptors) has long been known, though the exact sequential roles of all of these protein types are still not clear, even in direct genomic response pathways. It is likely that both nuclear receptor-like membrane steroid receptors, and also other unique steroid-binding membrane proteins (such as serpentine receptors and others [19;26–30]), play subtly different roles. It is also important to remember that downstream, rapid membrane-initiated steroid effects can ultimately impinge upon nuclear actions via post-translational modifications of transcription factors (including nuclear receptors themselves). Our past studies in both a pituitary tumor cell line selected for robust nongenomic estrogenic responses, and similarly selected MCF-7 breast cancer cells, clearly indicate that a membrane version of ERα is involved. We demonstrated this via antibody (Ab)-elicited responses, increased or decreased receptor expression linked to responses, antisense knockdown of ERα, and the absence of other estrogen receptor types in these cells [31–38].
Nongenomic effects in the pituitary, and in our cell model
In pituitary, estrogens facilitate both genomic (synthesis) and nongenomic (regulated secretion) of PRL [39]. The numerous functional consequences of PRL activity include coordination of the female hormonal cycle with preparation of various tissues for reproduction by inducing protein synthesis and secretion, the growth of new tissue (e.g. mammary gland), and the control of reproductive behavior. In this scenario many different functional endpoints are thus candidates for mis-regulation by xenoestrogens. Our clonal cell line GH3/B6/F10 was selected for its natural (not transfection-driven) expression of high levels of a membrane form of the estrogen receptor-α (mERα). Expression of mERα was correlated with very sensitive responses to E2, including those for ERK activation [40], Ca++ entry [41], and rapid PRL release [41]. We first observed changes in mERα levels detected in the membrane when cells were treated with low concentrations of xenoestrogens just before fixation for immunocytochemistry. E2 caused rapid loss (by 3 min) and a slower return (~15 min) of the mERα epitope. (Whether that be actual exit and return of the protein from the membrane, or a change in epitope recognition, we are not sure.) Xenoestrogens also caused this rapid change in epitope recognition, with a slightly different time course of the slow reversal [42]. This initiated a series of studies comparing physiological vs. nonphysiological estrogens and their use of membrane-initiated signaling mechanisms. The evidence that we will review here summarizes the arguments for believing that xenoestrogens also effect signaling changes leading to functional endpoints via the same nongenomic pathways as E2, but with altered pathway kinetics and use preferences.
EXPERIMENTAL
Materials and reagents
We purchased phenol red-free Dulbecco modified Eagle medium (DMEM) from Mediatech (Herndon, VA); horse serum from Gibco BRL (Grand Island, NY); defined supplemented calf sera and fetal bovine sera from Hyclone (Logan, UT); endosulfan and DDE from Ultra Scientific (North Kingstown, RI); and all other XEs from Sigma (St. Louis, MO). Paraformaldehyde and glutaraldehyde were purchased from Fisher Scientific (Pittsburgh, PA). We purchased Fura-2/AM from Molecular Probes (Eugene, OR). From Vector Laboratories (Burlingame, CA), we purchased biotinylated universal antimouse/rabbit IgG, Vectastain ABC-AP (avidin:biotinylated enzyme complex with alkaline phosphatase) detection systems, levamisol (endogenous alkaline phosphatase subtype inhibitor), and para-nitrophenol phosphate (pNpp; the substrate for our alkaline phosphatase reaction). Phospho-p44/42 ERK (pERK) monoclonal Ab, and lysis buffer were obtained from Cell Signaling Technology (Beverly, MA). ICI 182,780 (ICI) was purchased from Tocris (Ellisville, MO) and nifedipine, from Calbiochem (San Diego, CA). All other reagents were purchased from Sigma Chemical Company (St. Louis, MO) or mentioned in individual protocols below.
Cell culture
Our clonal rat prolactinoma cell line GH3/B6/F10 was selected for high expression of mER-α, and the/D9 subline for very low mERα expression (Pappas et al. 1994). Cells were routinely subcultured in DMEM growth medium containing 12.5% horse serum, 2.5% defined supplemented calf serum, and 1.5% fetal calf serum. For individual experiments, cells were deprived of steroids for 48 hr after plating by substituting DMEM containing 1% charcoal-stripped (4×) serum or DMEM containing 5 μg/ml insulin and transferrin, 5 ng/ml selenium, 0.1% BSA, 20 nM sodium pyruvate, and 25 mM HEPES (DMEM/ITS). Immediately before the experiments, cells were incubated in DMEM alone for 1 hr. All test estrogens were dissolved in ethanol (EtOH) at a 10−2 M concentration to create a stock solution and then diluted into experimental media to yield final concentrations from 10−8 to 10−12 M. The EtOH concentration used as the vehicle control was 0.0001%.
Ca++ measurements
GH3/B6 cell sublines were plated on poly-D-lysine–coated coverslips in wells of a six-well plate (105 cells/well). After serum deprivation in DMEM/ITS and then DMEM, the cells were washed in Ringer’s solution (120 mM NaCl, 1.25 mM CaCl2, 4.7 mM KCl, 1.2 mM MgCl2, 20 mM HEPES, 10 mM glucose, 0.1% BSA; pH 7.4), loaded with 2 μM Fura-2/AM diluted in Ringer’s, wrapped in aluminum foil, and incubated at room temperature (RT) for 1 hr. The cells were washed twice and left to equilibrate in Ringer’s for 20 min at RT before imaging. E2 and XEs were administered using a perfusion pump system at a rate of 2 mL/min. Although responses to E2 continue during a 5-min hormonal treatment, these effects are reversible, taking about 5 min to wash out [41]. Imaging was performed using a TE200-IUC Quantitative Fluorescence Live-Cell and Multidimensional Imaging System equipped with a digital monochrome cooled CCD camera (Roper Scientific, Tucson, AZ). Ca++ measurements were collected using the MetaFluor program (Universal Imaging, Downingtown, PA), making sure that only single cells were used as the region of interest. Data were recorded every second. Signals were obtained in dual excitation mode (340/380 nm), and the intracellular Ca++ was calculated as a ratio (R340/380) of emission data collected at 510 nm after background subtraction. Intracellular Ca2+ was quantified by calculating the change in fluorescence ratio (R – R0) during a 5-min treatment period, normalized to the basal fluorescence value (R0) for each individual cell. These calculations for individual cells were then averaged to calculate the means and SEs for the population. Test and calibration solutions included Ca2+-free solution (Ringer’s without CaCl2 and with 2 mM EGTA), Ringer’s–20 mM KCl (Ringer’s with NaCl decreased to 105 mM and KCl increased to 20 mM), and maximum Ca2+ solution (Ringer’s with NaCl decreased to 112 mM and CaCl2 increased to 10 mM). KCl treatments were used at the end of each experiment to establish cell viability. Cells that did not respond transiently to KCl depolarization at the end of the experiment were eliminated from the composite calculations.
PRL release and radioimmunoassay
Cells (0.5–0.7 × 106) were plated in poly-D lysine–coated six-well plates. After serum deprivation in DMEM/ITS, this medium was removed and new DMEM/0.1% BSA with or without the appropriate reagent or vehicle control (ethanol) was added. The cells were incubated for 1, 3, 6, 10, or 15 min and centrifuged at 4°C, 350 × g, for 5 min. The supernatant was then collected and stored at −20°C until radioimmunoassay (RIA). Concentrations of PRL were determined using components of the rat PRL RIA kit from the National Institute of Diabetes and Digestive and Kidney Disease and the National Hormone and Pituitary Program (Baltimore, MD). Briefly, RIA buffer [80% phosphate-buffered saline (PBS), 20% DMEM, 2% normal rabbit serum], 100 μL cold standard (rat PRL-RP-3) or unknown sample, rPRL-s-9 antiserum (final dilution of 1:437,500 in RIA buffer), and [125I]-rat-PRL (PerkinElmer, Wellesley, MA, USA; using 15,000 counts per tube diluted in RIA buffer) were combined and incubated with shaking overnight at 4°C. Anti-rabbit IgG (R-0881; Sigma) was added to a final dilution of 1:9, and the samples were incubated with shaking at RT for 2 hr. One milliliter of polyethylene glycol solution [1.2 M polyethylene glycol (P-6667; Sigma), 50 mM Tris, pH 8.6] was then added, and the samples were incubated with shaking at RT for 15 min. The samples were then centrifuged at 4,000 × g for 10 min at 4°C, the supernatant was decanted, and the pellet was counted in a gamma counter. The PRL concentration was then calculated and normalized to the crystal violet values representing cell number.
Fixed cell–based ELISA for ERK
To estimate ERK phosphorylation quantitatively, we used a cell-based ELISA, which we previously developed and described [40]. Briefly, cells (104 cells/well) were plated in 96-well plates (Corning Incorporated, Corning, NY) and withdrawn from serum hormones by incubation in medium containing 1% charcoal-stripped serum for 48 hr before experiments began. The cells were next treated with hormones and estrogen mimetics for 3–30 min (or vehicle, 0.0001%EtOH), and then fixed with 2% paraformaldehyde/0.2% picric acid at 4°C for 48 hr. After fixation, the cells were incubated with phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA) and 0.1% Triton X-100 for 1 hr at room temperature (RT), and then with primary Ab against pERK (1:400 in PBS/1% BSA/0.1% Triton X-100) overnight at 4°C. After a wash with PBS, biotin-conjugated secondary Ab (1:300) in PBS/1% BSA was added for 1 hr at RT. The cells were again washed in PBS and incubated with Vectastain ABC-AP solution (100 μL/well) for 1 hr at RT, and then Vectastain alkaline phosphatase substrate (pNpp solution) with levamisole was added to each well (100 μL). Plates were incubated in the dark for 30 min at 37°C, and the signal from para-nitrophenol (pNp) was read at A405. The pNp signal was normalized to cell number, determined by using the crystal violet (CV) assay [31].
Crystal violet assay
After fixing (2% paraformaldehyde/0.1% glutaraldehyde in PBS for 30 min at RT with shaking) and then washing with double-distilled H2O, the plates were completely dried at RT. CV solution (0.1% in water, filtered) was added, incubated for 30 min at RT, and washed out with double-distilled H2O. Dye was released from the cells with acetic acid (10% in water) at RT for 30 min. The A590 signal was then read in the microplate reader.
Statistics
Data were compared for significance of differences using Sigma Stat 3 (Jandel Scientific, San Rafael, CA) and oneway analysis of variance (significance accepted at p ≤ 0.05).
RESULTS
E2 and xenoestrogens can rapidly and potently elicit Ca++ influx and PRL secretion
We developed quantitative assays for both signaling and functional endpoints for nongenomic xenoestrogen activity. First, we directly examined the ability of E2 and xenoestrogens to raise intracellular Ca++ levels, as Ca++ signaling is likely to be involved in other downstream events, including both ERK activation [40] and regulation of secretion of peptide hormones like PRL [43]. E2, E2-P (E2 conjugated to peroxidase to impede its entry into cells), and all xenoestrogens caused increased Ca++ spikes at low concentrations [24;41]. Individual traces of Ca++ cellular levels over time show that Ca++ spikes usually began ~30 seconds after application of all of these compounds [24;41]. To quantitate these Ca++ responses, and thus measure the response amplitudes and xenoestrogen potencies, we calculated the change in FURA-2 ratio values during a 5 min treatment period, and expressed them as a percentage of the basal fluorescence values (in the absence of estrogens, as in [19]). DES, coumestrol, dieldrin, and bisphenol A all elicited Ca++ responses, to some extent, at 10−12 to 10−8 M concentrations (Fig. 2A; also nonylphenol and DDE, not shown). Some compounds did not elicit as large a maximum response as did E2 (for example DES and coumestrol). Others gave equivalent responses compared to E2 at their maximally effective concentrations, yet did not respond as well at lower concentrations (see endosulfan). The D9 cell subline (which has very low mERα levels) did not respond to any concentration of E2, or to any of the xenoestrogens [24;41].
Fig. 2. Xenoestrogens at low doses cause Ca++ influx and PRL release.
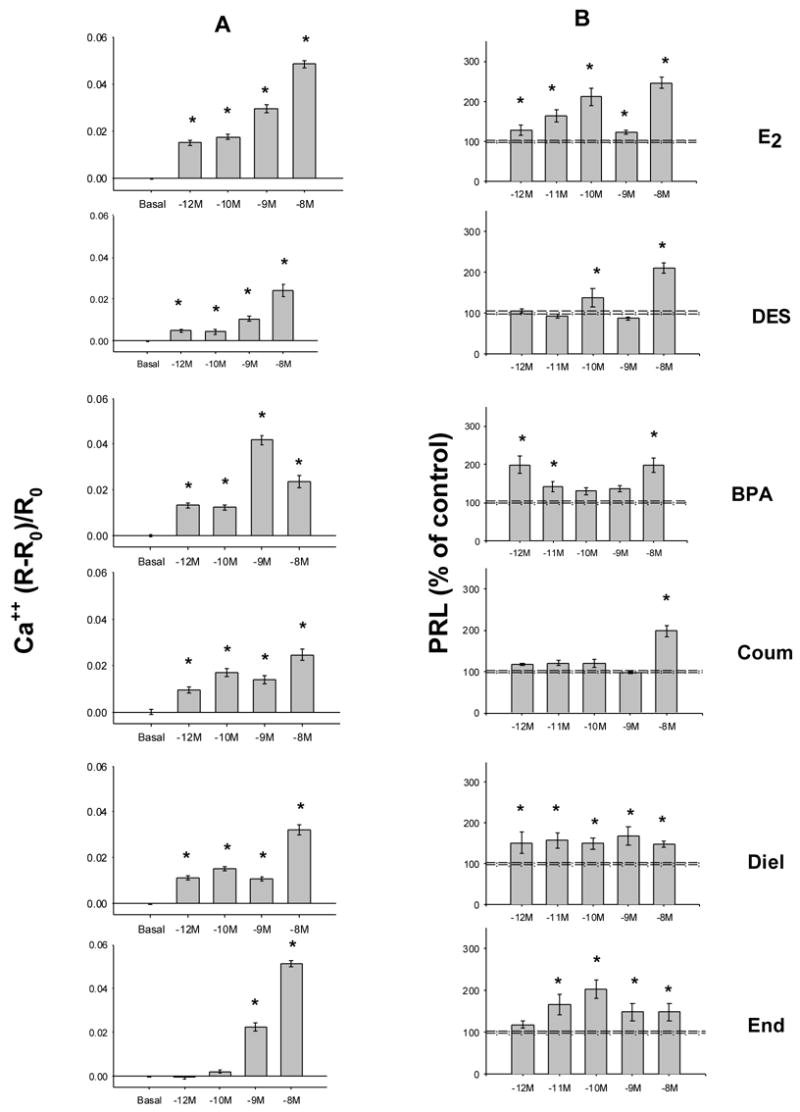
Values are mean ± S.E. * indicates significance at the P<0.05 level. A. Ca++ was measured over a 5 min time block. For each value 12–28 individual cells were imaged over 3 to 4 different experiments. B. PRL release measured by RIA was normalized to the number of cells for each assay value. Release into the medium was assessed after a 3 min exposure to estrogens. An average of 18 cell-containing wells were assessed per point, spread over 3–7 experiments. Dashed lines indicate the error range around the basal level.
We next examined a functional response that could be a consequence of elevated Ca++ levels -- secretion of the pituitary peptide hormone PRL. E2 and most xenoestrogen treatments caused significant PRL release by 3 min at pM-nM concentrations (Fig. 2B). However, xenoestrogens elicit PRL release with different potencies than does E2. Note the slightly lower potency response by DES and endosulfan, and the requirement of 10−8 M coumestrol for a response. Also note the characteristic "bimodal" dose-responses (inactive doses between active doses) that we and others had seen originally for E2 [44;45], and now see repeatedly for some (DES, BPA)., but not all, xenoestrogens A time course (not shown, see [24]) revealed that although this response was essentially finished for most compounds by 1 min, the polychlorinated biphenyl compounds (DDE, dieldrin, and endosulfan) required up to 15 min to cause cells to dump their entire store of releasable PRL. So again, responses to xenoestrogens can differ among themselves, and from E2.
We then examined the Ca++ response and its consequence, PRL release, in more detail at the mechanistic level [24;41;46]. Conditions that prevented the influx of Ca++ through the plasma membrane (cells in Ca++-free medium) blocked our responses to E2 and all xenoestrogens tested. Thapsigargin, a drug which empties internal Ca++ stores so that they are subsequently not available for a response, did not dampen these E2 or xenoestrogen-induced Ca++ responses; therefore, in these instances, the Ca++ entered the cytoplasm from outside the cells. To directly correlate this functional response of PRL release with Ca++ signaling, BAPTA (which chelates free intracellular Ca++) was shown to block PRL release [23]. Nifedipine (and nimodipine, not shown), both L-type channel blockers, prevented both Ca++ elevation (Fig. 3A) and PRL release (Fig. 3B), by E2 and all tested xenoestrogens (Fig. 3C).
Fig. 3. L-type channels are responsible for Ca++ elevation leading to PRL release evoked by both E2 and xenoestrogens.
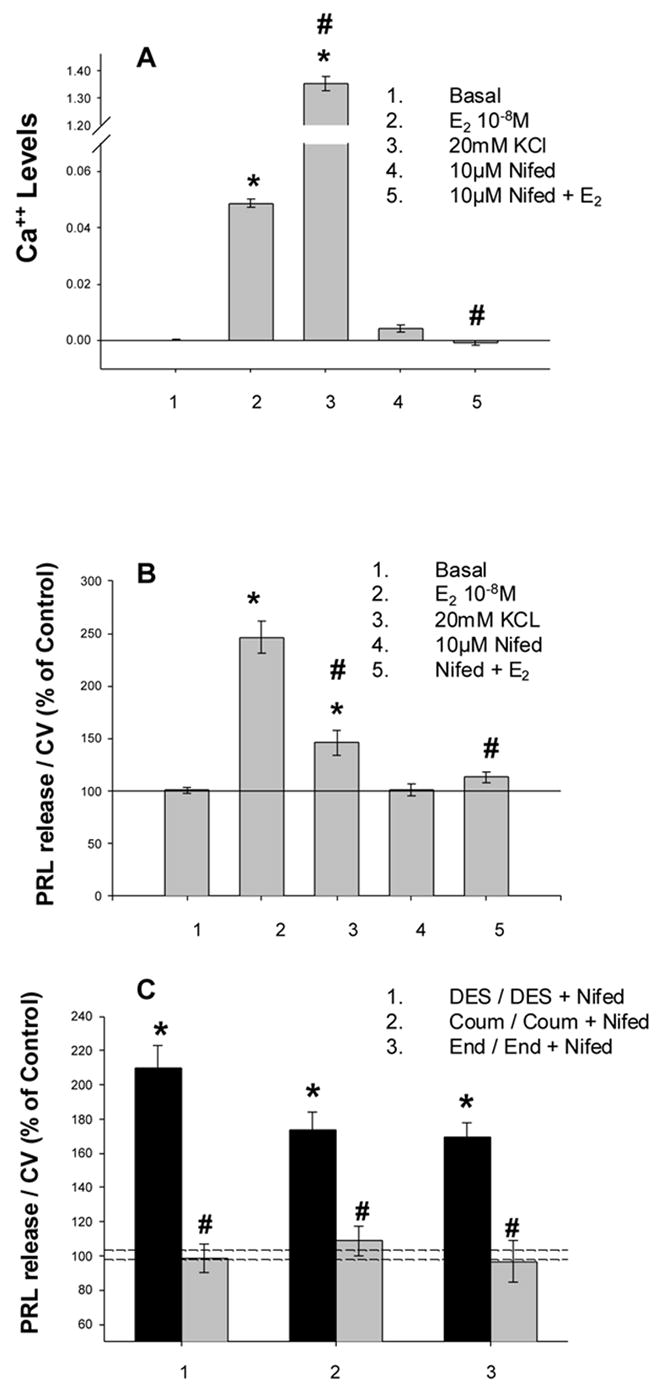
Ca++ and PRL levels were determined as for Fig. 2. Values are the mean ± S.E. Symbols indicate significance at the P<0.05 level compared to vehicle treated controls (*) or to estrogen or xenoestrogen-induced levels (#). A. Ca++ levels evoked by E2, KCl, and blocked by Nifedipine. B. PRL release elicited by the same conditions applied in A. C. Nifedipine inhibits the PRL release caused by three xenoestrogens.
Although in general all compounds that raised Ca++ levels also caused a PRL release, the quantitative correlations for this signal vs. functional response are strikingly different. For example, treating the cells with KCl caused a very large Ca++ level increase, but only a modest PRL release, whereas a large PRL release was caused by E2, while it only modestly raised Ca++ levels, at least compared to KCl (Fig. 3 A vs. B). Similar differential action was also seen when comparing the signaling vs. secretory response effectiveness of different xenoestrogens (Fig 2).
While nM BPA caused a large Ca++ response, it evoked no PRL release. The intracellular Ca++ concentrations produced at lower coumestrol concentrations caused no PRL release, while similar levels in response to other compounds did evoke PRL release. While nM DES caused Ca++ influx, it caused no PRL response. These findings lead us to conclude that estrogens must control some additional responses, other than just elevation of Ca++ levels, that contribute to PRL release [41]. Though the Ca++ elevation is necessary (blocking it blocks the secretory response), the additional signaling contribution(s) elicited by estrogens are necessary for an optimal response. Such results suggest nongenomic effects of estrogens on other parts of the secretory machinery, and that even low doses of xenoestrogens may alter responses.
Application of KCl to the D9 subline (with very low mERα levels) showed that they could release PRL in comparable levels when their Ca++ levels were elevated [41], even though they couldn't respond to E2 or xenoestrogens. Therefore their defect in this response is low mER levels, and not a defective PRL production or secretion mechanism.
ICI182780, a specific ER antagonist, blocked E2-generated Ca++ elevation as well as PRL release [41]. A 1nM concentration of the 17α-E2 stereoisomer of 17ß E2 did not cause a significant rise in Ca++ levels, nor in PRL release [41], though a 10 nM 17α-E2 concentration did [36]. Therefore, we have demonstrated a spectrum of important ligand specificity and signaling characteristics of estrogen and xenoestrogen nongenomic signaling. From these studies it is clear how this signaling pathway leading to a functional endpoint is subject to xenoestrogen interference.
Estrogens and xenoestrogens can rapidly activate oscillating mitogen-activated kinase activities
We also investigated the ability of xenoestrogens to affect another common pathway in nongenomic estrogenic responses: activation of the mitogen-activated kinases ERK 1 and 2. E2 activated ERKs at low concentrations, but in comparison to the large responses induced by EGF, the actions of estrogens were more subtle [41]. For this reason we developed a fixed cell-based 96-well plate immunoassay with a colorimetric readout, using the same phospho-specific ERK Abs that are generally used to assay this response by immunoblots. In this way we could easily normalize the data to cell number in individual wells and quantify our responses without the errors involved in choosing backgrounds for density measurements. Using this very efficient assay (which quantifies both p44 and p42 ERK activation together), we have now tested a collection of xenoestrogens over extensive time courses and dose-response ranges, examples of which are shown in Figs. 4 and 5. Time-course patterns basically fell into four categories (one of each type is shown in Fig. 4). E2 (and DES, not shown) caused a bimodal temporal response with both early (~3 min) and late (~15 min) activation peaks, with apparent deactivation in between. Nonylphenol (and endosulfan, not shown) caused only one later (30 min) activation. Dieldrin (and DDE, not shown) caused a single activation peak at ~6 min. Coumestrol evoked a unique pattern of activation at 6 min, which never declined throughout the 30 min assessment period. Data from other labs have demonstrated that the length of time that an ERK activation is sustained is relevant to functional responses such as cell proliferation [47]. Our D9 subline, which expresses very low levels of mERα, did not respond in these assays (see E2 panel, Fig. 4); this is consistent with the involvement of mERα in these responses, as this cell line also does not express ERß [31] or GPR30 [48]. Such activations have also been shown to occur with the impeded ligand E2-P, and several other xenoestrogens at 10−9 M concentrations, but not with bisphenol A [22].
Fig. 4. E2 - and xenoestrogen-induced time dependence of changes in ERK phosphorylation.
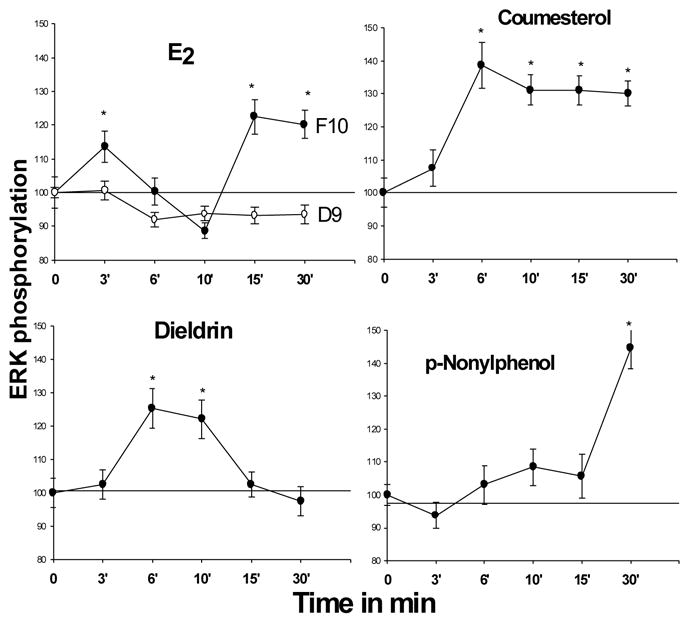
Compounds were applied at 10−9M concentrations; *=statistical significance at the P<0.05 level, compared to vehicle-treated controls. Data were normalized to cell number and presented as % of control values, which were set to 100. n = 48–60 wells/point, taken from 3 different experiments.
Fig. 5. E2 - and xenoestrogen-induced dose responses for ERK activation.
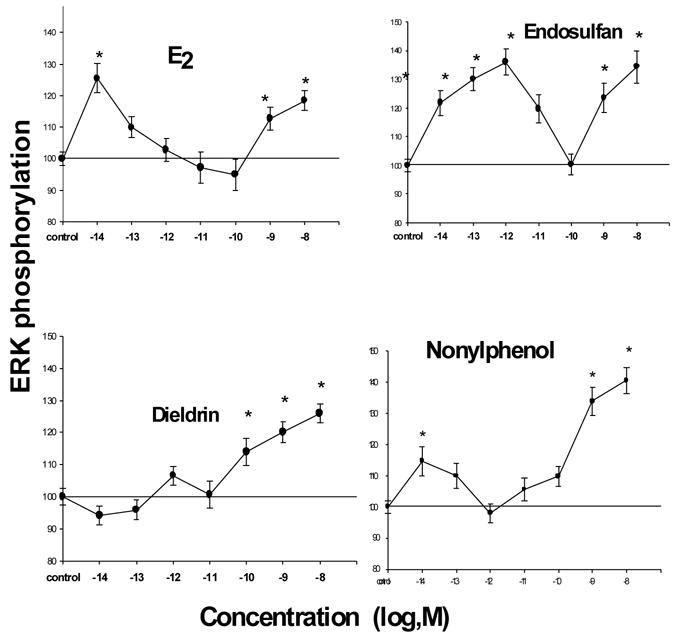
Cells were treated at the optimal time for each compound (3 min for E2, 6 min for dieldrin, and 30 min for endosulfan and nonylphenol). *=statistical significance at the P<0.05 level, compared to vehicle-treated controls. Data were normalized to cell number and presented as % of control values, which were set to 100. n = 48–60 wells/point, taken from 3 different experiments.
The dose-response pattern for ERK activation also differed between estrogenic compounds (Fig.5). Similar to the response to E2, xenoestrogens caused multiple peaks of activation at different doses, separated by inactive or inhibitory doses. The reason(s) for this bimodal pattern of effective dose ranges, and the above oscillating temporal activation, have long puzzled us. We have seen indications that there may be two populations of membrane steroid receptors in our collaborative studies on membrane glucocorticoid receptors, where a single Ab recognizes cells with two different signal-intensity membrane receptor populations [49]. This could reflect membrane steroid receptors in two different lipid compartments in the plasma membrane (such as rafts vs. invaginated caveolae or rafts vs. non-raft membrane [37;50;51]), but this has not been directly demonstrated. These potent responses (at the pM to nM concentrations range, [40]) contrast dramatically to the μM-mM effective concentrations required for genomic responses, and also used for studies on ERK activation by others [25]. In other studies we also showed that inhibitors of signaling pathways that lead to ERK activation inhibit both E2- and xenoestrogen-induced kinase phosphorylation [22]. However, xenoestrogen-induced responses were affected differentially by these specific pathway inhibitors (temporal and dose differences), again suggesting subtle differences in pathway usage by estrogen mimetics.
To compare these compounds for activity in genomic assays (from the literature) to our three nongenomic responses, Table 1 collates this information into a score based on the maximum value obtainable. The 17ß form of E2 is consistently a highly active compound in all responses, and all other compounds were compared to this level of activity. Some nongenomic responses (ERK activation, PRL release) are additionally graded on the rapidity/latency of the response where this was variable, since this is one of the characteristics defining nongenomic responses. (All Ca++ elevation responses were very rapid, and thus not scored for latency.)
Table 1. A comparison of estrogenic activities across genomic vs. three nongenomic responses.
The number at the far right of each table cell is a tally of an activity score across several categories. This number summarizes the strength or weakness of these compounds in these different pathways.
| Responses →
Compounds ↓ |
Nuclear assays (taken from the literature as reviewed in the text)
sensitivity/amplitude maximum score 6 |
ERK activation
rapidity/sensitivity/amplitude maximum score 9 |
Ca++ elevation
sensitivity/amplitude maximum score 6 |
PRL release
rapidity/sensitivity/amplitude maximum score 9 |
|---|---|---|---|---|
| 17ß estradiol | +++/+++ 6 | +++/+++/++ 8 | +++/+++ 6 | +++/++/+++ 8 |
| 17α estradiol | - 0 | +//+ (br ca) (3) | /+ (2) | +//+ (3) |
| E2-P or E2-BSA | - 0 | (++//+++) (7.5) | +++/+++ 6 | +++/+++/+++ 9 |
| DES | +++/+++ 6 | (++//+++) (7.5) | ++/+ 3 | +++/+/++ 6 |
| coumestrol | +++/+++ 6 | +++/+++/+++ 9 | +++/++ 5 | +++/+/++ 6 |
| bisphenol A | +/++ 3 | - 0 | +++/+++ 6 | +++/+++/++ 8 |
| nonylphenol | +/++ 3 | +/+++/+++ 7 | +++/+++ 6 | +++/+++/+ 7 |
| DDE | - 0 | ++/+/++ 5 | +/+ 2 | +/+/+/++ 5 |
| endosulfan | - 0 | +/+++/++ 6 | +/+++ 4 | +/++/+ 4 |
| dieldrin | - 0 | ++/+/++ 5 | +++/++ 5 | ++/+++/+ 6 |
All nongenomic studies summarized here were done with GH3/B6/F10 pituitary tumor cells, with the exception of one done with MCF-7 mER++ breast cancer cells (labeled br ca in column 2) and those for impeded ligands (E2-P or E2-BSA) on Ca++ elevation, which were taken from the literature [59;60]. Scored components are shown separated by "/". The highest rating is +++ for each component scored. The lowest rating is -, indicating no activity. The first data column is the activity of these compounds in nuclear transcription assays, gleaned from the literature (referenced in the text). For each compound a score was totaled, awarding 1 numerical increment for each score of +, yielding numerical "overall activity" scores for a final comparison. Where not all parameters were tested, those that were, were prorated as a percentage of the total and the score placed in parentheses.
The 17α-steroisomer of highly active 17β-E2 is inactive in genomic assays, but sometimes slightly active in nongenomic assays [36]. The impeded ligands E2-P or E2-BSA are active only in nongenomic responses, as would be expected of a compound that cannot enter cells by passing through the plasma membrane [52]; this is corroborated by reports of these impeded ligand activities in many other studies (reviewed in [53;54]).
DES, while a potent mediator of genomic responses, is not maximally effective at Ca++-driven secretory responses. The phytoestrogen coumestrol is an effective and potent estrogen in all responses, both genomic and nongenomic. Members of the structurally related category of compounds including the detergent (nonyphenol) and plastics monomer (BPA) are weakly active in genomic responses [4;6], relatively strong in Ca++ elevation leading to PRL release, but differ dramatically in their ability to activate ERKs. DDE is a weak evoker of Ca++ entry, but with unexpectedly stronger ability to release PRL [24]. However, as we have noted above (Figs. 2 and 3), the ability to generate a Ca++ signal is not always directly correlated to the strength in PRL release. As a group, the organochlorine pesticides have little if any genomic activity, but are moderately strong secretagogues for PRL, and have moderate strength for eliciting ERK activations.
DISCUSSION
Studies of multiple xenoestrogens will eventually allow us to decipher the structural requirements for nongenomic estrogenic signaling. Many xenoestrogens originally deemed "weak" appear to be potent via some nongenomic signaling pathways, and could contribute to these compounds’ ability to disrupt endocrine functions. While xenoestrogens can disrupt several signaling pathways, these structurally heterogeneous compounds affect estrogenic responses via diverse types of signaling pattern changes. These compounds could act on organisms at various stages of development or adult life, and in combination with stage-specific physiological estrogens to disrupt membrane-initiated signaling pathways.
Though both E2 and xenoestrogens potently elicit nongenomic responses, individual compounds (and sometimes structural categories of xenoestrogens) differ in their potencies and temporal response patterns. These differences could be the basis of both their inappropriate estrogenic activities and their interference with the activities of endogenous estrogens. The temporal pattern of kinase activation oscillates for both E2 and some xenoestrogens, though with quite different phasing. If compounds are experienced by the cell/organism simultaneously, as is likely to happen in the complex mixtures at contamination sites, or in combination with endogenous estrogens (typical for most exposures), then phasing differences in multiple responses could add up to an inappropriate (not oscillating) sustained response [22;40]. Compounds with different temporal patterns of activation may in fact be active in a time frame in which their co-stimulant (endogenous estrogens or other xenoestrogens) is inactive, and vice versa. We currently have very little information about how these compounds act in combination, but clearly such actions are opportunities for endocrine disruption of various kinds, leading to a variety of diseases of both reproductive and other tissues. In the future quantitative assays like these described here can be used to test combinations of physiological and xenoestrogenic compounds, and show how they might interact via the temporal and dose-dependent response patterns in our model system.
Another possibility for effects of xenoestrogen combinations, or combinations of xenoestrogens and physiological estrogens, could be additive overstimulation resulting in disruption. Such sustained responses may also exceed a maximum stimulation, and thus inappropriately trigger compensatory inhibitory responses, usually seen in steroid dose-response curves (for example [55]). This consideration is also pertinent to the important biological issue of exposures during different developmental and reproductive status windows. This may explain differing potential dangers of exposures during times when endogenous estrogen levels are particularly high or low in both males and females (infant, pubertal, reproductive, or post-reproductive stages). These exposures may also interact with ingested phytoestrogens (such as soy isoflavones, coumestrol, or resveratrol), or therapeutic estrogen exposures (e.g. patches for hormone replacement or birth control).
Specific xenoestrogens have been previously investigated extensively via experimental systems that only monitor genomic steroid mechanisms. Our results for participation in nongenomic signaling pathways clearly differ from those conclusions. Xenoestrogens also showed different time- and dose-dependent patterns of activation, also different from that of E2. Xenoestrogens also differentially utilized the multiple signaling pathways within the web of signaling possibilities, to impinge upon final responses or signaling summation mechanisms (like ERK activation, [56]) and functional endpoints. It is very interesting that bisphenol A is active in one nongenomic signaling pathway (Ca++ flux leading to PRL release), but not at all in another (ERK activation). Clearly, each xenoestrogen must be tested for activity (including sensitivity and temporal pattern) separately in all likely signaling cascades to determine its potential for endocrine disruption of specific functions.
Nuclear versions of steroid receptors are affected by different tissue backgrounds, and the responses of membrane steroid receptors will probably also be affected similarly. This is because the functionally interacting repertoire of proteins (co-regulators in the case of transcription factors, and signaling partners in the case of the membrane steroid receptors) will be different in each cell type [18;57;58]. The chemical environment of the nuclear receptors (aqueous) vs. that of the membrane receptors (lipidaceous) will probably affect the shape of the protein, and consequently alter the shape of its binding pocket, and its ligand specificity. Therefore, focused studies will be required to resolve all of these complex issues in understanding xenoestrogenic responses in the whole organism. Finding answers to these questions has great implications for avoiding harmful effects of xenosteroids, but perhaps also for the development of therapeutic drugs that utilize these pathways [53].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singleton DW, Khan SA. Xenoestrogen exposure and mechanisms of endocrine disruption. Front Biosci. 2003;8:s110–s118. doi: 10.2741/1010. [DOI] [PubMed] [Google Scholar]
- 2.Carson R. Silent Spring. Greenwich, Conn, Fawcett. 1962 [Google Scholar]
- 3.Witorsch RJ. Endocrine disruptors: can biological effects and environmental risks be predicted? Regul Toxicol Pharmacol. 2002;36(1):118–130. doi: 10.1006/rtph.2002.1564. [DOI] [PubMed] [Google Scholar]
- 4.Gaido KW, Leonard LS, Lovell S, Gould JC, Babai D, Portier CJ, McDonnell DP. Evaluation of chemicals with endocrine modulating activity in a yeast- based steroid hormone receptor gene transcription assay. Toxicol Appl Pharmacol. 1997;143(1):205–212. doi: 10.1006/taap.1996.8069. [DOI] [PubMed] [Google Scholar]
- 5.Gutendorf B, Westendorf J. Comparison of an array of in vitro assays for the assessment of the estrogenic potential of natural and synthetic estrogens, phytoestrogens and xenoestrogens. Toxicology. 2001;166(1–2):79–89. doi: 10.1016/s0300-483x(01)00437-1. [DOI] [PubMed] [Google Scholar]
- 6.Jorgensen M, Vendelbo B, Skakkebaek NE, Leffers H. Assaying estrogenicity by quantitating the expression levels of endogenous estrogen-regulated genes. Environ Health Perspect. 2000;108(5):403–412. doi: 10.1289/ehp.108-1638061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kloas W, Lutz I, Einspanier R. Amphibians as a model to study endocrine disruptors: II. Estrogenic activity of environmental chemicals in vitro and in vivo. Sci Total Environ. 1999;225(1–2):59–68. doi: 10.1016/s0048-9697(99)80017-5. [DOI] [PubMed] [Google Scholar]
- 8.Petit F, Legoff P, Cravedi JP, Valotaire Y, Pakdel F. Two complementary bioassays for screening the estrogenic potency of xenobiotics - recombinant yeast for trout estrogen receptor and trout hepatocyte cultures. J Mol Endocr. 1997;19(3):321–335. doi: 10.1677/jme.0.0190321. [DOI] [PubMed] [Google Scholar]
- 9.Sheeler CQ, Dudley MW, Khan SA. Environmental estrogens induce transcriptionally active estrogen receptor dimers in yeast: activity potentiated by the coactivator RIP140. Environ Health Perspect. 2000;108(2):97–103. doi: 10.1289/ehp.0010897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinmetz R, Brown NG, Allen DL, Bigsby RM, Benjonathan N. The environmental estrogen bisphenol A stimulates prolactin release in vitro and in vivo. Endocr. 1997;138(5):1780–1786. doi: 10.1210/endo.138.5.5132. [DOI] [PubMed] [Google Scholar]
- 11.Watson CS. STKE E1, Science's Signal Transduction Knowledge Environment. 1999. Signaling themes shared between peptide and steroid hormones at the plasma membrane [Review] Ref Type: Electronic Citation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watson CS. The Identities of Membrane Steroid Receptors.... and Other Proteins Mediating Nongenomic Steroid Action. Boston: Kluwer Academic Publishers; 2003. [Google Scholar]
- 13.Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. 1977;265:69–72. doi: 10.1038/265069a0. [DOI] [PubMed] [Google Scholar]
- 14.Baker ME. Co-evolution of steroidogenic and steroid-inactivating enzymes and adrenal and sex steroid receptors. Mol Cell Endocrinol. 2004;215(1–2):55–62. doi: 10.1016/j.mce.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Sonnenschein C, Soto AM. An updated review of environmental estrogen and androgen mimics and antagonists. J Steroid Biochem & Mol Biol. 1998;65(1–6):143–150. doi: 10.1016/s0960-0760(98)00027-2. [DOI] [PubMed] [Google Scholar]
- 16.Antignac JP, Cariou R, Le BB, Cravedi JP, Andre F. Identification of phytoestrogens in bovine milk using liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(12):1256–1264. doi: 10.1002/rcm.1052. [DOI] [PubMed] [Google Scholar]
- 17.Herbst AL, Green TH, Jr, Ulfelder H. Primary carcinoma of the vagina. An analysis of 68 cases. Am J Obstet Gynecol. 1970;106(2):210–218. doi: 10.1016/0002-9378(70)90265-6. [DOI] [PubMed] [Google Scholar]
- 18.Singleton DW, Feng Y, Burd CJ, Khan SA. Nongenomic activity and subsequent c-fos induction by estrogen receptor ligands are not sufficient to promote deoxyribonucleic acid synthesis in human endometrial adenocarcinoma cells. Endocr. 2003;144(1):121–128. doi: 10.1210/en.2002-220625. [DOI] [PubMed] [Google Scholar]
- 19.Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(21):11603–11608. doi: 10.1073/pnas.97.21.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.onso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ Health Perspect. 2006;114(1):106–112. doi: 10.1289/ehp.8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.onso-Magdalena P, Laribi O, Ropero AB, Fuentes E, Ripoll C, Soria B, Nadal A. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ Health Perspect. 2005;113(8):969–977. doi: 10.1289/ehp.8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulayeva NN, Watson CS. Xenoestrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Environ Health Perspect. 2004;112(15):1481–1487. doi: 10.1289/ehp.7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-alpha: estrogens, xenoestrogens, and phytoestrogens. Steroids. 2005;70(5–7):364–371. doi: 10.1016/j.steroids.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Wozniak AL, Bulayeva NN, Watson CS. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-alpha-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect. 2005;113(4):431–439. doi: 10.1289/ehp.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Zhang S, Safe S. Activation of kinase pathways in MCF-7 cells by 17beta-estradiol and structurally diverse estrogenic compounds. J Steroid Biochem Mol Biol. 2005 doi: 10.1016/j.jsbmb.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Y, Bond J, Thomas P. Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc Natl Acad Sci U S A. 2003;100(5):2237–2242. doi: 10.1073/pnas.0436133100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerdes D, Wehling M, Leube B, Falkenstein E. Cloning and tissue expression of two putative steroid membrane receptors. Biol Chem. 1998;379(7):907–911. doi: 10.1515/bchm.1998.379.7.907. [DOI] [PubMed] [Google Scholar]
- 28.Luconi M, Bonaccorsi L, Bini L, Liberatori S, Pallini V, Forti G, Baldi E. Characterization of membrane nongenomic receptors for progesterone in human spermatozoa. Steroids. 2002;67(6):505–509. doi: 10.1016/s0039-128x(01)00173-8. [DOI] [PubMed] [Google Scholar]
- 29.Filardo EJ, Quinn JA, Frackelton AR, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 30.Berdal A, Mesbah M, Papagerakis P, Nemere I. Putative membrane receptor for 1,25(OH)2 vitamin D3 in human mineralized tissues during prenatal development. Connect Tissue Res. 2003;44 (Suppl 1):136–140. [PubMed] [Google Scholar]
- 31.Campbell CH, Watson CS. A comparison of membrane vs. intracellular estrogen receptor-α in GH3/B6 pituitary tumor cells using a quantitative plate immunoassay. Steroids. 2001;66:727–736. doi: 10.1016/s0039-128x(01)00106-4. [DOI] [PubMed] [Google Scholar]
- 32.Norfleet AM, Clarke C, Gametchu B, Watson CS. Antibodies to the estrogen receptor-α modulate prolactin release from rat pituitary tumor cells through plasma membrane estrogen receptors. FASEB J. 2000;14:157–165. doi: 10.1096/fasebj.14.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norfleet AM, Thomas ML, Gametchu B, Watson CS. Estrogen receptor-α detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary cells by enzyme-linked immunocytochemistry. Endocr. 1999;140(8):3805–3814. doi: 10.1210/endo.140.8.6936. [DOI] [PubMed] [Google Scholar]
- 34.Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptor-enriched GH3/B6 cells have an enhanced non-genomic response to estrogen. Endocrine. 1995;3:743–749. doi: 10.1007/BF03000207. [DOI] [PubMed] [Google Scholar]
- 35.Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995;9(5):404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- 36.Pappas TC, Gametchu B, Yannariello-Brown J, Collins TJ, Watson CS. Membrane estrogen receptors in GH3/B6 cells are associated with rapid estrogen-induced release of prolactin. Endocrine. 1994;2:813–822. [Google Scholar]
- 37.Zivadinovic D, Watson CS. Membrane estrogen receptor-alpha levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005;7(1):R130–R144. doi: 10.1186/bcr959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zivadinovic D, Gametchu B, Watson CS. Membrane estrogen receptor-alpha levels in MCF-7 breast cancer cells predict cAMP and proliferation responses. Breast Cancer Res. 2005;7(1):R101–R112. doi: 10.1186/bcr958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dannies PS. Control of prolactin production by estrogen. Biochemical Actions of Hormones. In: Litwack G, editor. Orlando: Academic Press, Inc; 1985. pp. 289–310. [Google Scholar]
- 40.Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids. 2004;69(3):181–192. doi: 10.1016/j.steroids.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulayeva NN, Wozniak A, Lash LL, Watson CS. Mechanisms of membrane estrogen receptor-{alpha}-mediated rapid stimulation of Ca2+ levels and prolactin release in a pituitary cell line. Am J Physiol Endocrinol Metab. 2005;288:E388–E397. doi: 10.1152/ajpendo.00349.2004. [DOI] [PubMed] [Google Scholar]
- 42.Watson CS, Campbell CH, Gametchu B. Membrane estrogen receptors on rat pituitary tumor cells: Immunoidentification and responses to estradiol and xenoestrogens. Experimental Physiology. 1999;84(6):1013–1022. doi: 10.1111/j.1469-445x.1999.01903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kits KS, Mansvelder HD. Regulation of exocytosis in neuroendocrine cells: spatial organization of channels and vesicles, stimulus-secretion coupling, calcium buffers and modulation. Brain Res Brain Res Rev. 2000;33(1):78–94. doi: 10.1016/s0165-0173(00)00023-0. [DOI] [PubMed] [Google Scholar]
- 44.Watson CS, Norfleet AM, Pappas TC, Gametchu B. Rapid actions of estrogens in GH3/B6 pituitiary tumor cells via a plasma membrane version of estrogen receptor-α. Steroids. 1999;64:5–13. doi: 10.1016/s0039-128x(98)00107-x. [DOI] [PubMed] [Google Scholar]
- 45.Picotto G, Massheimer V, Boland R. Acute stimulation of intestinal cell calcium influx induced by 17 beta-estradiol via the cAMP messenger system. Mol Cell Endocrinol. 1996;119(2):129–134. doi: 10.1016/0303-7207(96)03799-9. [DOI] [PubMed] [Google Scholar]
- 46.Wozniak AL, Bulayeva NN, Watson CS. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-α-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Env Health Perspect. 2005;113(4):431–439. doi: 10.1289/ehp.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xing C, Imagawa W. Altered MAP kinase (ERK1,2) regulation in primary cultures of mammary tumor cells: elevated basal activity and sustained response to EGF. Carcinogenesis. 1999;20(7):1201–1208. doi: 10.1093/carcin/20.7.1201. [DOI] [PubMed] [Google Scholar]
- 48.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocr. 2005;146(2):624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 49.Gametchu B, Watson C, Shih C, Dashew B. Studies on the arrangement of glucocorticoid receptors in the plasma membrane of S-49 lymphoma cells. Steroids. 1991;56:411–419. doi: 10.1016/0039-128x(91)90029-u. [DOI] [PubMed] [Google Scholar]
- 50.Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, Mendelsohn ME, Anderson RG, Shaul PW. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res. 2000;87(11):E44–E52. doi: 10.1161/01.res.87.11.e44. [DOI] [PubMed] [Google Scholar]
- 51.Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. ERs associate with and regulate the production of caveolin: Implications for signaling and cellular actions. Mol Endocrinol. 2002;16(1):100–115. doi: 10.1210/mend.16.1.0757. [DOI] [PubMed] [Google Scholar]
- 52.Watson CS, Pappas TC, Gametchu B. The other estrogen receptor in the plasma membrane: implications for the actions of environmental estrogens. Environ Health Perspect. 1995;103 (Suppl 7):41–50. doi: 10.1289/ehp.95103s741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watson CS, Gametchu B. Proteins of multiple classes participate in nongenomic steroid actions. Exp Biol Med. 2003;228(11):1272–1281. doi: 10.1177/153537020322801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson CS, Gametchu B. Membrane-initiated steroid actions and the proteins that mediate them. Proc Soc Exp Biol Med. 1999;220(1):9–19. doi: 10.1046/j.1525-1373.1999.d01-2.x. [DOI] [PubMed] [Google Scholar]
- 55.Kushner PJ, Hort E, Shine J, Baxter JD, Greene GL. Construction of cell lines that express high levels of the human estrogen receptor and are killed by estrogens. Mol Endocrinol. 1990;4(10):1465–1473. doi: 10.1210/mend-4-10-1465. [DOI] [PubMed] [Google Scholar]
- 56.Watson CS, Lange CA. Steadying the boat: integrating mechanisms of membrane and nuclear-steroid-receptor signalling. EMBO Rep. 2005;6(2):116–119. doi: 10.1038/sj.embor.7400336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13(2):55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- 58.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29(14):2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benten WPM, Lieberherr M, Giese G, Wunderlich F. Estradiol binding to cell surface raises cytosolic free calcium in T cells. FEBS Lett. 1998;422(3):349–353. doi: 10.1016/s0014-5793(98)00039-8. [DOI] [PubMed] [Google Scholar]
- 60.Doolan CM, Harvey BJ. A Galphas protein-coupled membrane receptor, distinct from the classical oestrogen receptor, transduces rapid effects of oestradiol on [Ca(2+)](i) in female rat distal colon. Mol Cell Endocrinol. 2003;199(1–2):87–103. doi: 10.1016/s0303-7207(02)00303-9. [DOI] [PubMed] [Google Scholar]