

Abstract
The classic neuropathological diagnostic markers for AD are amyloid plaques and neurofibrillary tangles, but their role in the etiology and progression of the disease remains incompletely defined. Research over the last decade has revealed that cell cycle abnormalities also represent a major neuropathological feature of AD. These abnormalities appear very early in the disease process, prior to the appearance of plaques and tangles; and it has been suggested that neuronal cell cycle regulatory failure may be a significant component of the pathogenesis of AD. The amyloid precursor protein (APP) is most commonly known as the source of the β-amyloid (Aβ) peptides that accumulate in the brains of patients with AD. However, a large body of work supports the idea that APP is also a signaling receptor. Most recently, it has been shown that familial AD (FAD) mutations in APP or simple overexpression of wild type APP cause dysfunction of APP signaling, resulting in initiation of DNA synthesis in neurons and consequent apoptosis. In this article, we review the evidence that APP has the potential to activate aberrant neuronal cell cycle re-entry in AD, and we describe a signal transduction pathway that may mediate this abnormal activation of the cell cycle.
Keywords: Alzheimer’s disease, amyloid precursor protein, p21 activated kinase, APP-BP1, cell cycle, apoptosis
1. Introduction
AD is a progressive, incurable disease that always ends in death. Its predominant clinical manifestation is memory loss, but a number of other changes in brain function, including disoriented behavior and impairments in language, comprehension, and visual-spatial skills, also characterize this disorder. The diagnostic hallmarks of AD, namely neuronal attrition, amyloid deposits, and neurofibrillary tangles, have not as yet explained the etiology and pathogenesis of the disease. However, additional markers of AD have been described that may give some clues to the mechanism by which neurons die in AD brain. Notably, aberrant expression of cell cycle proteins and DNA tetraploidy in neurons in pathologically affected regions of AD brain have been described. It has been shown also that those neurons that have entered the cell cycle subsequently undergo a form of programmed cell called apoptosis. By now, the evidence that neuronal death in AD may be at least partly due to cell cycle entry followed by apoptosis, is considerable. This has clear therapeutic implications: understanding the molecular pathways underlying this cell cycle-mediated neurodegeneration will reveal new therapeutic targets and lead to novel strategies for slowing or even blocking the onset and progression of AD. One such pathway, that is mediated by APP, will be described.
2. Cell cycle abnormalities in AD
2.1 The cell cycle is activated in neurons in AD brain
A decade ago, Vincent and Davies showed definitively that activation of cell cycle components occurred in AD brain (1). Subsequently, abnormal expression of such cell cycle molecules as cdc2, cdk4, p16, Ki-67, cyclin B1, and cyclin D was reported in pathologically affected or vulnerable neurons in AD brain (2–6). Busser et al. (6) found ectopic expression of cell cycle proteins in regions of AD brain displaying extensive cell death, and Chow et al. (7) detected increased expression of genes encoding cell cycle proteins in single neurons in late-stage relative to early-stage AD brain. A number of these cell cycle regulators have been reported to appear in vulnerable neurons prior to lesion formation (6,8,9).
These data showed correlations between the expression of cell cycle markers and AD neuropathology, but did not reveal the consequences for AD neurodegeneration of such activation of cell cycle proteins. Yang et al. (10) showed that one of these consequences appears to be that vulnerable neurons in AD brain re-enter the cell cycle. This report demonstrated that a significant number of hippocampal pyramidal (4% vs. 0% in control hippocampus) and basal forebrain neurons in AD brain had undergone full or partial DNA replication, evidence that these neurons had completed the S phase of the cell cycle. Such DNA synthesis is not normally seen in neurons; and indeed, it was not detected in regions of AD brain untouched by pathology or in control brains. Yang et al. (10) suggested that the state of tetraploidy eventually is lethal to neurons. In a follow-up report, this same group (11) described the emergence of cell cycle-related proteins prior to neuronal cell death not only in the brains of individuals with advanced AD, but also in the brains of individuals with mild cognitive impairment, which usually progresses to AD. Yang and co-authors went on to describe the reexpression of cell cycle markers in four different plaque-bearing mouse models of AD (12). In one of these models, cell cycle events were detected many months before the first appearance of plaques. These in vivo observations supported reports showing that at least two in vitro models for AD neurodegeneration featured re-activation of cell cycle entry in neurons (13,14).
2.2. Cell cycle activation may link to tangles in AD
Activation of the cell cycle may also contribute to the formation of neurofibrillary tangles, which are pathological markers for AD that contain hyperphosphorylated microtubule associate protein tau. The phosphoepitope S214 of tau, that appears in neurofibrillary tangles in AD, is a prominent phosphorylation site in metaphase but not in interphase of dividing cells expressing tau (15). The tau TG3 epitope (phosphorylated T231) has been detected in mitotic cells but not in quiescent cells (1). Moreover, tau is hyperphosphorylated during mitosis in neuroblastoma cells (16), and a cdc2-related kinase is associated with paired helical filaments, the ultrastructural correlates of neurofibrillary tangles, in AD brain (17). Patrick et al. (1999) showed that activation of the p25/Cdk5 complex results in hyperphosphorylation of tau and reduces the ability of tau to associate with microtubules. Mitotic phosphorylation of tau likely causes conformational changes, which may be associated with the development of neurofibrillary tangles. These data support the idea that reactivation of the cell cycle machinery not only causes DNA synthesis but also may cause tau hyperphosphorylation in AD brain.
2.3. Cell cycle activation may lead to apoptosis in AD brain
Activation of cell cycle proteins in neurons also can, under certain circumstances, lead to a form of cell suicide called apoptosis (reviewed in ref. 18). The possibility that such a process occurs in AD brain was first suggested by Su et al. (19), when they reported evidence for DNA fragmentation in neurons in AD brain. This same group showed that β-amyloid induces neuronal apoptosis in vitro (20), a finding that has been replicated in many laboratories. In support of the idea that apoptosis may be one means by which neurons die in AD, several groups have reported the presence of activated caspases in the postmortem brains of individuals with AD (21–23), and have described apoptotic neurons in the brains of transgenic mouse models for AD (24–27). Apoptosis normally proceeds to completion within 16–24 hours, which seems incompatible with the relatively slow progression of AD. However, Cotman (28) and Raina et al. (23) have both proposed that the induction of compensatory responses to apoptosis in the AD brain protects the neurons from terminal apoptosis, and that a dynamic balance between cell death processes and compensatory responses exists in AD brain.
3. APP Signaling Pathways That Mediate Cell Cycle Activation
3.1. APP is a signaling protein
The amyloid precursor protein (APP) is the source of the β-amyloid peptides that accumulate in the brains of patients with Alzheimer’s disease (AD). The primacy of APP in the etiology of AD is underscored by the fact that virtually all individuals with Down syndrome (DS), who as a consequence of chromosome 21 trisomy overexpress APP, develop AD by the age of 40. This finding, together with the observation that either specific mutations in APP or simple duplication of the wild type APP gene can cause some forms of FAD (29,30), suggests the possibility that an alteration in the normal function of APP may occur in AD, and has refocused attention on the delineation of the function that APP subserves in the brain.
The notion that APP may be a signaling receptor was originally proposed on the basis of the predicted amino acid sequence of APP, which suggested that APP was a type 1 intrinsic membrane protein whose structure was consistent with that of a “cell surface receptor” (31). The first direct evidence in support of this idea was the finding that the APP cytodomain interacted with and activated the heterotrimeric G protein Go (32), a finding that was subsequently confirmed independently (33). During the last decade, a multitude of additional cytosolic proteins that interact with the APP cytodomain have been described (14,34–43), suggesting that APP has versatile signaling roles.
To define neuronal signaling pathways that may be mediated by APP, we established an in vitro paradigm for AD neurodegeneration, in which wild type or FAD APP cDNAs were expressed as transgenes in primary cortical neurons (14, 44). Using this model, we showed that overexpression of wild type or even modest expression of FAD mutants of APP in neurons leads to neuronal DNA synthesis and apoptosis (14,44), suggesting that an APP-mediated signaling pathway may play a role in the cell cycle activation and neurodegeneration seen in AD.
3.2. APP-BP1, which binds to APP, is a cell cycle protein
As a first step in defining neuronal signaling pathways that may be initiated by APP, a search of brain cDNA libraries was carried out to detect genes encoding proteins that interact with the cytodomain of APP (14,43). One of these, APP-BP1, was found to be an essential cell cycle protein that drives the cell cycle through the S-M checkpoint in dividing cells, but which causes apoptosis when overexpressed in neurons (44). Overexpression of wild type APP or expression of FAD mutants of APP in neurons, using the paradigm described above, resulted in an increase in expression of APP-BP1 in lipid rafts, followed by entry of the neurons into the S phase of the cell cycle, and neuronal apoptosis (44). As predicted by these in vitro results, APP-BP1 was shown to be overexpressed in lipid rafts in at-risk regions of human AD brain relative to cognitively intact controls (45). Consistent with these observations, modest overexpression of APP-BP1 in neurons, sufficient to mimic the observed increases of this protein in lipid rafts in AD brains, caused neuronal cell cycle entry and apoptosis (45).
The interaction of APP with APP-BP1 activates a pathway leading to the conjugation of NEDD8, a ubiquitin-like protein, to its target (Fig. 1, ref. 44). APP-BP1, together with hUba3, is functionally analogous to the ubiquitin activating enzyme E1, with hUba3 containing the active cysteine and ATP binding site. When NEDD8 is activated by the APP-BP1/hUba3 complex, it forms a thiol ester bond with hUbc12, which has a function parallel to that of ubiquitin-conjugating enzyme Cdc32. Subsequently, NEDD8 is covalently coupled to lysine residues in its target proteins (46). So far, the proteins known to be neddylated via this pathway are a family of proteins called cullins (47) and the Mdm2 oncogene product, which in turn regulates neddylation of the cell cycle protein p53 (48). Cullins are scaffold proteins for the E3 ubiquitin ligase complex, and neddylation of cullin enhances its ability to promote ubiquitination (49,50). Indeed, NEDD8 has been found in ubiquitinated neurofibrillary tangles in AD brain (51). NEDD8 signalling has been shown to regulate protein degradation pathways participating in cell cycle progression (52–55). The discovery of a novel protein, NUB1, which recruits NEDD8-conjugates to the proteasome for degradation, provides a direct link between these 2 systems (56,57). Inhibition of the neddylation pathway in neurons by expression of a dominant negative mutant of hUbc12 prevents FAD APP-mediated cell cycle entry and apoptosis (44,45). Thus, elements of this pathway are attractive targets for potential therapies aimed at preventing neurons in AD brain from entering the cell cycle.
Figure 1.
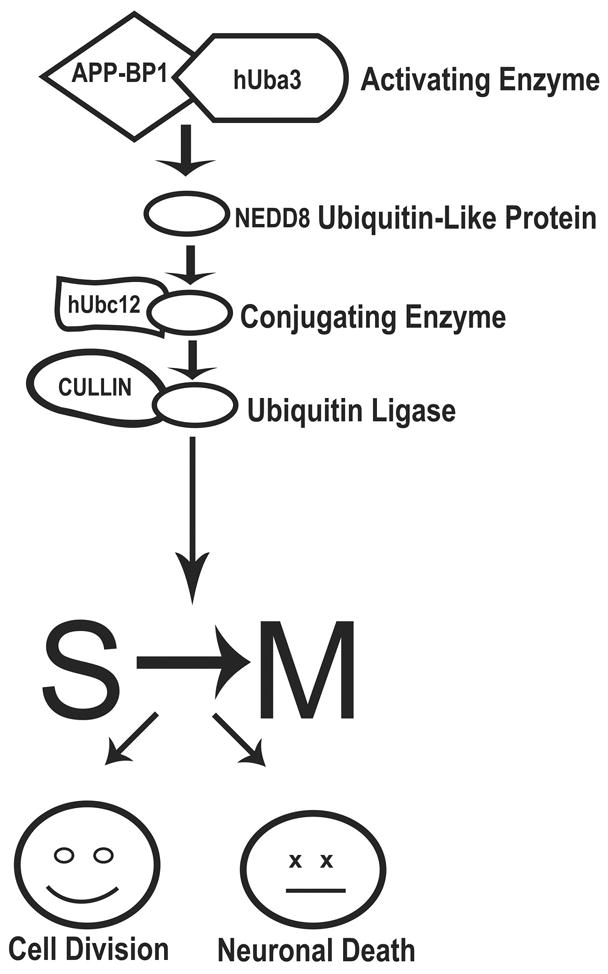
Graphic representation of the neddylation pathway activated by the interaction of APP with APP-BP1. Steps in the cascade that are potential therapeutic targets include the interaction between APP and APP-BP1, and the interaction of APP-BP1/hUba3 with the conjugating enzyme hUbc12.
3.3. p21 activated kinase 3 mediates neuronal DNA synthesis caused by FAD mutants of APP
In addition to APP-BP1, the p21-activated kinase PAK3 also was shown to bind to the C-terminus of APP (14). PAK3 is a serine/threonine kinase whose activity is regulated by the p21 family of small GTPases. Mutations in this molecule, which is expressed selectively in the nervous system (14), cause one form of X chromosome-linked mental retardation (58,59). PAK3 is involved in the control of cytoskeleton dynamics, possibly affecting cognition by regulating the shape of neuronal synapses. Consistent with this idea, mice lacking the Pak3 gene are impaired in both synaptic plasticity and cognition (60).
PAK3 also has been implicated in the abnormal DNA synthesis and apoptosis caused by overexpression of wild type APP or ectopic expression of FAD mutants of APP in neurons in vitro (14). A graphic representation of the neuronal signaling pathway activated by interaction of FAD APP with PAK3 is shown in Figure 2. A dominant negative kinase-dead mutant of PAK3 inhibits wild type or FAD APP-mediated neuronal apoptosis and DNA synthesis in our in vitro model; this effect is abolished by deletion of the PAK3 APP-binding domain or by co-expression of a peptide representing this binding domain (14). These data suggest that both the kinase activity of PAK3 and also its interaction with APP are important for the FAD APP signaling pathway. The specificity of PAK3 involvement in FAD APP-mediated apoptosis rather than in general apoptotic pathways is suggested by the facts that a constitutively active mutant of PAK3 does not alone cause neuronal apoptosis, and that the dominant negative mutant of PAK3 does not inhibit chemically-induced apoptosis. In vitro, FAD APP-mediated DNA synthesis precedes FAD APP-mediated apoptosis in neurons; and inhibition of neuronal entry into the cell cycle blocks the apoptosis (14).
Figure 2.
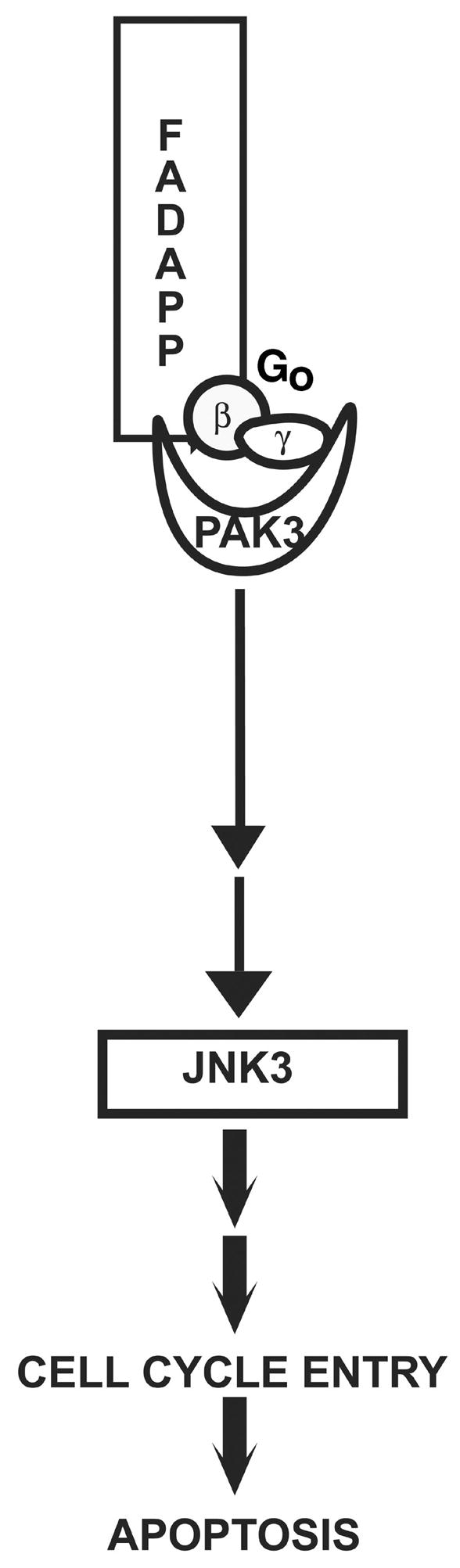
Graphic representation of the signaling pathway activated by the interaction of PAK3 with APP and Go. Steps in the cascade that are potential therapeutic targets include interactions among APP, Go and PAK3 or the activity of JNK3.
Pertussis toxin inhibits DNA synthesis and apoptosis caused in vitro by FAD APP mutants, implicating Go in the process as well (14). These data are consistent with the reports by Nishimoto and colleagues that the His657-Lys676 domain of APP-695 activates the heterotrimeric GTP-binding protein Go in a GTPγS-inhibitable manner (32,61). Their demonstration that an antibody to the extracellular domain of APP that acts as a ligand mimetic (62) causes activation of Go, argues that APP may be a G protein-coupled receptor. Notably, neuronal expression of FAD APP deleted for residues His657-Lys676 does not cause cell cycle entry (14).
These data support a model in which APP normally is part of a Go protein-centered complex including PAK3 that transduces extracellular signals to the cytoplasm (Figure 2), and in which overexpression of wild type APP or expression of FAD APP causes dysfunction of this pathway. How might this complex be related to the pathway activated by APP-BP1? At least one of the downstream molecules in the PAK3-mediated pathway is the c-Jun N-terminal kinase 3 (JNK3). A dominant negative mutant of JNK3 inhibits FAD APP-mediated cell cycle entry, while a constitutively active mutant of JNK3 alone causes neuronal DNA synthesis (McPhie, D.L. and Neve, R.L., unpublished data). The effect of the constitutively active JNK3 is not blocked by a dominant negative mutant of PAK3, implying that the action of JNK3 is downstream of PAK3. However, inhibition of the APP-BP1-mediated neddylation pathway in neurons by expression of a dominant negative mutant of hUbc12 prevents constitutively active JNK3 activation of neuronal cell cycle entry (Chen, Y. and Neve, R.L., unpublished data). Whereas pertussis toxin blocks neuronal DNA synthesis caused by ectopic expression of FAD APP, it does not block neuronal DNA synthesis caused by APP-BP1. These data suggest that the order of events in the FAD APP-mediated pathway leading to neuronal cell cycle entry and apoptosis consists of formation of the APP-Go-PAK3 complex, followed by activation of JNK3, which is then followed by activation of the APP-BP1 neddylation pathway. Intermediate events are likely to occur.
These findings also implicate JNK3 in FAD APP-mediated neuronal DNA synthesis and apoptosis, and are consistent with the finding that JNK3 is highly expressed and activated in postmortem brains of individuals with AD (63). JNK3 is associated with neurofibrillary tangles, and JNK upregulation colocalizes with phosphorylated tau (63), a microtubule associated protein which has been shown to be phosphorylated by JNK (64). Abnormal phosphorylation of tau by JNK3 causes the formation of oligomeric tau fibrils that have been termed “pretangles” (65). In a transgenic mouse model of AD, JNK activation is associated with amyloid deposits and phospho-tau. Age-dependent increased JNK activity is correlated not only with increased amyloid deposition in this mouse model, but also loss of functional synapses similar to that observed in AD brain (66).
We and others have hypothesized that APP and its C-terminal binding proteins – among them Go, PAK3, and APP-BP1 -- have a regulated interaction that activates signaling pathways important for normal brain function, perhaps mediating synaptic remodeling or neurogenesis during learning. In analogy with the cell proliferation and resultant tumorigenesis caused either by a constitutively active mutant of the epidermal growth factor receptor (EGFR) or by sustained overexpression of wild type EGFR (Lorimer, 2002), neuronal cell cycle entry and consequent apoptosis may be caused either by constitutive activation of APP signaling pathways in neurons due to FAD mutations in APP or by sustained overexpression of wild type APP. Thus, therapeutic reagents that target these signaling pathways may have efficacy in treating AD neurodegeneration.
3.4. APP-C31 may mediate neuronal cell cycle entry and neurodegeneration in AD brain
All but one of the binding proteins for the APP cytodomain interact with APP within the last 31 amino acids of this domain. Why is this important? For one thing, C31 can be generated from APP by caspase cleavage (67,68). Furthermore, this cleavage has functional significance, in that expression of C31 alone has been shown to cause neuronal DNA synthesis and apoptosis (14, 45, 68). Recently, inhibition of C31-producing caspase cleavage of APP was shown to prevent the development of AD-like pathology and behavior caused by two FAD mutations of APP (69). A D664A mutation (which prevents the generation of C31) was introduced into the background of a human APP minigene carrying the K670N/M671L (Swedish) and V717F (Indiana) mutations. Both the original FAD mutant minigene (PDAPP) and also the D664A version of it [PDAPP(D664A)] were expressed in transgenic mice under the control of the PDGF B-chain promoter.
Interestingly, the authors showed that the D664A mutation neither altered the net in vivo production of Aβ40 and Aβ42, nor affected the extent of amyloid plaque deposition in the brains of PDAPP(D664A) mice compared to PDAPP mice. However, the D664A mutation did have an effect on neurodegeneration and on behavior. The PDAPP mice without the D664A mutation displayed decreased hippocampal presynaptic density number relative to controls at 8–10 months of age, a pronounced increase in GFAP immunoreactivity in the hippocampus by 12 months, a loss in dentate gyrus volume at 3 months, learning and spatial impairments at 12 months, and an increase in the number of proliferating cells present in the subgranular zone of the dentate gyrus in both 3- and 12-month. In contrast, the PDAPP(D664A) mice, with the D664A mutation, were indistinguishable from controls in every one of these parameters.
It can be inferred from these data that the mutation Asp664 “rescues” multiple aspects of neuropathology and impaired learning that are normally caused by the Swedish and Indiana mutations in APP. In other words, if C31 cannot be generated, the FAD APP mutations cannot cause certain pathological and behavioral abnormalities. What are the implications of these findings? First of all, note that Asp664 selectively rescues the neurodegeneration and the learning abnormalities of the PDAPP mice without decreasing the production of Aβ40 or Aβ42. Thus, the rescue is independent of the production of Aβ. Secondly, the C31 region of APP encompasses the binding sites for nearly all of the signaling proteins, including APP-BP1 and PAK3, that have been shown to bind to the intracellular domain of APP.
The data suggest a scenario in which C31, when removed from APP, abnormally activates or disrupts signaling pathways mediated by APP. One possibility is that the signaling initiated by interaction of the APP C-terminal domain with signal transduction proteins normally is tightly regulated by binding of ligand(s) to its extracellular domain [variously identified as F-spondin (70), notch (71, 72), TGF-β2 (73), and even Aβ (74, 75)]. In such a scenario, as suggested by McPhie et al. (14), C31, when not attached to APP, is relieved of the normal constraints imposed on it when the extracellular domain of APP is not occupied by a ligand, and becomes constitutively active or else takes on signaling functions that it does not normally possess (Figure 3).
Figure 3.
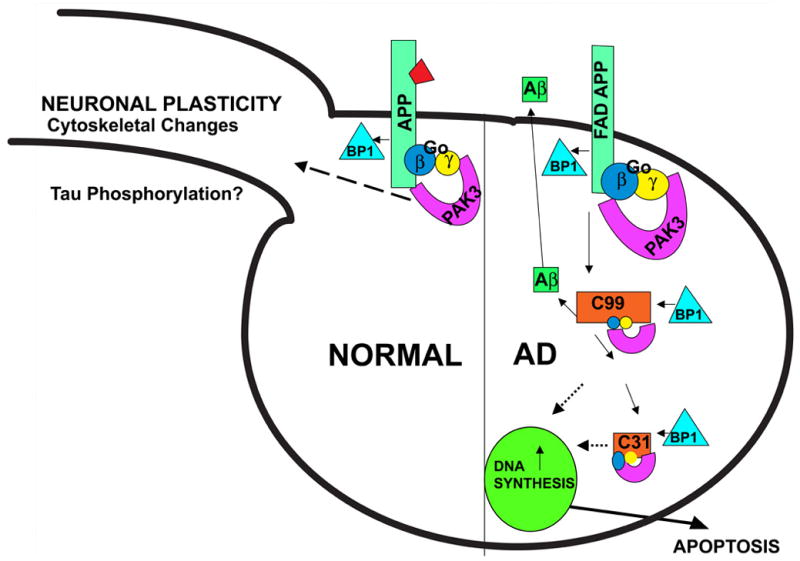
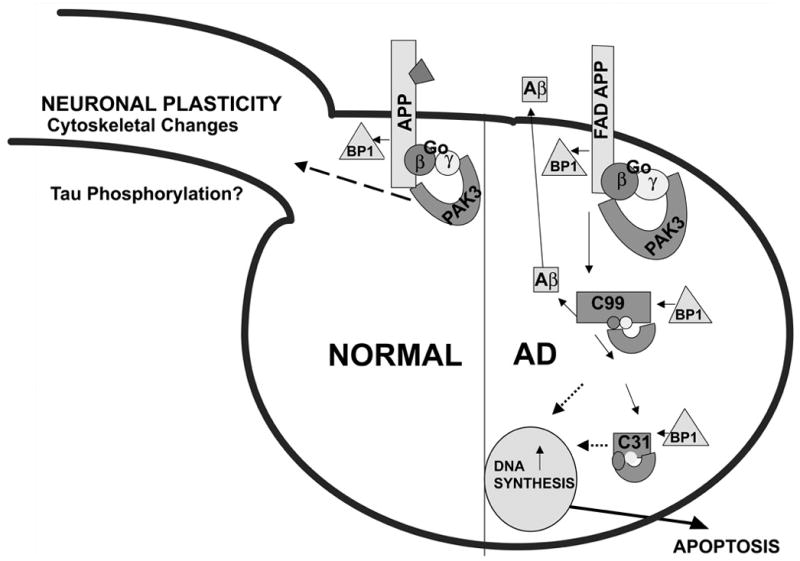
Schematic of a signal transduction pathway activated by FAD mutations of APP. In the normal cell, the function of APP is tightly regulated by the binding of its ligand (portrayed in the figure as a trapezoid). FAD APP is relieved of such constraints, and, together with PAK3, Go, and APP-BP1, causes aberrant neuronal DNA synthesis and apoptosis.
To test the idea that C31 becomes constitutively active, one may ask whether causing APP signaling to become tonically active results in the same consequences for the neuron that expression of C31 alone does. It has been found that exposure of neuronal cells to the antibody 22C11, raised against the extracellular domain of APP, causes constitutive activation of Go (62) which is known to bind to and be activated by APP (32, 33). In this context, 22C11 could be considered to be an APP ligand mimetic of sorts. Indeed, sustained exposure of neurons to 22C11 causes DNA synthesis (14) and neuronal apoptosis (14, 76, 77) via the APP-BP1- and PAK3- mediated signal transduction pathway that is activated by C31 alone (14).
4. Concluding remarks
We have proposed that, in neurons, APP is a signaling receptor whose normal function is tightly regulated by ligand binding (Figure 3). We hypothesize that this normal, regulated function is altered by overexpression of wild type APP or by expression of FAD mutants of APP, leading to neuronal cell cycle entry and consequent apoptosis. We have reported previously that, in contrast to FAD mutant APPs, neither wild type nor FAD mutant presenilin enhance apoptosis in neurons (78). In addition, we have shown (McPhie, D.L. and Neve, R.L., unpublished data) that neither wild type or FAD mutant presenilin causes aberrant neuronal cell cycle entry. It is worth noting that expression of FAD APP leads to increased levels of secreted α-APP and β-APP, and to elevation of levels of specific C-terminal fragments of APP in addition to Aβ, while the only known effect of FAD presenilins on APP processing is alteration of the ratio of Aβ (42) to Aβ (40). These data suggest that fragments of APP other than Aβ may play a role in abnormal neuronal cell cycle entry and consequent apoptosis, and also suggest that there is more than one pathway to neurodegeneration in AD. These data highlight the importance of understanding the normal function of APP in the brain and how this function may be disturbed in AD.
Acknowledgments
I thank Nikolaos Robakis and the members of my lab for many helpful discussions. Much of the work described in this review was supported by National Institutes of Health grants AG12954 and AG021185 to R.L.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent I, Jicha G, Rosado M, Dickson D. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arendt T, Rodel L, Gartner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport. 1996;7:3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- 4.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 5.Nagy Z, Esiri M, Cato A, Smith A. Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol (Berl) 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- 6.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chow N, Cox C, Callahan LM, Weimer JM, Guo L, Coleman PD. Expression profiles of multiple genes in single neurons of Alzheimer’s disease. Proc Natl Acad Sci USA. 1998;95:9620–9625. doi: 10.1073/pnas.95.16.9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondratick CM, Vandre DD. Alzheimer’s disease neurofibrillary tangles contain mitosis-specific phosphoepitopes. J Neurochem. 1996;67:2405–2416. doi: 10.1046/j.1471-4159.1996.67062405.x. [DOI] [PubMed] [Google Scholar]
- 9.Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P. Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease. Neurobiol Aging. 1998;19:287–296. doi: 10.1016/s0197-4580(98)00071-2. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Slzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–784. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging. 2000;21:797–806. doi: 10.1016/s0197-4580(00)00219-0. [DOI] [PubMed] [Google Scholar]
- 14.McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, Kozlowski MR, Neve KA, Neve RL. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci. 2003;23:6914–6927. doi: 10.1523/JNEUROSCI.23-17-06914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Illenberger S, Zhen-Fischhofer Q, Preuss U, Stamer K, Baumann K, Trinczek B, Biernat J, Godemann R, Mandelkow EM, Mandelkow E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: Implications for Alzheimer’s disease. Mol Biol Cell. 1998;9:1495–1512. doi: 10.1091/mbc.9.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pope W, Lambert M, Leypole B, Seupaul R, Sletten L, Krafft G, Klein W. Microtubule-associated protein tau is hyperphosphorylated during mitosis in the human neuroblastoma cell line SH-SY5Y. Exp Neurol. 1994;126:185–194. doi: 10.1006/exnr.1994.1057. [DOI] [PubMed] [Google Scholar]
- 17.Liu WK, Williams R, Hall F, Dickson D, Yen SH. Detection of a cdc2-related kinase associated with Alzheimer paired helical filaments. Am J Pathol. 1995;146:228–238. [PMC free article] [PubMed] [Google Scholar]
- 18.Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Merno M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- 19.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for DNA fragmentation in neurons in the AD brain. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 20.Loo DT, Copani AG, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM, Jr, Roth KA. In situ immunodetection of neuronal caspase-e activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassman H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raina AK, Hochman A, Zhi X, Rottkamp CA, Nunomura A, Siedlak SL, Boux H, Castellani RJ, Perry G, Smith MA. Abortive apoptosis in Alzheimer’s disease. Acta Neuropathol. 2001;101:305–310. doi: 10.1007/s004010100378. [DOI] [PubMed] [Google Scholar]
- 24.Hwang DY, Cho JS, Lee SH, Chae KR, Lim HJ, Min SH, Seo SJ, Song YS, Song CW, Paik SG, Sheen YY, Kim YK. Aberrant expressions of pathogenic phenotype in Alzheimer’s diseased transgenic mice carrying NSE-controlled APPsw. Exp Neurol. 2004;186:20–32. doi: 10.1016/j.expneurol.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 25.Xie J, Chang X, Zhang X, Guo Q. Aberrant induction of Par04 is involved in apoptosis of hippocampal neurons in presenilin-1 M146V mutant knock-in mice. Brain Res. 2001;915:1–10. doi: 10.1016/s0006-8993(01)02803-7. [DOI] [PubMed] [Google Scholar]
- 26.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 27.LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s Aβ peptide induces neurodegeneration and apoptotic cell death in transgenice mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- 28.Cotman CW. Apoptosis decision cascades and neuronal degeneration in Alzheimer’s disease. Neurobiol Aging. 1998;19:S29–S32. doi: 10.1016/s0197-4580(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 29.Rovelet-Lecrus A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid antiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 30.Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006 doi: 10.1093/brain/aw1203. [DOI] [PubMed] [Google Scholar]
- 31.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 32.Nishimoto I, Okamoto T, Matsuura Y, Okamoto T, Murayama Y, Ogata E. Alzheimer amyloid protein precursor complexes with brain GTP-binding protein G(o) Nature. 1993;362:75–79. doi: 10.1038/362075a0. [DOI] [PubMed] [Google Scholar]
- 33.Brouillet E, Trembleau A, Galanaud D, Volovitch M, Bouillot C, Valenza C, Prochiantz A, Allinquant B. The amyloid precursor protein interacts with Go heterotrimeric protein within a cell compartment specialized in signal transduction. J Neurosci. 1999;19:1717–1727. doi: 10.1523/JNEUROSCI.19-05-01717.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taru H, Iijima K, Hase M, Kirino Y, Yagi Y, Suzuki T. Interaction of Alzheimer’s β-amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J Biol Chem. 2002;277:20070–20078. doi: 10.1074/jbc.M108372200. [DOI] [PubMed] [Google Scholar]
- 35.Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- 36.Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomita S, Ozaki T, Taru H, Oguchi S, Takeda S, Yagi Y, Sakiyama S, Kirino Y, Suzuki T. Interaction of a neuron-specific protein containing PDZ domains with Alzheimer’s amyloid precursor protein. J Biol Chem. 1999;274:2243–2254. doi: 10.1074/jbc.274.4.2243. [DOI] [PubMed] [Google Scholar]
- 38.Tanahashi H, Tabira T. X11L2, a new member of the X11 protein family, interacts with Alzheimer’s β-amyloid precursor protein. Biochem Biophys Res Commun. 1999;255:663–667. doi: 10.1006/bbrc.1999.0265. [DOI] [PubMed] [Google Scholar]
- 39.Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Niraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto I. C-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer’s amyloid precursor protein with JNK. J Neurosci. 2001;21:6957–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:3556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 41.Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 42.Cao X, Südhof TC. A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 43.Chow N, Korenberg JR, Chen XN, Neve RL. APP-BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem. 1996;271:11339–11346. doi: 10.1074/jbc.271.19.11339. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y, McPhie DL, Hirschberg J, Neve RL. The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem. 2000;275:8929–8935. doi: 10.1074/jbc.275.12.8929. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Liu W, McPhie DL, Hassinger L, Neve RL. APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. J Cell Biol. 2003;163:27–33. doi: 10.1083/jcb.200304003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- 47.Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. Cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- 48.Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, Pierce JW, Podust VN, Luo RS, Chau V, Palombella VJ. Nedd8 modification of cul-1 activates SCFβTrCP dependent biquitination of IκBα. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu K, Chen A, Pan ZQ. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1-CUL1 complex to promote ubiquitin polymerization. J Biol Chem. 2000;275:32317–32324. doi: 10.1074/jbc.M004847200. [DOI] [PubMed] [Google Scholar]
- 51.Mori F, Nishie M, Piao YS, Kito K, Kamitani T, Takahashi H, Wakabayashi K. Accumulation of NEDD8 in neuronal and glial inclusions of neurodegenerative disorders. Neuropathol Appl Neurobiol. 2005;31:53–61. doi: 10.1111/j.1365-2990.2004.00603.x. [DOI] [PubMed] [Google Scholar]
- 52.Liakopoulos D, Busgen T, Brychzy A, Jentsch S, Pause A. Conjugation of the ubiquitin-like protein NEDD8 to cullin-2 is linked to von Hippel-Lindau tumor suppressor function. Proc Natl Acad Sci USA. 1999;96:5510–5515. doi: 10.1073/pnas.96.10.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freed E, Lacey KR, Huie P, Lyapina SA, Deshaies RJ, Stearns T, Jackson PK. Components of an SCF ubiquitin ligase localize to the centrosome and regulate the centrosome duplication cycle. Genes Dev. 1999;13:2242–2257. doi: 10.1101/gad.13.17.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Podust VN, Brownell JE, Gladysheva TB, Luo RS, Want C, Coggins MB, Pierce JW, Lightcap ES, Chau V. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci USA. 2000;97:4579–4584. doi: 10.1073/pnas.090465597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tateishi K, Omata M, Tanaka K, Chiba T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001;155:571–579. doi: 10.1083/jcb.200104035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kito K, Yeh ET, Kamitani T. NUB1, a NEDD8-interacting protein, is induced by interferon and down- regulates the NEDD8 expression. J Biol Chem. 2001;276:20603–20609. doi: 10.1074/jbc.M100920200. [DOI] [PubMed] [Google Scholar]
- 57.Kamitani T, Kito K, Fukuda-Kamitani T, Yeh ET. Targeting of NEDD8 and its conjugates for proteasomal degradation by NUB1. J Biol Chem. 2001;276:46655–46660. doi: 10.1074/jbc.M108636200. [DOI] [PubMed] [Google Scholar]
- 58.Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998;20:25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- 59.Bienvenu T, des Portes V, McDonell N, Carrie A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walth CA, Boue J, Kahn A, Chelly J, Beldjord C. Missense mutation in PAKd, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet. 2000;93:294–298. doi: 10.1002/1096-8628(20000814)93:4<294::aid-ajmg8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 60.Meng J, Meng Y, Hanna A, Janus C, Jia Z. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J Neurosci. 2005;25:6641–6650. doi: 10.1523/JNEUROSCI.0028-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lang J, Nishimoto I, Okamoto T, Regazzi R, Kiraly C, Weller U, Wollheim CB. Direct control of exocytosis by receptor-mediated activation of the heterotrimeric GTPases Gi and G(o) or by the expression of their active G alpha subunits. EMBO J. 1995;14:3635–3644. doi: 10.1002/j.1460-2075.1995.tb00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okamoto T, Takeda S, Murayama Y, Ogata E, Nishimoto I. Ligand-dependent G protein coupling function of amyloid transmembrane precursor. J Biol Chem. 1995;270:4205–4208. doi: 10.1074/jbc.270.9.4205. [DOI] [PubMed] [Google Scholar]
- 63.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 64.Reynolds CH, Utton MA, Gibb GM, Yates A, Anderton BH. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem. 1997;68:1736–1744. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- 65.Sato S, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A. Aberrant tau phosphorylation by glycogen synthase kinase-3β and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem. 2002;277:42060–42065. doi: 10.1074/jbc.M202241200. [DOI] [PubMed] [Google Scholar]
- 66.Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhi Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thorneberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 68.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 69.Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl AcadSci UDS. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho A, Südhof TC. Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci USA. 2004;101:2548–53. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oh SY, Ellenstein A, Chen CD, Hinman JD, Berg EA, Costello CE, Yamin R, Neve RL, Abraham CR. Amyloid precursor protein interacts with notch receptors. J Neurosci Res. 2005;82:32–42. doi: 10.1002/jnr.20625. [DOI] [PubMed] [Google Scholar]
- 72.Fassa A, Mehta P, Efthimiopoulos S. Notch 1 interacts with the amyloid precursor protein in a Numb-independent manner. J Neurosci Res. 2005;82:214–24. doi: 10.1002/jnr.20642. [DOI] [PubMed] [Google Scholar]
- 73.Hashimoto Y, Chiba T, Yamada M, Nawa M, Kanekura K, Suzuki H, Terashita K, Aiso S, Nishimoto I, Matruoka M. Transforming growth factor beta2 is a neuronal death-inducing ligand for amyloid-beta precursor protein. Mol Cell Biol. 2005;25:9304–17. doi: 10.1128/MCB.25.21.9304-9317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA. Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nat Neurosci. 2000;3:460–4. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- 75.Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597–624) FASEB J. 2006;20:1254–6. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sudo H, Jiang H, Yasukawa T, Hashimoto Y, Niikura T, Kawasumi M, Matsuda S, Takeuchi Y, Aiso S, Matsuoka M, Murayama Y, Nishimoto I. Antibody-regulated neurotoxic function of cell-surface beta-amyloid precursor protein. Mol Cell Neurosci. 2000;16:708–23. doi: 10.1006/mcne.2000.0910. [DOI] [PubMed] [Google Scholar]
- 77.Rohn TT, Ivins KJ, Bahr BA, Cotman CW, Cribbs DH. A monoclonal antibody to amyloid precursor protein induces neuronal apoptosis. J Neurochem. 2000;74:2331–2342. doi: 10.1046/j.1471-4159.2000.0742331.x. [DOI] [PubMed] [Google Scholar]
- 78.Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, Neve RL. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J Neurosci. 1998;18:9790–9. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]