

Abstract
Pituitary function has been shown to be regulated by an increasing number of intra-pituitary factors including cytokines. Here we show that the important cytokine TNF-α activates prolactin gene transcription in pituitary GH3 cells stably expressing luciferase under control of 5 kb of the human prolactin promoter. Similar regulation of the endogenous rat prolactin gene by TNF-α in GH3 cells was confirmed using real time PCR. Luminescence microscopy revealed heterogeneous dynamic response patterns of promoter activity in individual cells. In GH3 cells treated with TNF-α, western blot analysis showed rapid IκBα degradation and phosphorylation of p65. Confocal microscopy of cells expressing fluorescence labelled p65 and IκBα fusion proteins showed transient cytoplasmic-nuclear translocation and subsequent oscillations in p65 localisation and confirmed IκBα degradation. This was associated with increased NF-κB mediated transcription from an NF-κB responsive luciferase reporter construct. Disruption of NF-κB signaling by expression of dominant negative variants of IKKs or truncated IκBα abolished TNF-α activation of the prolactin promoter, suggesting that this effect was mediated by NF-κB. TNF-α signaling was found to interact with other endocrine signals to regulate prolactin gene expression, and is likely to be a major paracrine modulator of lactotroph function.
Keywords: prolactin, TNF-α, NF-κB, pituitary, gene transcription, single cell imaging
Introduction
The human prolactin gene is located on the short arm of chromosome 6 (1) and consists of five coding exons within a region of 10 kb. It is expressed in lactotroph cells in the anterior pituitary, but is also expressed in the endometrium (2;3) and myometrium (4) of the uterus and in lymphoid cells (5;6). In pituitary cells, prolactin gene expression is regulated in response to multiple hormonal signals by an extensive 5’-flanking region extending 5000 bp upstream from the transcriptional start site, containing multiple binding sites for Pit-1 (7-10). Prolactin mRNA transcribed in extra-pituitary tissues contains an additional non-coding exon 1a, and expression is regulated by an alternate upstream promoter. Pituitary prolactin expression in vivo is regulated by dopamine and other hypothalamic factors such as TRH and by an increasing number of intrapituitary factors (11). In addition to growth factors, such as FGF-2 or EGF and other proteins like galanin, recent evidence has shown that cytokines are also expressed in the pituitary gland and are likely to act as important paracrine or autocrine regulators of pituitary function (11;12).
Tumor necrosis factor-α (TNF-α) is a cytokine that plays a role in a variety of biological processes including cell proliferation, differentiation and apoptosis. Furthermore it can activate transcription factors such as NF-κB and AP-1 (13). NF-κB transcription factors consist of homo- or heterodimers assembled from subunits including p50, p52, p65, c-Rel and Rel B (14) and are involved in the regulation of many genes (15). In the cytoplasm NF-κB is bound by inhibitory proteins (IκB proteins) that block its nuclear localization domain (14). Activation of IκB kinases IKKα and IKKβ (16) leads to phosphorylation of IκBs which targets them for rapid degradation by the proteasome (17). The released NF-κB dimers can then translocate to the nucleus and regulate target gene transcription.
TNF-α is expressed in multiple tissues including brain and pituitary (18). In the pituitary, expression of TNF-α has been found in the intermediate lobe and in somatotroph cells (19). The presence of TNF-α receptors in the pituitary cells has also been reported in vitro and in vivo (20;21), suggesting that TNF-α might well play a role as an autocrine or paracrine regulator.
Studies on the effect of TNF-α on the secretion of pituitary hormones have revealed an effect on ACTH, GH, TSH and prolactin release (12;22). The results regarding the effect on prolactin are conflicting. Using mouse hemipituitaries, Milenkovic et al. showed no effect (23), whereas others using dispersed primary rat pituitary cells showed an decrease (24;25) or an increase (26;27) in prolactin release after TNF-α treatment.
Here we report that TNF-α is a potent activator of prolactin promoter activity in pituitary cells, with a biphasic effect over 6-24h. Heterogeneous patterns of response in individual cells led to the observed averaged population response. The effect of TNF-α is associated with activation of NF-κB and could be blocked by co-expressing dominant negative isoforms of components of the NF-κB signaling pathway. The effects of TNF-α were amplified by simultaneous treatment with a range of other hormonal stimuli, suggesting a physiological role as a major modulator of other signaling pathways in determining physiological outcome.
Material and Methods
Plasmids
Plasmids expressing the transdominant negative IKKα, IKKβ and NEMO/IKKγ were a gift from Dr. N. Inohara (University of Michigan, USA; (28). The expression plasmid for truncated IκBα (missing the 40 NH2-terminal amino acids) was a kind gift from Dr. E. Costello (University of Liverpool), and the wild type IKKβ was from Dr. C. Paya (Mayo Clinic, Rochester, USA (29). The pNF-κB-luc plasmid was from Stratagene (La Jolla, USA). The fluorescent CMV-IκBα-EGFP, NF-IκBα-EGFP and CMV-p65-dsRedXP fusions protein vectors were described previously (30;31).
Cell culture and luciferase assays
The construction of the GH3/hPrl-luc cell line was described previously (32). Cells were maintained in DMEM with pyruvate/glutamax (Life Technologies Inc., Paisley, UK) and the addition of 10% FCS (Harlan Sera-Lab, Crawley Down, UK). Except for TNF-α and FGF-2 (Merck Biosciences Ltd., Nottingham, UK) all other reagents were obtained from Sigma (Sigma Aldrich Corp. Ltd., Dorset, UK).
For transient transfections 1.2 × 106 GH3 cells were grown in 10-cm dishes for 24h and then transfected using Fugene 6 (Boehringer Mannheim/Roche, Germany) according to the manufacturer’s protocol. For cotransfection studies the total amount of DNA used was held constant by addition of empty expression vector. After 24h cells were trypsinized and seeded onto 24-well plates.
For luciferase assays 105 cells/well were grown in 24-well plates, serum starved for 24h and then stimulated as indicated. Cells were washed twice with ice cold PBS and lysates (Tris/PO4 25 mM, MgCl2 10 mM, EDTA 5 mM, 15% glycerol, 0.1% Triton X100, 0.1 mg/ml BSA) were prepared. The luciferase activity was measured using a Berthold Mithras 960 luminometer (Berthold Technologies, Redbourn, UK). 4-8 replicates were analysed for each treatment group and experiments were performed at least 3 times. Results are shown as mean fold induction ± standard deviation of at least three independent experiments. For statistical analysis P-values were calculated using Student’s t-test.
Real-time PCR
For real-time PCR analysis GH3 cells were grown in 25cm2 flasks (∼ 5 × 106 cells), serum starved for 24h and then stimulated with 10 ng/ml TNF-α for the indicated times. Cells were harvested by trypsinization and washed twice in ice cold PBS. Total RNA was isolated using the QIAGEN RNAeasy Kit (QIAGEN, Hilden, Germany) and cDNA was generated with the Omniscript RT system (QIAGEN). Quantitative real time PCR using the Stratagene Mx3000 P thermocycler (Stratagene) and the SYBRgreen Jump Start Taq Ready Mix (Sigma) was performed as follows: 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C, 1 min at 56 °C and 30 s at 72°C. The following primers were used for cyclophilin, TTTTCGCCGCTTGCTGCAGAC and CACCCTGGCACATGAATCC-TGGA, and prolactin, AGCCAAGTGTCAGCCCGGAAAG and TGGCCTTGGCAATAAACTCACGA and resulted in amplicons of 217 bp and 227 bp, respectively. The dissociation curves of used primer pairs showed a single peak and samples after PCR reactions had a single DNA band of the expected size in an agarose gel. Cyclophilin was used as a housekeeping gene for normalization. Data are shown as average of three independent experiments ± standard deviation.
Real time luminescence imaging
GH3/hPrl-luc cells (105) were seeded onto 35-mm glass coverslip-based dishes(IWAKI, Tokyo, Japan) and cultured in 10% FCS and then serum starved for 24h before imaging. Luciferin (final concentration,1 mM; Bio-Synth, Inc., Staad, Switzerland) was added at least 10h prior to the start of the experiment, and the cells were transferred to the stage of a Zeiss Axiovert 100M (Carl Zeiss, Welwyn Garden City, UK) in a dark room. The cells on the stage were maintained at 37°C in a Zeiss incubator M with 5% CO2-95% air. Bright field images were taken before and after luminescence imaging to allow localisation of the cells. Luminescence images were obtained using a Fluar x20, 0.75 numerical aperture, dry objective and captured using a photon counting charge coupled device camera (Orca II, Hamamatsu Photonics, Welwyn Garden City, UK). Sequential images, integrated over 30 min, were taken using 4 by 4 binning and acquired using Kinetic Imaging software AQM6 (Andor, Belfast, UK). 10 ng/ml TNF-α was added directly to the dish immediately before commencing the experiment.
Analysis was carried out using Kinetic Imaging software AQM6. Regions of interest were drawn around each single cell and total photon counts for individual cell areas were integrated over 30 min intervals. Mean intensity data was collected after the average instrument dark count (corrected for the number of pixels being used) was subtracted from the luminescence signal.
Data were analyzed using the Cluster peak detection algorithm (33) to detect significant peaks [>12 (background + 2 SD)]in luciferase activity. Further data analysis was performed with Excel and SPSS statistics package (SPSS, Inc., Chicago, IL, USA).
Western blotting
GH3 cells were seeded in 60mm dishes at a density of 2×106 cells in 6ml and serum starved for 24h before stimulation. Separate dishes were stimulated with 10 ng/ml TNF-α for the indicated times. Cells were washed with ice cold PBS and lysed with ice cold lysis buffer (1% sodium dodecyl sulphate [SDS]; 10% glycerol; 1% β-mercaptoethanol; 62.5 mM Tris-HCl, pH 6.8) containing a protease and phosphatase inhibitor cocktail (Sigma, Poole, UK). Samples were collected, heated at 100°C for 5 mins and sonicated for 10 sec. Proteins were separated by electrophoresis on SDS-polyacrylamide (7.5%) gels and electro-transferred onto nitrocellulose. Non-specific binding sites were blocked with 5% dried skimmed milk in 0.05% Tween-20 supplemented Tris-buffered saline (TTBS; 25 mM Tris HCl, pH 8.0, 150 mM NaCl). Nitrocellulose membranes were incubated overnight at 4°C with either phospho-p65, p65 or IκBα monoclonal antibodies (Cell Signaling, Beverly, MA, USA). The membranes were then washed extensively in TTBS and incubated with goat anti-rabbit immunoglobulin G conjugated to horseradish peroxidase for 1h at room temperature, following by extensive washing in TTBS. Positive immunoreactive bands were detected by chemiluminescence using SuperSignal® West Pico according to the manufacturer’s instructions (Pierce Biotechnology, Rockford, IL, USA).
Detection of transcription factor activation
GH3 cells were seeded in 60mm dishes at a density of 2×106 cells in 6ml and serum starved for 24h before stimulation. Separate dishes were stimulated with 10 ng/ml TNF-α for the indicated times. A TransAM NF-κB p65 Chemi transcription factor assay kit (Activemotif, Belgium) was used according to the manufacturer’s instructions. Briefly, the kit comprised a 96-well plate with immobilised NF-κB consensus binding site oligonucleotide. Cells were lysed with 100 μl of the provided lysis buffer and 10μl of lysed sample was used per well. Activated NF-κB from the whole cell extracts specifically bound to the oligonucleotide and was detected using an antibody against the p65 subunit. A secondary antibody conjugated to horseradish peroxidase provided a chemiluminescent readout. Jurkat nuclear extract was used as a positive control (data not shown), and a blank well was run for background luminescence subtraction. All reactions were performed in duplicate, and results are shown as the average of 2 independent experiments ± SEM.
Real time fluorescence imaging
GH3 cells (2×105) were seeded onto 35-mm glass based dishes(IWAKI) and cultured in 10% FCS. After 24h cells were transiently transfected with CMV-p65-dsRedXP and either CMV-IκBα-EGFP or NF-IκBα-EGFP (driven by 5 tandem repeats of a consensus NF-κB binding site (30) using Fugene 6. Cells were imaged 24h after transfection on a Zeiss Axiovert 200 equipped with an XL incubator (maintained at 37C, 5% CO2, in humid conditions) through a 63x objective (NA = 1.4, Zeiss). Excitation of EGFP was performed using an Argon ion laser at 488nm. Emitted light was captured through a 505-550 nm bandpass filter from a 545 nm dichroic mirror. DsRed-XP fluorescence was excited using a Helium Neon laser (543 nm) and detected through a 560 nm longpass filter. Images were taken every 5min. Data was captured and analysed using LSM510 software.
Results
TNF-α activates the human prolactin promoter
To evaluate the role of TNF-α in the transcriptional regulation of pituitary prolactin expression we have used GH3 cells stably expressing luciferase under the control of a 5000 bp fragment of the human prolactin promoter (32). This promoter is activated by TNF-α in a dose dependent manner, with maximal effects seen at 2ng/ml (P < 0.05), with significantly greater induction at 24h compared to 6h (Fig 1A). GH3 cell lines expressing luciferase under the control of a 500 bp GH promoter fragment showed no response to TNF-α, confirming the specificity of this response, and that it was not an artefact due to cryptic response elements in the reporter vector (Fig 1A, inset panel). Detailed time course experiments showed a clear biphasic response of transcription, with a transient plateau between 6-12h (P < 0.05 after 2h) and a second plateau reached by 24h (Fig. 1B). To test whether the endogenous rat prolactin gene is also activated by TNF-α, GH3 cells were treated with TNF-α, total RNA prepared and subjected to real time PCR analysis. These experiments revealed that the endogenous rat prolactin gene is activated by TNF-α in a similar manner to the human prolactin promoter reporter gene construct, with a similar magnitude and time-course of response (Fig. 1C).
Fig. 1.
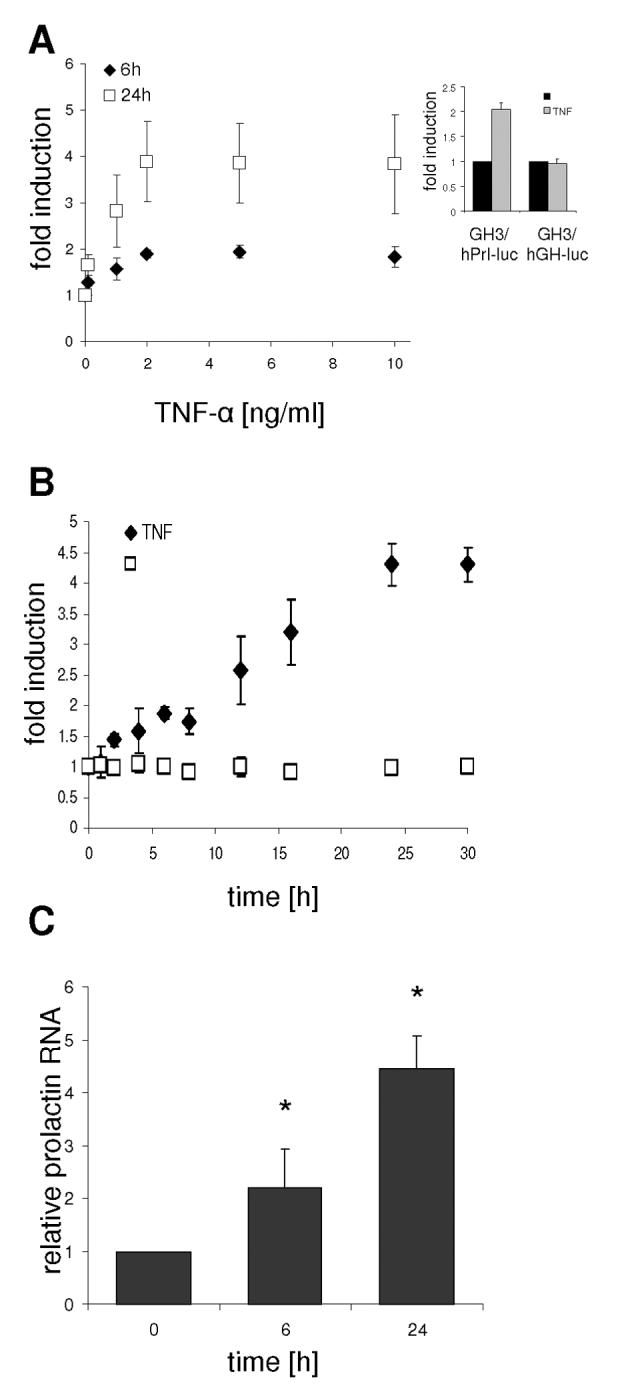
Dose and time dependent activation of the prolactin promoter by TNF-α. GH3/hPrl-luc cells were serum starved for 24h. Cells were then treated with the indicated doses of TNF-α for 6h (◆) and 24h (□) (A) or with 10 ng/ml TNF-α (◆) or not (□) for the indicated time periods (B). (Inset A) GH3 cells expressing luciferase under the control of 5000 bp of the hPrl or 500 bp of the hGH promoter were treated with TNF-α for 6h. Basal expression levels for the hPrl promoter construct under these conditions were 5-10 times higher than for the hGH promoter. Cells were lysed and luciferase activity measured by luminometry. (C) GH3 cells were treated as above with TNF-α for the indicated times. Cells were harvested, RNA prepared and subjected to real time PCR analysis. Data were normalized using expression of the housekeeping gene cyclophilin. Results shown are the means ± SD from at least three independent experiments; * P < 0.05.
Dynamics of transcriptional response in single cells
Previous work has shown that gene promoter activity in individual living cells is highly unstable and frequently pulsatile (32;34;35). Therefore, in order to analyse the contribution of single cells to the overall population response that was seen using analysis by luminometry, GH3/prl-luc cells were imaged for 48h either in resting conditions or after stimulation with TNF-α, and Cluster analysis of the transcriptional response in single cells was performed (n = 116 cells from 7 experiments for TNF-α treatment, and n= 75 cells from 4 experiments for resting conditions). Microscopic analysis of whole fields of cells confirmed the biphasic response pattern observed using luminometry (Fig. 2A). Single cell analysis revealed a heterogeneous response of individual cells to the TNF-α treatment that could be classified into three predominant patterns, an early peak, a late peak or more than one peak in promoter activity. Examples of individual cells showing the three typical patterns observed are shown in Fig. 2C-E. Cluster analysis of the single cell data showed a significant peak in transcription after TNF-α treatment between 0-14h (termed period 1) for 41 ± 4% of the cells (compared to 20 ± 8% under resting conditions); 20-34h after addition of TNF-α (termed period 2), 59 ± 7% of cells showed a pulse in transcription (compared to 20 ± 4% control). 16 ± 5% of TNF-α treated cells (compared to 55 ± 8% control) showed no peak during the whole experiment (Fig. 2B). Significant peaks in both time periods were observed in 30 ± 3% (compared to 4 ± 1%) of the cells. These data suggest that the increase in transcription after TNF-α treatment is due to activation of more cells rather than due to a simultaneous graded increase of expression levels in all cells.
Fig. 2.

Real time luminescence imaging showed heterogeneous transcriptional responses in individual cells after TNF-α treatment.
GH3/hPrl-luc cells were cultured in 35 mm glass coverslip culture dishes, serum starved for 24h and then imaged for 48h following stimulation with 10 ng/ml TNF-α (◆) or not (□). (A) The mean of the total photon counts obtained from a whole microscopic field of cells (about 20 per field): shaded areas indicate the two time periods used to categorise cells into different patterns of expression. (B) Single cell transcriptional responses with or without TNF-α stimulation were classified into four groups using Cluster analysis software. The groups were defined as cells showing no significant response, all cells showing significant responses in period 1, all cells showing significant responses in period 2, and cells showing significant responses in both periods. Data shown are means ± SD from four or seven independent experiments. (C-E) Examples of single cells representing the predominant response patterns observed.
TNF-α activates the NF-κB signaling pathway in GH3 cells
To test whether the activation of the prolactin promoter could be mediated by NF-κB we first wanted to establish whether TNF-α can activate NF-κB in pituitary GH3 cells. Western blot analysis for p65, phospho-p65 and IκBα was performed with lysates from GH3 cells treated with TNF-α for 0-2h (Fig. 3A). There was no change in the level of total p65, but treatment with TNF-α led to a rapid transient increase in phosphorylation of p65 that lasted for approximately 30 min and was followed by a decrease below basal levels. Degradation of IκBα was observed after 20-30 min with subsequent recovery to pre-treatment level after 2h. Using a DNA/protein interaction assay for p65 we found an increase in complex formation using lysates from GH3 cells treated with TNF-α for 30-60 min (Fig. 3B). To show that IκBα degradation and p65 phosphorylation after TNF-α stimulation was associated with transcriptional activation, GH3 cells were transiently transfected with a reporter construct containing 5 repeats of the NF-κB consensus binding sequence linked to the luciferase gene (pNF-κB-luc). Treatment of these cells with TNF-α caused a time- (Fig. 3C) and dose-dependent (data not shown) increase in luciferase activity (P < 0.05 at 2h and later). These data indicate that TNF-α is indeed able to stimulate gene transcription via activation of the NF-κB signaling pathway in GH3 cells. We then transiently transfected GH3 cells with expression vectors encoding p65-DsRedXP and IκBα-EGFP under the control of the strong human Cytomegalovirus promoter (CMV) (30;31) and used dual fluorescence time-lapse imaging to observe and quantitate dynamic changes in the nuclear : cytoplasmic localisation ratio (N:C) of p65 and the level of cytoplasmic IκBα in single transfected cells. TNF-α treatment led to rapid degradation of IκBα and nuclear translocation of p65 (Fig. 3D). Subsequent oscillations in the level of nuclear p65 were seen in ∼60% of cells (11 out of 18 cells) with a period of around 90 minutes (Fig. 3E), similar to those reported in other cell lines (31). We repeated these experiments using NF-IκBα-EGFP (driven by 5 tandem repeats of a consensus NF-κB binding motif; as previously described (31) alongside CMV-p65-DsRedXP. Addition of TNF-α to the medium led to a rapid decrease in cytoplasmic IκBα reaching a minimum after 1h, and subsequent recovery to pre-treatment levels after approximately 2h. The degradation of IκBα was accompanied by a transient nuclear translocation of p65 as before (Fig. 3F-G).
Fig. 3.

Analysis of NFκB activation by TNF-α in GH3 cells.
GH3 cells were serum starved for 24h before stimulation with 10 ng/ml TNF-α for the indicated times. (A) Endogenous p65, phospho-p65 and IκBα levels were assessed by Western blotting cell extracts using anti-p65, anti-phosho-p65 and anti-IκBα antibodies. (B) DNA/protein interaction for p65 was analysed in lysates from TNF-α stimulated GH3 cells using a TransAM NFkB p65 Chemi transcription factor assay kit. Results are shown as mean ± SEM of four replicates from two independent experiments. (C) GH3 cells were transfected with the pNFκB-luc reporter plasmid, serum starved for 24h and treated with 10 ng/ml TNF-α for the indicated times. Results are shown as means ± SD from at least three independent experiments. (D) Time course showing co-expression of p65-DsRedXP and CMV-IκBα-EGFP, in cells stimulated with TNF-α at time zero. The movement of p65 is presented as the ratio of nuclear to cytoplasmic fluorescent signal. The IκBα signal was normalised to resting levels at time zero. (E) Examples of single cells showing oscillatory movement of p65 after TNF-α stimulation. (F-G) GH3 cells were transfected with p65-dsRedXP and NF-IκB-EGFP, stimulated with TNF-α and confocal images were taken every 5 min from living cells: quantitative results (F) and images of typical cells (G) are shown at 0-120 min after addition of TNF-α.
TNF-α activation of the prolactin promoter is mediated by the NF-κB signaling pathway
To test whether the observed activation of the human prolactin promoter is mediated via this pathway, we co-transfected the 5000 bp human prolactin promoter reporter construct into GH3 cells together with a well characterised set of expression vectors encoding wild type and dominant negative components of the NF-κB signaling pathway (Fig. 4) that have previously been used to investigate the role of NF-κB in Cyclin D1 transcription (36). Cotransfection of wild type IKKβ led to an increase in basal expression levels from the prolactin promoter but no change in fold induction (Fig. 4A). On the other hand, dominant negative IKKβ abolished the induction by TNF-α completely and slightly decreased basal expression (Fig. 4B). Expression of dominant negative NEMO/IKKγ (16) or truncated IκBα (Δ IκBα) also abolished the increase in transcription after TNF-α treatment but had no effect on basal expression levels (Fig. 4C-D). Dominant negative IKKα also had no effect on basal levels and had a reduced inhibitory effect on the response to TNF-α (Fig. 4E). Co-transfection of these constructs did not alter activation of the prolactin promoter by FGF-2 or forskolin (Fig. 4F). Taken together, these data suggest that the TNF-α stimulation of the prolactin promoter is mediated via the NF-κB pathway.
Fig. 4.

Cotransfection of dominant negative components of the NFκB signaling pathway with a human prolactin promoter-luciferase construct.
Using GH3 cells the 5000 bp prolactin promoter-luc construct (1μg) was cotransfected with the indicated amounts (0.1, 0.5, 1 μg) of expression plasmid for wildtype IKKβ, dnIKKβ, dnIKKα, dnNEMO/IKKγ and a truncated IκBα(Δ IκBα). The total amount of DNA for each transfection was kept constant by addition of empty expression vector (A-E). For (F), cells were transfected with 1μg of 5000 bp prolactin promoter-luc construct and 1μg dnIKKβ, ΔIκBα or dnNEMO. Cells were serum starved for 24h and then treated for 6h with 10 ng/ml TNF-α (A-E) or the indicated stimuli (F); FGF 10 ng/ml, forskolin 1μM. Results are shown as means ± SD of at least three independent experiments; * P < 0.05.
TNF-α interacts with other signaling pathways to modulate prolactin gene expression
As TNF-α is a potent stimulus of the human prolactin promoter we analysed its interactions with other physiologically important positive or negative regulators of prolactin gene expression such as FGF-2 and glucocorticoids (9;37). GH3/prl-luc cells were treated with FGF-2, dexamethasone or estradiol, alone or in combination with TNF-α (Fig. 5). Treatment of GH3/hPrl-luc cells with 10 ng/ml FGF-2 alone led to an approximately 2 and 8 fold increase in luciferase activity after 6h and 24h respectively. TNF-α and FGF-2 in combination led to a further increase in promoter activation (Fig. 5A), suggesting that FGF-2 and TNF-α act independently and utilize different signaling pathways. This is further supported by the fact that whereas TNF-α was able to induce expression of luciferase from the pNF-κB-luc reporter construct, FGF-2 was not (data not shown), indicating that FGF-2 unlike TNF-α does not activate NF-κB.
Fig. 5.
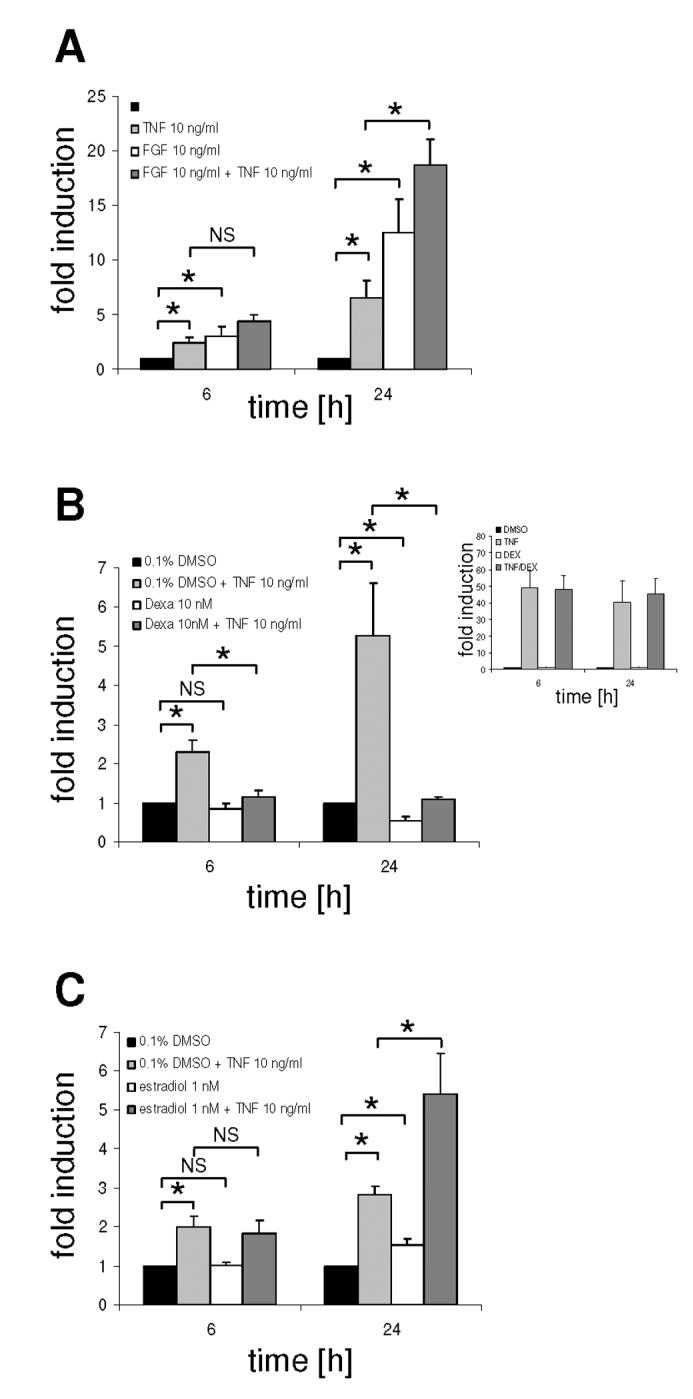
Combinatorial effect of TNF-α with FGF-2, dexamethasone or estradiol on prolactin promoter activity.
GH3/hPrl-luc cells were serum starved for 24h and then stimulated for 6h or 24h with (A) FGF-2 (10 ng/ml), (B) dexamethasone (10 nM), or (C) estradiol (0.1 nM) alone, in combination with TNF-α (10 ng/ml), or TNF-α alone. Inset (B): GH3 cells were transfected with the pNFκB-luc reporter construct, serum starved for 24h and treated with TNF-α and/or dexamethasone for 6h. Results are shown as means ± SD of at least three independent experiments; * P < 0.05.
Glucocorticoids are known to decrease prolactin expression (9;38) and have been shown to interact with NF-κB signaling in a variety of systems (39;40). In GH3/hPrl-luc cells treatment with 10 nM dexamethasone had no effect on luciferase activity after 6h but decreased gene expression by about 50% after 24h. When given in combination with TNF-α, dexamethasone completely abolished the TNF-α induction of the human prolactin promoter not only after 24h but also after 6h (Fig. 5B). In contrast, in GH3 cells transiently transfected with the pNF-κB-luc reporter construct dexamethasone failed completely to repress TNF-α activation (inset Fig. 5B). This is in marked contrast to the inhibitory effect of GR stimulation on TNF-α induction of this promoter in other cells such as HeLa cells (39).
Whereas previous studies have revealed an estrogen response element in the rat prolactin promoter, studies on the human prolactin promoter so far have not shown major effects of estrogen (9;41). Treatment of GH3/hPrl-luc cells with 10 nM estradiol alone or in combination with 10 ng/ml TNF-α had no effect on the activity of the human prolactin promoter after 6h. After 24h, estradiol gave a consistent but small 1.5-fold induction, however in combination with 10 ng/ml TNF-α, estradiol led to a synergistic 2-3 fold increase in gene activation (Fig. 5C).
Discussion
Here we have reported that TNF-α activates the human prolactin gene promoter in GH3 pituitary cells, and significantly modulates the effects of other important physiological regulators of prolactin expression such as estrogen, FGF-2 and glucocorticoids. Cotransfection studies with dominant negative proteins of the NF-κB signaling pathway have shown that this regulation is mediated via NF-κB.
Prolactin gene expression is regulated by various hypothalamic factors, but in recent studies an increasing number of intrapituitary factors have also been identified as important regulators (11). Here we show for the first time that in pituitary GH3 cells the cytokine TNF-α is able to activate the prolactin promoter in a dose- and time-dependent manner, and also that expression of the endogenous rat prolactin gene is similarly stimulated by TNF-α. As prolactin is thought to play a role in the stress response, our data, together with findings showing expression of TNF-α (18;19) and its receptors (20;21) in the pituitary, suggest that TNF-α might be an important paracrine or autocrine factor involved in the modulation of prolactin expression. Studies of TNF-α effects on prolactin using primary pituitary cells have so far focused on hormone secretion and shown conflicting results (23-27). The discrepancy in these studies is most likely due to differences in the experimental design and model systems used. A recent study on primary lactotrophs from female rats showed an apoptotic effect of TNF-α (42), but used much higher (50 ng/ml) concentrations than we have employed (2-10ng/ml). Using flow cytometry we have seen no evidence of increased apoptosis in GH3 cells with 10ng/ml TNF-α for periods up to 30h (data not shown). However, the effect of TNF-α on prolactin gene expression in primary cell cultures or in vivo remains the subject of further investigation.
We and others have found that gene promoter activity in clonal pituitary cell lines is highly dynamic and variable among different individual cells, with rapid fluctuations in transcriptional activity in resting cells as well as in hormone treated cells (32;43). This is a phenomenon also found in normal pituitary cells using microinjection of reporter plasmids (44) or transfection with recombinant adenoviruses (34). Here we show that stimulation of GH3/hPrl-luc cells with TNF-α is accompanied by heterogeneous changes in prolactin promoter activity, with increased proportions of cells displaying transient peaks or pulses in transcription, with the overall effect that more cells become transcriptionally active. The cells in our study were a clonal cell line, as opposed to a mixed population of cells seen in the normal pituitary gland: thus the heterogeneity we have seen arises from different transcriptional patterns amongst individual cells in a single cell population. All of the cells in the population can express the transgene, but at varying levels, so that some cells exhibiting very low levels of gene expression at a given time may then become transcriptionally active at different rates and time points after a stimulus.
There are two theoretical mechanisms to regulate transcription from a gene promoter. Transcriptional enhancers might increase the probability that a promoter is active, meaning that genes are either ‘off’ or ‘on’ (binary behaviour), or they might increase the rate of gene transcription modulating a continuous range of activity (45;46). Studies in yeast suggest that a promoter can adopt either binary or graded behaviour depending on the signalling pathway involved (47). The data we report here would favour a stochastic (or binary) model of gene transcription, as we have found an increased number of transcriptionally active cells after the stimulation with TNF-α rather than a simultaneous graded increase in promoter activity in all cells. Furthermore, the cells showed transient peaks of activity with a substantial number of cells showing more than one peak rather than constant transcription levels, underlining the stochastic nature of this process. These stochastic changes in transcription phenotype in single cells are most likely caused by the relative instability of transcription complexes. This would be consistent with recent findings of dynamic recruitment and release of transcription regulators to chromatin (48;49). Stochastic patterns of transcription may be physiologically important in determining the way in which cells integrate their patterns of response to combinations of physiological regulators (50).
TNF-α can activate transcription factors such as AP-1 and NF-κB, which may override the apoptotic pathways (13). Stimulation of NF-κB activates the IKK signalsome which phosphorylates IκB and NF-κB proteins, leading to ubiquitination and degradation of IκB and subsequent translocation of free NF-κB dimers to the nucleus (14;17). In agreement with this we found rapid phosphorylation and transient nuclear translocation of p65 as well as degradation of cytoplasmic IκBα in TNF-α stimulated GH3 cells. In agreement with our previous studies in other cell types (30) we observed highly damped oscillations in the level of nuclear NF-κB which had a period of around 90 minutes. These cycles of NF-κB translocation have been associated with cycles of re-activation of NF-κB (measured by Ser536 phosphorylation) that maintain NF-κB-dependent gene expression. As described in other cell types (30) the use of a consensus NF-κB promoter in front of the IκBα-EGFP reporter led to oscillations in cytoplasmic EGFP fluorescence level and an increase in p65 oscillation period (data not shown). Indeed, using NF-κB responsive reporter gene constructs we showed that NF-κB translocation and IκBα-degradation is accompanied by dose- and time-dependent increase in NF-κB-mediated transcription. These findings are supported by a previous study showing increased protein/DNA complex formation in electrophoretic mobility shift assays using nuclear extracts from GH3 cells treated with TNF-α (51). Taken together, our evidence strongly suggests that TNF-α is able to activate the NF-κB signaling pathway in GH3 cells.
Activity of the IKK complex is crucial for the activation of NF-κB (16). Disruption of its activity by expression of dominant negative variants of IKKα or IKKβ has been used in previous studies to show NF-κB involvement in transcriptional regulation of the Cyclin D1 gene (36). Here we found that expression of dominant negative IKKs abolished induction of the prolactin promoter activity by TNF-α in GH3 cells, suggesting that the TNF-α effect specifically is mediated by NF-κB. This is further supported by our finding that expression of a truncated form of IκBα (ΔIκBα) that cannot be phosphorylated/degraded (and therefore abolishes NF-κB activation by preventing its translocation to the nucleus) also blocks activation of the prolactin promoter by TNF-α. Forskolin and FGF-2 regulation of the prolactin promoter were independent of NF-κB. This was expected from previous reports showing that regulation by FGF-2 involves a Rac1, PLCγ, PKCδ and ERK dependent mechanism (52), while forskolin increases intracellular cAMP levels to activate PKA.
Our results show that TNF-α not only activates the human prolactin promoter, but also interacts with other well-known regulators of prolactin expression. Simultaneous treatment of GH3/hPrl-luc cells with TNF-α and FGF-2 led to a significant additive effect on promoter activation. FGF-2 did not activate NF-κB in pituitary GH3 cells, and therefore independent activation of distinct signaling pathways may explain the additional effect. Dexamethasone blocked activation of the prolactin promoter by TNF-α. The repressive effect of glucocorticoids on the expression of prolactin involves interaction between Pit-1 and GR but binding of GR to DNA is not required (38). The loss of activation by TNF-α in the presence of dexamethasone could therefore be explained by either loss of NF-κB transactivation due to negative cross-talk with GR, or by independent glucocorticoid repression of the prolactin promoter overriding the TNF-α activation. In contrast to results in other cell types (39) dexamethasone did not alter transcriptional activation of a reporter gene construct containing 5 NF-κB response elements by TNF-α in GH3 cells, arguing against a direct effect of GR on NF-κB signaling pathways in these cells, and suggesting that this effect was characteristic of the prolactin promoter but not of a consensus NF-κB promoter. Bioinformatic analysis has revealed two potential NF-κB binding sites at -1300/-1309 and -3810/-3819bp in the human prolactin promoter, but preliminary experiments suggest that they may not be involved in these effects of TNF-α (data not shown), suggesting that a more complex mechanism may be involved, as shown for other genes such as IL-6 and COX-2 (53-55). NF-κB regulation of prolactin might therefore involve other factors, and the discrepancy in dexamethasone repression seen here may be explained by GR blocking interaction of NF-κB with some yet unknown factor. Understanding of this interesting behaviour requires further more detailed study.
Despite the effect of estrogen on the rat prolactin promoter (41) and the well known in vivo effect on prolactin production in man, previous studies have shown little or no estrogen effect on human prolactin promoter activity (9;41). Here we show a small but consistent activation of the prolactin promoter by estrogen after 24h, but not after 6h. Interestingly, compared to treatment with either TNF-α or estrogen alone, stimulation of GH3/hPrl-luc cells with a combination of both stimuli led to a synergistic large increase in promoter activity. Recent studies have demonstrated molecular cross talk between estrogen receptor (ER) and NF-κB (52). Most studies have found inhibition of NF-κB signaling by ER and suggested a series of molecular mechanisms for this antagonism including direct inhibition of DNA binding, effects of the ER on IκB processing, or interference with coactivator recruitment (56). Synergistic interaction between estrogen and NF-κB signaling appears to be unusual, but has previously been shown for the serotonin 5HT1A receptor promoter (57). It will be interesting to determine the molecular mechanisms by which ER and NF-κB may differentially cross-regulate their effects in different cell types and at different promoters. This interaction between TNF-α and estrogen is likely to be biologically important, given the major role of estrogen in normal pituitary physiology, and possibly in pituitary adenoma formation.
In conclusion, we have shown for the first time that TNF-α can activate the human prolactin promoter and that this effect is mediated by NF-κB. TNF-α and TNF-α receptors are produced locally in the pituitary gland (18-21) suggesting that this effect could be important in vivo. We show that other important physiological regulators of prolactin expression, such as estrogen, interact with TNF-α in order to give rise to synergistic regulation of prolactin expression. Taken together, these data suggest that TNF-α and NF-κB regulation of the prolactin gene is likely to be important for pituitary physiology in vivo.
Acknowledgements
We would like to acknowledge the help of Angharad Bidder for assistance with preliminary imaging experiments and Andrea Norris for help with Cluster analysis. We thank N. Inohara, C. Paya and E. Costello for the generous gift of plasmids. This work was supported by Programme grant #67252 from the Wellcome Trust, and through collaborations with Hamamatsu Photonics and Carl Zeiss Ltd.
Footnotes
This work was funded by the Wellcome Trust (programme grant #067252)
Reference List
- 1.Owerbach D, Rutter WJ, Cooke NE, Martial JA, Shows TB. The prolactin gene is located on chromosome 6 in humans. Science. 1981;212:815–816. doi: 10.1126/science.7221563. [DOI] [PubMed] [Google Scholar]
- 2.DiMattia GE, Gellersen B, Duckworth ML, Friesen HG. Human prolactin gene expression. The use of an alternative noncoding exon in decidua and the IM-9-P3 lymphoblast cell line. J Biol Chem. 1990;265:16412–16421. [PubMed] [Google Scholar]
- 3.Wu WX, Brooks J, Glasier AF, McNeilly AS. The relationship between decidualization and prolactin mRNA and production at different stages of human pregnancy. J Mol Endocrinol. 1995;14:255–261. doi: 10.1677/jme.0.0140255. [DOI] [PubMed] [Google Scholar]
- 4.Gellersen B, Bonhoff A, Hunt N, Bohnet HG. Decidual-type prolactin expression by the human myometrium. Endocrinology. 1991;129:158–168. doi: 10.1210/endo-129-1-158. [DOI] [PubMed] [Google Scholar]
- 5.Pellegrini I, Lebrun JJ, Ali S, Kelly PA. Expression of prolactin and its receptor in human lymphoid cells. Mol Endocrinol. 1992;6:1023–1031. doi: 10.1210/mend.6.7.1508218. [DOI] [PubMed] [Google Scholar]
- 6.O'Neal KD, Montgomery DW, Truong TM, Yu-Lee LY. Prolactin gene expression in human thymocytes. Mol Cell Endocrinol. 1992;87:R19–R23. doi: 10.1016/0303-7207(92)90251-z. [DOI] [PubMed] [Google Scholar]
- 7.Nelson C, Albert VR, Elsholtz HP, Lu LI, Rosenfeld MG. Activation of cell-specific expression of rat growth hormone and prolactin genes by a common transcription factor. Science. 1988;239:1400–1405. doi: 10.1126/science.2831625. [DOI] [PubMed] [Google Scholar]
- 8.Lemaigre FP, Peers B, Lafontaine DA, Mathy-Hartert M, Rousseau GG, Belayew A, Martial JA. Pituitary-specific factor binding to the human prolactin, growth hormone, and placental lactogen genes. DNA. 1989;8:149–159. doi: 10.1089/dna.1.1989.8.149. [DOI] [PubMed] [Google Scholar]
- 9.Berwaer M, Monget P, Peers B, Mathy-Hartert M, Bellefroid E, Davis JR, Belayew A, Martial JA. Multihormonal regulation of the human prolactin gene expression from 5000 bp of its upstream sequence. Mol Cell Endocrinol. 1991;80:53–64. doi: 10.1016/0303-7207(91)90142-f. [DOI] [PubMed] [Google Scholar]
- 10.Iverson RA, Day KH, d'Emden M, Day RN, Maurer RA. Clustered point mutation analysis of the rat prolactin promoter. Molecular Endocrinology. 1990;4:1564–1571. doi: 10.1210/mend-4-10-1564. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz J. Intercellular communication in the anterior pituitary. Endocr Rev. 2000;21:488–513. doi: 10.1210/edrv.21.5.0408. [DOI] [PubMed] [Google Scholar]
- 12.Ray D, Melmed S. Pituitary cytokine and growth factor expression and action. Endocr Rev. 1997;18:206–228. doi: 10.1210/edrv.18.2.0297. [DOI] [PubMed] [Google Scholar]
- 13.Leong KG, Karsan A. Signaling pathways mediated by tumor necrosis factor alpha. Histol Histopathol. 2000;15:1303–1325. doi: 10.14670/HH-15.1303. [DOI] [PubMed] [Google Scholar]
- 14.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 15.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 16.Israel A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 2000;10:129–133. doi: 10.1016/s0962-8924(00)01729-3. [DOI] [PubMed] [Google Scholar]
- 17.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 18.Nadeau S, Rivest S. Regulation of the gene encoding tumor necrosis factor alpha (TNF-alpha) in the rat brain and pituitary in response in different models of systemic immune challenge. J Neuropathol Exp Neurol. 1999;58:61–77. doi: 10.1097/00005072-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Arras M, Hoche A, Bohle R, Eckert P, Riedel W, Schaper J. Tumor necrosis factor-alpha in macrophages of heart, liver, kidney, and in the pituitary gland. Cell Tissue Res. 1996;285:39–49. doi: 10.1007/s004410050618. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi H, Fukata J, Murakami N, Usui T, Ebisui O, Muro S, Hanaoka I, Inoue K, Imura H, Nakao K. Tumor necrosis factor receptors in the pituitary cells. Brain Res. 1997;758:45–50. doi: 10.1016/s0006-8993(96)01437-0. [DOI] [PubMed] [Google Scholar]
- 21.Wolvers DA, Marquette C, Berkenbosch F, Haour F. Tumor necrosis factor-alpha: specific binding sites in rodent brain and pituitary gland. Eur Cytokine Netw. 1993;4:377–381. [PubMed] [Google Scholar]
- 22.Arzt E, Pereda MP, Castro CP, Pagotto U, Renner U, Stalla GK. Pathophysiological role of the cytokine network in the anterior pituitary gland. Front Neuroendocrinol. 1999;20:71–95. doi: 10.1006/frne.1998.0176. [DOI] [PubMed] [Google Scholar]
- 23.Milenkovic L, Rettori V, Snyder GD, Beutler B, McCann SM. Cachectin alters anterior pituitary hormone release by a direct action in vitro. Proc Natl Acad Sci U S A. 1989;86:2418–2422. doi: 10.1073/pnas.86.7.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaillard RC, Turnill D, Sappino P, Muller AF. Tumor necrosis factor alpha inhibits the hormonal response of the pituitary gland to hypothalamic releasing factors. Endocrinology. 1990;127:101–106. doi: 10.1210/endo-127-1-101. [DOI] [PubMed] [Google Scholar]
- 25.Theas MS, De Laurentis A, Lasaga M, Pisera D, Duvilanski BH, Seilcovich A. Effect of lipopolysaccharide on tumor necrosis factor and prolactin release from rat anterior pituitary cells. Endocrine. 1998;8:241–245. doi: 10.1385/endo:8:3:241. [DOI] [PubMed] [Google Scholar]
- 26.Koike K, Hirota K, Ohmichi M, Kadowaki K, Ikegami H, Yamaguchi M, Miyake A, Tanizawa O. Tumor necrosis factor-alpha increases release of arachidonate and prolactin from rat anterior pituitary cells. Endocrinology. 1991;128:2791–2798. doi: 10.1210/endo-128-6-2791. [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi M, Koike K, Yoshimoto Y, Ikegami H, Miyake A, Tanizawa O. Effect of TNF-alpha on prolactin secretion from rat anterior pituitary and dopamine release from the hypothalamus: comparison with the effect of interleukin-1 beta. Endocrinol Jpn. 1991;38:357–361. doi: 10.1507/endocrj1954.38.357. [DOI] [PubMed] [Google Scholar]
- 28.Tang ED, Inohara N, Wang CY, Nunez G, Guan KL. Roles for homotypic interactions and transautophosphorylation in IkappaB kinase beta IKKbeta) activation [corrected] J Biol Chem. 2003;278:38566–38570. doi: 10.1074/jbc.M304374200. [DOI] [PubMed] [Google Scholar]
- 29.Trushin SA, Pennington KN, Algeciras-Schimnich A, Paya CV. Protein kinase C and calcineurin synergize to activate IkappaB kinase and NF-kappaB in T lymphocytes. J Biol Chem. 1999;274:22923–22931. doi: 10.1074/jbc.274.33.22923. [DOI] [PubMed] [Google Scholar]
- 30.Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 31.Nelson G, Paraoan L, Spiller DG, Wilde GJ, Browne MA, Djali PK, Unitt JF, Sullivan E, Floettmann E, White MR. Multi-parameter analysis of the kinetics of NF-kappaB signalling and transcription in single living cells. J Cell Sci. 2002;115:1137–1148. doi: 10.1242/jcs.115.6.1137. [DOI] [PubMed] [Google Scholar]
- 32.Takasuka N, White MRH, Wood CD, Robinson WR, Davis JRE. Dynamic Changes in Prolactin Promotor Activation in Single Living Lactotrophic Cells. Endocrinology. 1998;129:1361–1368. doi: 10.1210/endo.139.3.5826. [DOI] [PubMed] [Google Scholar]
- 33.Veldhuis JD. New modalities for understanding dynamic regulation of the somatotropic (GH) axis: explication of gender differences in GH neuroregulation in the human. J Pediatr Endocrinol Metab. 1996;9(Suppl 3):237–253. [PubMed] [Google Scholar]
- 34.Stirland JA, Seymour ZC, Windeatt S, Norris AJ, Stanley P, Castro MG, Loudon AS, White MR, Davis JR. Real-time imaging of gene promoter activity using an adenoviral reporter construct demonstrates transcriptional dynamics in normal anterior pituitary cells. J Endocrinol. 2003;178:61–69. doi: 10.1677/joe.0.1780061. [DOI] [PubMed] [Google Scholar]
- 35.Shorte SL, Leclerc GM, Vazquez-Martinez R, Leaumont DC, Faught WJ, Frawley LS, Boockfor FR. PRL gene expression in individual living mammotropes displays distinct functional pulses that oscillate in a noncircadian temporal pattern. Endocrinology. 2002;143:1126–1133. doi: 10.1210/endo.143.3.8682. [DOI] [PubMed] [Google Scholar]
- 36.See V, Rajala NK, Spiller DG, White MR. Calcium-dependent regulation of the cell cycle via a novel MAPK--NF-kappaB pathway in Swiss 3T3 cells. J Cell Biol. 2004;166:661–672. doi: 10.1083/jcb.200402136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Black EG, Logan A, Davis JR, Sheppard MC. Basic fibroblast growth factor affects DNA synthesis and cell function and activates multiple signalling pathways in rat thyroid FRTL-5 and pituitary GH3 cells. J Endocrinol. 1990;127:39–46. doi: 10.1677/joe.0.1270039. [DOI] [PubMed] [Google Scholar]
- 38.Nalda AM, Martial JA, Muller M. The glucocorticoid receptor inhibits the human prolactin gene expression by interference with Pit-1 activity. Mol Cell Endocrinol. 1997;134:129–137. doi: 10.1016/s0303-7207(97)00176-7. [DOI] [PubMed] [Google Scholar]
- 39.Nelson G, Wilde GJ, Spiller DG, Kennedy SM, Ray DW, Sullivan E, Unitt JF, White MR. NF-kappaB signalling is inhibited by glucocorticoid receptor and STAT6 via distinct mechanisms. J Cell Sci. 2003;116:2495–2503. doi: 10.1242/jcs.00461. [DOI] [PubMed] [Google Scholar]
- 40.Garside H, Stevens A, Farrow S, Normand C, Houle B, Berry A, Maschera B, Ray D. Glucocorticoid ligands specify different interactions with NF-kappaB by allosteric effects on the glucocorticoid receptor DNA binding domain. J Biol Chem. 2004;279:50050–50059. doi: 10.1074/jbc.M407309200. [DOI] [PubMed] [Google Scholar]
- 41.Gellersen B, Kempf R, Telgmann R, DiMattia GE. Pituitary-type transcription of the human prolactin gene in the absence of Pit-1. Molecular Endocrinology. 1995;9:887–901. doi: 10.1210/mend.9.7.7476971. [DOI] [PubMed] [Google Scholar]
- 42.Candolfi M, Zaldivar V, De Laurentiis A, Jaita G, Pisera D, Seilicovich A. TNF-alpha induces apoptosis of lactotropes from female rats. Endocrinology. 2002;143:3611–3617. doi: 10.1210/en.2002-220377. [DOI] [PubMed] [Google Scholar]
- 43.McFerran DW, Stirland JA, Norris AJ, Khan RA, Takasuka N, Seymour ZC, Gill MS, Robertson WR, Loudon AS, Davis JR, White MR. Persistent synchronized oscillations in prolactin gene promoter activity in living pituitary cells. Endocrinology. 2001;142:3255–3260. doi: 10.1210/endo.142.7.8252. [DOI] [PubMed] [Google Scholar]
- 44.Villalobos C, Faught WJ, Frawley LS. Dynamics of stimulus-expression coupling as revealed by monitoring of prolactin promoter-driven reporter activity in individual, living mammotropes. Molecular Endocrinology. 1999;13:1718–1727. doi: 10.1210/mend.13.10.0358. [DOI] [PubMed] [Google Scholar]
- 45.Fiering S, Whitelaw E, Martin DI. To be or not to be active: the stochastic nature of enhancer action. Bioessays. 2000;22:381–387. doi: 10.1002/(SICI)1521-1878(200004)22:4<381::AID-BIES8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 46.Walters MC, Fiering S, Eidemiller J, Magis W, Groudine M, Martin DI. Enhancers increase the probability but not the level of gene expression. Proc Natl Acad Sci U S A. 1995;92:7125–7129. doi: 10.1073/pnas.92.15.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biggar SR, Crabtree GR. Cell signaling can direct either binary or graded transcriptional responses. EMBO J. 2001;20:3167–3176. doi: 10.1093/emboj/20.12.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 49.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 50.Norris AJ, Stirland JA, McFerran DW, Seymour ZC, Spiller DG, Loudon AS, White MR, Davis JR. Dynamic patterns of growth hormone gene transcription in individual living pituitary cells. Molecular Endocrinology. 2003;17:193–202. doi: 10.1210/me.2002-0201. [DOI] [PubMed] [Google Scholar]
- 51.Grandison L, Nolan GP, Pfaff DW. Activation of the transcription factor NF-KB in GH3 pituitary cells. Mol Cell Endocrinol. 1994;106:9–15. doi: 10.1016/0303-7207(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 52.Jackson TA, Koterwas DM, Morgan MA, Bradford AP. Fibroblast growth factors regulate prolactin transcription via an atypical Rac-dependent signaling pathway. Molecular Endocrinology. 2003;17:1921–1930. doi: 10.1210/me.2003-0167. [DOI] [PubMed] [Google Scholar]
- 53.Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL. NF-kappaB Activates IL-6 Expression through cooperation with c-Jun and IL6-AP1 site, But Is Independent of Its IL6-NFkappaB Regulatory site in Autocrine Human Multiple Myeloma Cells. Cancer Biol Ther. 2004;3:1007–1017. doi: 10.4161/cbt.3.10.1141. [DOI] [PubMed] [Google Scholar]
- 54.Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL. Cooperative functions between nuclear factors NFkappaB and CCAT/enhancer-binding protein-beta (C/EBP-beta) regulate the IL-6 promoter in autocrine human prostate cancer cells. Prostate. 2004;61:354–370. doi: 10.1002/pros.20113. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Zhao M, Rao R, Inoue H, Hao CM. C/EBPbeta and its binding element are required for NFkappaB induced COX2 expression following hypertonic stress. J Biol Chem. 2005;279:16354–16359. doi: 10.1074/jbc.M411134200. [DOI] [PubMed] [Google Scholar]
- 56.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 57.Wissink S, van der B B, Katzenellenbogen BS, van der Saag PT. Synergistic activation of the serotonin-1A receptor by nuclear factor-kappa B and estrogen. Molecular Endocrinology. 2001;15:543–552. doi: 10.1210/mend.15.4.0629. [DOI] [PubMed] [Google Scholar]