

Abstract
Recently we showed that the three-dimensional structure of proteins can be investigated from a network perspective, where the amino acid residues represent the nodes in the network and the noncovalent interactions between them are considered for the edge formation. In this study, the dynamical behavior of such networks is examined by considering the example of T4 lysozyme. The equilibrium dynamics and the process of unfolding are followed by simulating the protein at 300 K and at higher temperatures (400 K and 500 K), respectively. The snapshots of the protein structure from the simulations are represented as protein structure networks in which the strength of the noncovalent interactions is considered an important criterion in the construction of edges. The profiles of the network parameters, such as the degree distribution and the size of the largest cluster (giant component), were examined as a function of interaction strength at different temperatures. Similar profiles are seen at all the temperatures. However, the critical strength of interaction (Icritical) and the size of the largest cluster at all interaction strengths shift to lower values at 500 K. Further, the folding/unfolding transition is correlated with contacts evaluated at Icritical and with the composition of the top large clusters obtained at interaction strengths greater than Icritical. Finally, the results are compared with experiments, and predictions are made about the residues, which are important for stability and folding. To summarize, the network analysis presented in this work provides insights into the details of the changes occurring in the protein tertiary structure at the level of amino acid side-chain interactions, in both the equilibrium and the unfolding simulations. The method can also be employed as a valuable tool in the analysis of molecular dynamics simulation data, since it captures the details at a global level, which may elude conventional pairwise interaction analysis.
INTRODUCTION
Understanding the process of protein folding has been a subject of study in both theoretical and experimental biophysics. Significant progress, such as the prediction of folding rates, free energies, and the structure of small proteins, has been achieved from simulations (1). During the folding/unfolding process, an intermediate state description at an atomic level is often unavailable from experiments due to poor stability of the intermediate state. In this regard, all-atom molecular dynamics simulation is particularly useful for obtaining a detailed view. Room temperature simulation at the timescale required to follow protein folding is of the order of microseconds and demands high computational capabilities. Hence, long-time simulations have been carried out on only a limited number of proteins (2). The longest folding simulation available (1 μs) is on a 36-residue peptide, villian headpiece (3). Multiple simulations on a 23-residue peptide, amounting to 700 μs, have also been carried out (4). Alternatively, one could study protein folding/unfolding dynamics at lower timescales by increasing the temperatures. It has been demonstrated that the increase in temperature accelerates protein unfolding without changing the pathway of unfolding (5), thereby justifying the use of high temperature for unfolding simulations. Further, the pathways of folding and unfolding have been shown to be similar and independent of temperature (6). Although folding has been investigated in many peptides and small proteins, only a few proteins of reasonably large size, such as hen egg- white lysozyme (7–9), dihydrofolate-reductase (10), and β-lactamase (11), have been investigated for unfolding at high temperatures.
Monitoring the process of folding/unfolding is also a challenging task. The changes in parameters such as secondary structures (helices and sheets), native contacts, root mean-square deviation (RMSD), and radius of gyration are generally some of the important ones measured in following the folding/unfolding process (5,7–17). However, there is no systematic way to monitor the interactions of side chains in a collective manner, which is crucial for the intactness of the 3-D structure of a protein. In this study, we explicitly considered side-chain interactions using the concept of protein structure networks, and the unfolding process has been examined by tracking down changes in the network properties. We chose the example of bacteriophage T4 lysozyme for this investigation. The equilibrium properties of the protein structure network are derived from the 300 K simulation. The process of unfolding at high temperatures (400 K and 500 K) has been investigated by comparing the changes in the network properties with respect to the 300 K simulation.
Real-world networks in varied fields have been investigated for their network properties and it has been shown that many of them exhibit a scale-free behavior. Further, these networks also have a small number of highly connected nodes, known as “hubs”, that play an important role in the stability of the network structure (18). The presence of such hubs in small-world networks makes the network robust against random attacks, because the hubs are capable of holding the network intact even when some nodes are attacked randomly (18). Network properties of protein structures have been studied to understand protein structure and folding (19–24). The conformations accessed during molecular dynamics simulation have also been represented as a network, from which the transition-state and denatured-state ensembles have been identified (25). Recently, a generalized computational method with fully transferable potential has been presented for folding proteins (26). The potential function in this study represents the backbone hydrogen bonds as well as side-chain interactions and graph theoretic analysis is used to cluster the conformations generated from the simulations.
Our group has described protein structures as graphs or networks of noncovalently interacting amino acid residues (27). We observed that the protein structure graphs (PSG) show complex-network behavior and their properties depend on the strength of noncovalent interactions between the amino acid residues, which is an important parameter used in the construction of the PSG. The network behavior was considered to be complex, since a variety of profiles, ranging from random graphs to decay-like curves, were obtained at varying interaction strengths. Further, a transition-like behavior was observed in all proteins when the size of the largest cluster was monitored as a function of interaction strength. We also noted that such a transition was due to the loss of a large number of hydrophobic interactions, which were generally formed at low interaction strengths. Additionally, we explored the residue preferences of the hubs on either side of the transition and found that the aromatic amino acids and arginine have a greater propensity to form hubs at high interaction strength and these residues along with hydrophobic residues like leucine, valine, and isoleucine are the preferred hubs at low interaction strength. This study provided valuable insight into protein structure and stability and thus it is quite evident that the network representation is a powerful way of studying the side-chain interactions within the protein in a systematic way. In this study, we have adopted some of these concepts to analyze equilibrium dynamics and protein unfolding. Here, we demonstrate that the fluctuations and changes in protein structure, particularly related to side-chain interactions during equilibrium and unfolding dynamics, can be captured effectively by following the changes in the network parameters.
The equilibrium and unfolding dynamics of T4 lysozyme at various temperatures have been investigated in this work. The network analysis of the molecular dynamics (MD) simulation data provides detailed information on the side-chain interactions through clusters and hubs occurring in the lysozyme structure. Particularly, it provides insight into the role of amino acid side chains in the unique topology of the proteins. Furthermore, network parameters such as the largest cluster provide an understanding beyond pairwise interaction and therefore can prove to be a powerful tool for the analysis of MD simulation data.
MATERIALS AND METHODS
MD simulations were performed using the AMBER8 package (28) with parm99 parameters (29) on the high-resolution (1.7-Å) crystal structure (2LZM) of bacteriophage T4 lysozyme (30). Explicit water molecules were used. The simulations at 300 K and 400 K were performed for 5 ns and three 500 K simulations were carried out for 10 ns in TIP3P water (31). The solvation box was 10 Å from the farthest atom along any axis for 300 K and 400 K simulations. At 500 K, a 12-Å solvation box was used for one of the simulations and a 10-Å box was used for two simulations (denoted as S2 and S3). The simulation details are consolidated in Table 1. The simulations were performed under NTP conditions. Particle mesh Ewald summation (32) was used for long-range electrostatics and the van der Waals cutoff was 10 Å. The pressure and temperature relaxations were set at 0.5 ps−1. A time step of 2.0 fs was employed with the integration algorithm, and structures were stored every 1 ps. Apart from the network analysis outlined below, conventional analyses such as RMSD and the radius of gyration, were also carried out on the MD snapshots. Compactness of the protein is measured by calculating the radius of gyration, Rg (33), which is defined as the root mean-square distance of the collection of atoms from their center of gravity. The schematic representation of the protein structures are drawn using PyMOL (39) and all other figures are drawn using MATLAB.
TABLE 1.
Summary of simulations performed at three different temperatures
| Trejectory no. | Temperature | Trajectory length (ns) | No of water's added | Density of water (gm/cc) |
|---|---|---|---|---|
| I | 300K | 5.0 | 7854 | 1.010 |
| II | 400K | 5.0 | 7854 | 0.908 |
| S1 | 500K | 10.0 | 9593 | 0.717 |
| S2 | 500K | 10.0 | 7854 | 0.728 |
| S3 | 500K | 10.0 | 7854 | 0.730 |
Construction and analysis of the protein structure graph
The protein structure graphs, or PSGs, were constructed from the atomic coordinates of the crystal structure and the snapshots from the simulations. The details of the method have been described previously (27). A brief description is given below.
Definition of nodes and edges
Each amino acid in the protein structure is represented as a node and these nodes (amino acids) are connected by edges based on the strength of noncovalent interaction between the interacting nodes. The strength of interaction between two amino acid side chains is evaluated based on the article by Kannan and Vishveshwara (34). The strength of interaction between residues i and j (Iij) is evaluated as a percentage given by
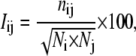 |
(1) |
where Iij is the percentage interaction between residues i and j; nij is the number of atom-atom pairs between residues i and j within a distance of 4.5 Å; and Ni and Nj are normalization values for residue types i and j (evaluated for 20 amino acid residues types) (27,34). This factor takes into account the differences in size of the side chains of the different residue types and their propensity to make the maximum number of contacts with other amino acid residues in protein structures. (The sequence neighbors (i ± 1 and i ± 2) have not been considered for the evaluation of Iij). An interaction cutoff Imin is then chosen and any residue pair ij for which Iij > Imin is considered to be interacting and hence is connected in the protein structure graph. Thus, we obtain different PSGs for the same protein structure based on the choice of Imin, and therefore, Imin can be varied to obtain graphs with strong or weak interactions forming the edges between the residues. At Imin = 0%, even residues with single atom-atom contact between them get connected in the graph, whereas at higher Imins, only strongly interacting residues with more atom-atom contacts get connected in the graph.
Hub definition
At a given Imin, different nodes make different numbers of edges. The residues making zero edges are termed as orphans and those that make four or more edges are referred to as hubs at that particular Imin. The definition of Iij for evaluating the hub character of a residue is slightly different from that given in Eq. 1. Here, the normalization value in the denominator is Ni instead of  , since the hub nature of the residue i is being evaluated (27).
, since the hub nature of the residue i is being evaluated (27).
Size of the largest cluster
The size of the largest cluster (or the giant component) in a graph is generally used to understand the properties of the graph (19,27). We used the depth first search graph algorithm (35) to identify the amino acid clusters in the PSG and then identify the size of the largest cluster in all the PSGs at different Imin values. For this purpose, the PSG is first represented as an adjacency matrix (A), where
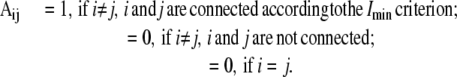 |
From the adjacency matrix, the depth first search method provides information on the nodes forming distinct clusters in the graph.
RESULTS AND DISCUSSION
Molecular dynamics simulations have been carried out at three different temperatures on bacteriophage T4 lysozyme. Simulations at 300 K and 400 K were performed for 5 ns. At 500 K, three simulations (S1, S2, and S3) of 10 ns were carried out to confirm the statistical significance of the results on unfolding by high-temperature simulations. The equilibrium properties were analyzed from the 300 K simulation. The results of high-temperature simulations were compared with the 300 K simulation to understand the process of unfolding/misfolding/refolding.
The overall fold and the secondary structural elements of the protein are stable at 300 K, as expected, and a typical snapshot is shown in Fig. 1. The tertiary structure of lysozyme is made up of two domains, D1 and D2. The smaller domain D1 includes all the three β strand and the helices α1 and α2, whereas the larger domain D2 consists of nine helices (α3–α11). The domains D1 and D2 are connected through the helix α3. The interactions between the secondary structures within and across domains have been investigated and the process of unfolding at high temperatures is monitored from the network perspective. These results are discussed below after the simulation profiles of some of the conventional parameters.
FIGURE 1.

A schematic representation of the structure of T4 lysozyme. It consists of 11 helices between residues 3–11 (α1), 39–50 (α2), 60–80 (α3), 82–90 (α4), 93–106 (α5), 108–113 (α6), 115–123 (α7), 126–134 (α8), 137–141 (α9), 143–155 (α10), 159–161 (α11) and three β-strands between residues 14 and 19, 25 and 28, and 31 and 34. The two domains are represented as D1 and D2.
Simulation profiles
RMSD
The time-dependent RMSD from the starting structure gives an estimation of the rate of unfolding at different temperatures (Fig. 2). At 300 K, Cα -RMSD fluctuates around 1 ± 0.5 Å. In the 400 K simulation, the RMSD is stable around 2 Å up to ∼4 ns and then increases to ∼3 Å. All the three simulations at 500 K behave in a similar manner, particularly until 4 ns. There is a sharp increase in RMSD between ∼1 ns and ∼2 ns. As the RMSD reaches a range of ∼6–8 Å around 3 ns, the rate of change of RMSD has diminished. The RMSD profiles of the three simulations, however, perceptibly vary during 4–10 ns, with values ranging from 7 Å to 12 Å. The increased RMSD clearly indicates a drastic conformational change in the structure of lysozyme in the 500 K simulations.
FIGURE 2.

MD trajectories of RMSD (Å) with reference to the minimized crystal structure.
Radius of gyration
The trajectories of the radius of gyration (Rg) of the protein at different temperatures are presented in Fig. 3. The value fluctuates around 13.4 Å in the 300 K simulation, Rg is slightly higher with increased fluctuation at 400 K. Large fluctuations, varying from 13 Å to 18 Å are seen in all three simulations at 500 K. However, it should be noted that the fluctuations are not so pronounced until around 2.0–2.4 ns, when a lower value of ∼14 Å is attained in all three simulations. The compactness at this point is related to the collapse of domains in the structure, which will be discussed later. The fluctuations in Rg are more drastic after this point, indicating large changes in the structure.
FIGURE 3.

Radius of gyration (Å) trajectories at different temperatures.
Network analysis
Protein structure network (PSN) analysis is carried out on the snapshots obtained from 300 K, 400 K, and 500 K simulations. Qualitative features of the network parameters along the trajectories are very similar in all three simulations at 500 K. Hence, most of the results are presented from only one of the simulations (S1) (some of the results from simulation S2 are presented in Supplementary Material). The interaction-strength-dependent analysis is an important feature of this work. First, general network properties such as the degree distribution and size of the largest cluster are presented, and then the trajectories are probed to gain insight into the details at the structural level.
Degree distribution profiles
The nonbonded connections made by amino acid residues in every snapshot were evaluated at different interaction strengths (Imin). The nodes with a given number of links were averaged over the snapshots obtained from the simulations. The number of nodes N(k) with k links were extracted from the simulation snapshots for interaction strengths ranging from 0% to 10%. The Imin-dependent plots (degree distribution plots) of N(k) as a function of the number of links (k) from the 300 K structures are given in Fig. 4 a. The profiles are very similar to the one observed in the static structures of proteins (27) obtained from the Protein Data Bank. The number of nodes with one or two links is higher than the number of nodes with zero links (orphans) for Imin values <5%. This gives rise to a bell-shaped curve at lower Imin values and a decay form at higher Imin values. Thus, the degree distribution profile is clearly dependent on Imin. It is interesting to compare similar plots obtained from the snapshots of the 500 K simulation. The 500 K trajectory is split up into two regions, A (0–2.2 ns) and B (2.2–10 ns), on the basis of Rg profile, and the corresponding degree distribution profiles from simulation S1 are presented in Fig. 4, b and c (similar profiles are presented for simulation S2 in Supplementary Material, Fig. S1, a and b). Although the plots look qualitatively similar to those obtained at 300 K, some important differences can be noted. First, the number of orphans is higher at 500 K at all Imin values, giving rise to a higher ratio of number of orphans to number of nodes with connections. Second, the transition from a bell-shaped curve to a decay-like curve takes place at a lower Imin of 3% and 2% for regions A and B, respectively, at 500 K. The profiles at the two temperatures are very similar for nodes with links between 2 and 7. There are very few nodes with links >7 at either temperature, and the number is close to zero at 500 K. Thus, the 500 K structures differ from the 300 K structures in terms of the increased number of orphans and a change in the degree distribution profile to a lower Imin value.
FIGURE 4.

Distribution of the number of nodes making k links, at various interaction strengths (Imin): (a) averaged over 300 K snapshots; (b) averaged over snapshots from 0–2.2 ns at 500 K; (c) averaged over snapshots from 2.2–8.0 ns at 500 K (unless specified, all results in this figure and in subsequent tables and figures for 500 K are presented for simulation S1).
Largest cluster profiles
The size of the largest cluster (or the giant component) is often used to understand the nature and properties of graphs (19,27) and to understand the phase transition from a percolation point of view (36). It has been observed that in a large number of globular proteins, there exists a critical Imin value below which the PSNs are almost completely connected, and above this Imin the PSNs split up into smaller clusters (27). Here, the profiles of the size of the largest cluster (averaged over simulation snapshots) as a function of Imin are presented in Fig. 5. Profiles at all the temperatures are sigmoidal in behavior and a critical Imin (Icritical, defined as the Imin at which the size of the largest cluster is half the size at Imin = 0%), ranging from 2.5% to 3.4%, can be identified. However, significant differences between the 300 K and all three 500 K simulations can be seen in the size of the largest cluster and the Icritical. The size of the largest cluster at Imin = 0% on average is 135 in the structures at 300 K and it reduces to ∼105 in all the 500 K simulations. Also, the sizes of the largest cluster at all Imins are smaller in the 500 K structures. Furthermore, the Icritical shifts from 3.4% at 300 K to 2.5% at 500 K. It is interesting to note that such a shift in Icritical is also correlated with the differences observed in the degree distribution plots of N(k) versus k (Fig. 4, a–c).
FIGURE 5.

The size (averaged over simulation snapshots) of the largest cluster as a function of the interaction strength Imin, at different temperatures.
Tertiary contacts
The extent of unfolding is generally measured in terms of the number of native contacts (37), as evaluated from noncovalent atom-atom contacts between amino acids in the protein. This corresponds to residue interactions at an Imin of 0% in our study. However, it is also important to consider the extent of interaction (captured by the parameter Imin in our analysis) in evaluating the number of contacts. Here, we present the tertiary interactions at Imin values of 0% and Icritical. The number of contacts at 300 K and 400 K is presented as a function of time in Fig. 6 a. At 300 K and 400 K, the average values for Imin at 0% are 210 and 150, respectively, and those for Imin at 3.4% are 100 and 75, respectively. The number of nonnative contacts (compared with the average from the 300 K trajectory) is also plotted for the 400 K simulation and the average values are 50 and 40 for Imin at 0% and 3.4%, respectively. Thus, the native contacts at both Imin values at 400 K are higher than the nonnative contacts, indicating a near-native structure in the simulation. The trajectories of the native and nonnative contacts at Imin = 0% and 2.5% from the 500 K simulation (S1) are presented in Fig. 6 b (similar trajectories for simulation S2 are presented in Fig. S2). The number of native contacts steeply decreases until ∼3 ns for both Imins and then attains a reasonably stable value. Concomitantly, the number of nonnative contacts increases until 3 ns. Although the curves have flattened after this point, the fluctuations are more for the nonnative contacts compared to that of the native contacts, which perhaps accounts for large fluctuations in Rg after 3 ns (Fig. 3). Interesting structural transformations seem to be taking place within 3 ns. The ratio of the native to nonnative contacts becomes 1 (which is generally associated with the folding/unfolding transition state (37)) at approximate time points 0.415 ns and 1.9 ns, respectively for Imin values at 2.5% and 0.0%. An RMSD-based conformational cluster analysis (7,38) was carried out, which showed a transition close to 0.415 ns (the results of such a cluster analysis of simulation S1 are presented in the Fig. S3). Based on network features at Icritical, structural analysis, and the clustering of conformations, we identify the transition from native state to unfolded state as associated with the structure at the time point when the native/nonnative contacts become equal at the interaction strength Icritical . Thus, our further analysis of the 500 K trajectory is divided into regions I (0–0.415 ns (time at which the native and nonnative contacts become equal at Imin = Icritical)), II (0.415–1.9 ns (time at which native/nonnative contacts become equal at Imin = 0%)), III (1.9–3 ns (time at which the native and nonnative curves flatten)), and IV (3–10 ns), as indicated in Fig. 6 b.
FIGURE 6.

The trajectories of native and nonnative contacts at different Imins: (a) 300 K and 400 K; (b) 500 K (profiles A and C represent the native contacts at 0.0% and 2.5%, respectively; profiles B and D represent the nonnative contacts at 0.0% and 2.5%, respectively). The numerals I–IV represent different regions of the simulation corresponding to different phases of unfolding.
Intersecondary structural interactions
Typical snapshots selected from the four regions shown in Fig. 6 b have been analyzed for specific details of native and nonnative contacts. The native contacts have been obtained from the 300 K simulation by evaluating the contacts at Imin = 3.4%, and those interactions present in >50% of the snapshots are plotted as a 2-D map in Fig. 7 i. The interactions within the β-sheets and intrahelical interactions are quite obvious. The intersecondary structural interaction regions are marked in rectangular boxes a–g in the figure. The long helix (α3), which separates the two domains, has more interactions with the regions of the smaller domain (D1), as shown in box a. The N-terminal helix (α1) interacts with helices α5, α9, and α10, (marked as regions b and c) of the larger domain (D2). A large number of interhelical interactions are seen in domain D2, and it is mainly dominated by interactions of helix α5 with other helices, as shown in boxes d–f in the figure. The residue interaction map (evaluated at Imin = 2.5%) for selected snapshots in region I (Fig. 6 b) from the 500 K simulation are presented in Fig. 7 ii. (Similar maps for snapshots (0.9 ns, 2.2 ns, and 8.0 ns) in the other regions are presented in Fig. S4.) The native and nonnative contacts are represented by different symbols. From these maps, we can see that the secondary structures, both the β-strands and the α–helices, are reasonably stable up to 0.9 ns. Although a fraction of native helical contacts are retained in 2.2-ns and 8.0-ns snapshots, the β-sheets (essentially from domain D1) seem to have completely melted away, losing the native contacts. Intersecondary structural contacts are also retained to a significant extent in the 0.415-ns and 0.9-ns snapshots. The nonnative interactions arise either in the regions closer to the native ones or between completely different regions absent in the native structure. For instance, the nonnative interaction of β-strands (residues 25–38) with residues of helices α3, α5, and α9 seems to take the conformation away from the native structure in all the 500 K snapshots. Finally, a part of the intersecondary structural contacts of helices α1 and α5 with α9/α10 are the only ones retained by 2.2 ns and the native interaction between helices α5 and α9/α10 are also lost by 8.0 ns. Thus, the compactness of the structure in region IV is mainly due to nonnative contacts (with the exception of fragments of local intrahelical contacts) and can be considered as misfolded states.
FIGURE 7.

Two-dimensional maps of residue-residue interactions evaluated for Imin = 3.4% and 2.5%, respectively, for 300 K and 500 K snapshots. (i) Contacts found in >50% of the 300 K snapshots. The secondary structures are marked below the diagonal. The rectangular boxes (a–g) correspond to different interactions across secondary structures/loops as given in the inset. (ii) (Open circles) native contacts; (*) nonnative contacts (compared with 300 K simulations). Two snapshots (points above the diagonal and below the diagonal correspond respectively to 0.387 ns and 0.415 ns snapshots) from 500 K simulation. The maps for a few other snapshots are given in supplementary Fig. S4.
Composition of the largest cluster
The largest cluster represents the extent of connectivity in the protein structure, which is not completely captured by pairwise contacts. Both the extent of connectivity and the residues contributing to the largest cluster can be compared across structures to evaluate their closeness with the native structure. The extent of connectivity was evaluated as the size of the largest cluster for different Imins and is presented in Fig. 5. The residue composition of the largest cluster is investigated in this section. The composition was identified in each of the snapshots of the simulations at Imin = Icritical (i.e., 3.4% for 300 K and 400 K simulations and 2.5% for 500 K simulations). The residues present in the largest cluster in >50% of the snapshots of 300 K simulations are listed in Table 2. The presence/absence of these residues in 400 K and 500 K simulations is also marked in this table. The results from 500 K simulation S1 are presented in two parts (0–3 ns and 3–8 ns). The appearance of new residues at higher temperatures is not listed, since our primary aim is to find out how close the structures are to the native state.
TABLE 2.
Composition of residues in the largest cluster during MD simulations (at Imin = Icritical)
| Hydrophobic
|
Polar
|
Aromatic
|
Acidic
|
Basic
|
Glycine
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | a | b | c | d | a | b | c | d | a | b | c | d | a | b | c | d | a | b | c | d |
| 1M | + | 2N | + | + | + | 4F | + | + | + | 5E | + | 96R | 12G | + | |||||||||
| 3I | + | + | 21T | + | + | 18Y | 10D | + | + | 119R | + | 23G | + | ||||||||||
| 6M | + | + | 38S | 25Y | 20D | 124K | 28G | + | + | ||||||||||||||
| 7L | + | + | + | 69Q | + | 104F | + | + | + | 61D | + | 137R | + | + | + | 30G | + | + | |||||
| 9I | + | + | + | 97C | + | 126W | + | 64E | 145R | + | 77G | ||||||||||||
| 13L | + | + | 101N | + | + | 138W | + | + | 70D | + | + | + | 147K | + | + | + | 110G | ||||||
| 29I | 105Q | + | + | 159D | 148R | + | + | 113G | + | ||||||||||||||
| 41A | 115T | + | |||||||||||||||||||||
| 66L | + | + | + | 116N | |||||||||||||||||||
| 87V | + | 132N | + | ||||||||||||||||||||
| 99L | 136S | + | |||||||||||||||||||||
| 100I | + | + | + | 140N | + | + | |||||||||||||||||
| 103V | + | + | 141Q | + | |||||||||||||||||||
| 131V | + | 144N | + | + | + | ||||||||||||||||||
| 143P | 151T | ||||||||||||||||||||||
| 149V | 152T | + | + | + | |||||||||||||||||||
| 150I | + | 157T | + | ||||||||||||||||||||
| 160A | + | + | |||||||||||||||||||||
The residues represented are part of the largest cluster and are present in >50% of the snapshots for Imin = 3.4% for 300 K and 400 K simulations and Imin = 2.5% for 500 K simulations. a: Residues in the largest cluster obtained from the 300 K simulation. b: Residues in the largest cluster obtained from the 400 K simulation and common with 300 K. c: Residues in the largest cluster obtained from the 500 K simulation for regions I–III and common with 300 K. d: Residues in the largest cluster obtained from the 500 K simulation for region IV and common with 300 K.
The residues in the largest cluster are grouped as hydrophobic, polar, etc. (Table 2). At 300 K, the cluster composition is largely made up of hydrophobic and polar residues. A few crucial aromatic, acidic, and basic residues (dominated by arginine residues) are also part of the cluster. Interestingly, several glycine residues are also part of the largest cluster and several of them are from the terminals of secondary structures. Most of the residues in the secondary structures contribute to the largest cluster; however, the contribution is greater from helices α1, α5, α9, and α10. Furthermore, the residues from both domains D1 and D2 are present in the largest cluster. Thus, the domain separation may be noticed only when the interactions are considered at Imin > Icritical. At 400 K, most of the residues of helices α1, α5, α9, and α10 are persistent, and several residues (29I, 41A, 38S, 18Y, 25Y, 20G, 12G, and 23G) from domain D1 detach themselves from the largest cluster. These residues are also absent from the largest cluster in the 500 K simulation. A considerable fraction of the residues retained in the 400 K simulation is also present for up to 3 ns of 500 K simulation and is reduced significantly after this time point. Interestingly, none of the glycine residues are part of the largest cluster after 3 ns at 500 K, although the secondary structures are reasonably intact. This indicates the lack of optimal packing of secondary structures, which is also evident from the contact map.
The cluster composition at Imin = 5% (interaction strengths greater than Icritical) was also investigated, and an analysis similar to the one presented above for Imin = Icritical was carried out at 300 K, 400 K, and for regions I–III (shown in Fig. 6 b) of the 500 K simulation. The residues that are present in >30% of the snapshots in simulations at 300 K are listed in Table 3, and the occurrence of these residues in 400 K and 500 K simulations has been checked. The results on a few specific snapshots in the chosen regions are also included in this table. In comparison with the cluster sizes at Icritical, the cluster sizes at Imin = 5% reduced considerably in all cases, as expected. Interestingly, the hydrophobic residues are almost completely absent in all cases. Further, a significant fraction of the 300 K residues is also part of the cluster at 400 K and before transition in the 500 K simulation. However, the average cluster size is drastically reduced after the transition (a list of both the native and nonnative residues in the largest cluster at Imin = 5% is provided in Table S1), and none of the residues of the largest cluster at Imin = 5% is common between 300 K and the 500 K trajectory after 3 ns. Thus, not only is the size of the strongly interacting cluster reduced, but its composition also bears no resemblance to the native residues after 3 ns in the 500 K simulation.
TABLE 3.
Composition of residues in the largest cluster during MD simulation (Imin = 5%)
| 300K
|
400K
|
500K (I)
|
500K(II)
|
500K(III)
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| avg | 4929ps | avg | 4177ps | avg | 415ps | avg | 2206ps | avg | 3000ps |
| 10D | + | + | + | + | + | + | + | ||
| 20D | + | ||||||||
| 21T | + | + | + | + | + | ||||
| 23G | + | + | |||||||
| 69Q | + | ||||||||
| 100I | + | + | + | + | + | ||||
| 101N | + | + | + | + | |||||
| 104F | + | + | + | + | + | ||||
| 105Q | + | + | + | + | + | + | |||
| 137R | + | + | + | + | + | ||||
| 138W | + | + | + | + | + | + | |||
| 140N | + | + | |||||||
| 141Q | + | + | + | + | |||||
| 143P | |||||||||
| 144N | + | + | + | + | + | + | + | ||
| 145R | + | + | + | + | + | ||||
| 147K | + | + | + | + | + | + | + | ||
| 148R | + | + | + | ||||||
| 153F | + | ||||||||
| 155T | + | ||||||||
The composition of the largest cluster is presented for different temperatures. avg indicates residues that are present in >30% of snapshots. The residues at 300 K (avg) are listed and those residues that are common with this list obtained from other simulations are marked. The composition of a few specific snapshots is also presented. For 500 K, I–III correspond, respectively, to the regions 0–415 ps, 0.5–1.9 ns, and 1.9–3.0 ns.
At Imin = 5%, the protein structure is made up of three clusters of significant size, with the top two corresponding to domain D2 and the third ne corresponding to domain D1. The three clusters are depicted on the crystal structure and on the structures selected from typical snapshots from different simulations in Fig. 8 (A list of residues in these clusters from selected snapshots are given in Table S2). In the crystal structure and a typical 300 K snapshot, it can be clearly seen that the top cluster is made of residues from helices α1, α5, α9, α10, and α11, and the second largest cluster is made up of helices α7 and α8. The third largest cluster represents domain D1, with residues mainly from helix α2 and the β-strands. The top two clusters of the 300 K snapshot (Fig. 8, second panel) and the 400 K snapshot (Fig. 8, third panel) are very similar; however, there is a reorganization of residues of domain D1 in the third-largest cluster. The snapshot around transition at 500 K (Fig. 8, fourth panel) also shows the top two clusters as similar to those of the snapshots at 300 K and 400 K. But the third-largest cluster size is considerably reduced, indicating the loss of compactness of domain D1 around the transition region. The 500 K snapshot around 2.2 ns (Fig. 8, fifth panel) shows a distorted backbone structure and a clear indication of the collapse of domains D1 and D2, with the largest cluster being composed of residues from both domains. However, a majority of residues come from domain D2, and also, the second-largest cluster is made up of residues exclusively from domain D2. Thus, domain D1 loses its structural identity long before domain D2. This feature is also evident from the residue contact maps, where the β-strands of domain D1 have melted away and the residues from this region have picked up nonnative contacts at an early stage of simulation. This is in agreement with the experimental finding that the domain D1 is formed later than D2 during the folding process (40). The 3-ns snapshot of the 500 K simulations (sixth panel) has completely lost the identity of the native structure. Here, the helices are wrongly oriented, only two clusters of small size appear. Although one of the clusters is composed of residues from domain D1, the residue composition has no resemblance to native clusters at Imin = 5% (Table S2). This result highlights the point that the folding process is cooperative and transition from the unfolded to the native state takes place only when appropriate contacts are established. Finally, the unfolded structure at 7.39 ns (seventh panel) clearly appears different and has moved completely away from the native structure.
FIGURE 8.

Representation of top clusters: The seven panels include the crystal structure, typical snapshots from 300 K and 400 K simulations, and four snapshots of 500 K simulations, representing different states during the unfolding processes. The times are given in parentheses below the structures. The N- and C-termini are colored blue and red, respectively. The top three clusters at Imin = 5.0% (residue composition given in supplementary Table S2) are depicted on crystal structure and simulation snapshots: orange, gray, and magenta represent first, second, and third largest clusters, respectively. Only two clusters were obtained in the last two snapshots.
Thus, the residue-level details corresponding to unfolding events can be effectively captured by the construction of interaction-strength-dependent PSNs and by examining the network parameters. Hen egg-white lysozyme, which has a structure very similar to that of T4 lysozyme, was earlier investigated by MD simulations to understand the unfolding events (7) and the results were correlated with experimental findings (41) .Our results agree with the unfolding events presented for hen egg-white lysozyme. Specifically, the collapse of the two domains, persistence of the α-domain and the early melting of the β-domain are some of the common features of the two studies. However, the network-based characterization described here is useful in following the structural changes at a global level.
Here we make a plausible assumption that the folding events can be reconstructed from the unfolding simulations, and based on our study, the following scenario can be presented for the folding process of T4-lysozyme: 1), formation of small helical segments, 2), protein chain fluctuation making random contacts; 3), transition step during which a major fraction of the strong native side-chain contacts are cooperatively established along with coevolution of complete secondary and near tertiary structure; and 4), hydrophobic residues join the core to give the final topology and to strengthen the native structure. These steps are entirely consistent with the existence of a folding funnel (42) guiding the protein to its native-state conformation. Partial secondary structures appear at the early stage in the proteins containing segments with greater helical propensities. A recent review has also focused on the role played by backbone hydrogen bonds in the formation of secondary structures and in the folding processes (43). The framework model (44) is adopted by proteins with high secondary structural propensities. Folding of T4 lysozyme seems to adopt the nucleation-condensation mechanism (45) with an element of the framework model. Such a mechanism was also observed in the protein c-Myb (46), which is a small protein made up of three helices.
Hubs
The hub-forming amino acid residues (those with four edges or more) in the PSN can belong to different secondary structural elements in the protein. Although the backbone hydrogen bonds give the information on the secondary structures, the hubs and their interactions provide information on connections across secondary structures, including residues from loops. Analysis of hubs and their connections can provide insight into the details of side-chain interactions, which are required for the structural integrity of the protein and further can be used as a tool to monitor the changes taking place in the high-temperature simulations. Here, each snapshot from the MD trajectory is examined for the residue capacity to form hubs, and those that are hubs for >50% of the simulation time are considered to be dynamically stable hubs. Such dynamically stable hubs have been identified from all the simulations, and the results for the 300 K and 400 K simulations, and for 500 K simulation S1, at Imins 0% and 3%, are presented in Tables 4 and 5, respectively.
TABLE 4.
Dynamically stable hub list for simulations at different temperatures for Imin = 0.0%
| 500K*
|
|||||||
|---|---|---|---|---|---|---|---|
| Crystal structure | 300K | 400K | R1 | R2 | |||
| 1M | 95R | 1M | 95R | 95R | 10D | 10D | |
| 3I | 99L | 3I | 99L | 3I | 99L | 25Y | 11E |
| 6M | 100I | 6M | 100I | 4F | 100I | 67F | 14R |
| 7L | 101N | 7L | 101N | 7L | 101N | 88Y | 138W |
| 10D | 102M | 10D | 102M | 13L | 102M | 95R | 145R |
| 11E | 103V | 13L | 103V | 17I | 103V | 101N | 148R |
| 17I | 104F | 17I | 104F | 25Y | 104F | 161Y | |
| 25Y | 106M | 25Y | 106M | 27I | 106M | 138W | 164L |
| 27I | 111V | 27I | 111V | 29I | 111V | 145R | |
| 29I | 114F | 29I | 114F | 31H | 114F | 148R | |
| 30G | 118L | 33L | 149V | ||||
| 33L | 120M | 33L | 120M | 120M | 153F | ||
| 39L | 121L | 121L | 42A | 121L | 161Y | ||
| 42A | 126W | 42A | |||||
| 43K | 133L | 81N | 133L | 46L | 133L | ||
| 46L | 138W | 46L | 138W | 50I | 138W | ||
| 50I | 139Y | 50I | 139Y | 139Y | |||
| 54C | 142T | 54C | 142T | ||||
| 56G | 145R | 145R | 58I | ||||
| 58I | 148R | 58I | 148R | 62E | |||
| 62E | 149V | 62E | 149V | 66L | 149V | ||
| 66L | 150I | 66L | 150I | 67F | 150I | ||
| 67F | 152T | 67F | 152T | 152T | |||
| 71V | 153F | 71V | 153F | 78I | 153F | ||
| 78I | 154R | 78I | 84L | ||||
| 84L | 158W | 84L | 158W | 158W | |||
| 87V | 161Y | 87V | 161Y | 88Y | 161Y | ||
| 88Y | 88Y | 91L | |||||
| 91L | 91L | ||||||
50% of snapshots in which the listed residues appear as hubs.
R1, regions I–III; and R2, region IV, as presented in Fig. 6 b.
TABLE 5.
Dynamically stable hub list for simulations at different temperatures for Imin = 3.0%
| 500K*
|
||||
|---|---|---|---|---|
| Crystal Structure | 300K | 400K | R1 | R2 |
| 3I | ||||
| 6M | 6M | 6M | 10D | |
| 7L | 7L | 7L | 145R | |
| 10D | 10D | 10D | 148R | |
| 25Y | 25Y | 25Y | 161Y | |
| 46L | 27I | 27I | ||
| 62E | 54C | 54C | ||
| 67F | 58I | 58I | ||
| 95R | 62E | 62E | ||
| 101N | 67F | 67F | ||
| 102M | 95R | 95R | ||
| 104F | 101N | 101N | ||
| 114F | 104F | 104F | ||
| 138W | 111V | 111V | ||
| 145R | 138W | 138W | ||
| 148R | 145R | 145R | ||
| 153F | 148R | 148R | ||
| 158W | 158W | 158W | ||
| 161Y | 161Y | 161Y | ||
50% of snapshots in which the listed residues appear as hubs.
R1=regions(I+II+III), R2=region IV as shown in figure 6 b.
About a third of the residues (56) in the protein are hubs in the crystal structure at Imin = 0%. Approximately 90% of them are retained at 300 K simulation, and the hydrophobic hubs are in the majority (60%). Aromatic and arginine residues form 26% of the hubs and the rest is made up of polar and acidic residues. It is obvious that the number of hubs is reduced in all the simulations at Imin = 3% (Table 5). However, it is to be noted (from the 300 K simulation) that the reduction is substantial in the hydrophobic hubs and there are only six hydrophobic hubs at Imin = 3%, as compared with 30 at Imin = 0%. All three arginines and six (out of 10) aromatic residues from the 300 K list of Imin = 0% are retained as hubs at Imin = 3%. This is consistent with our earlier results (27) on crystal structure analysis, i.e., that the hydrophobic hubs drastically reduce as interaction strength increases to around Icritical. Interestingly, this is also correlated with the decrease of hydrophobic residues in the largest cluster at interaction strengths beyond Icritical, as mentioned in the previous section. A comparison of the hub list in Table 4 with those residues in the largest cluster (Table 2) shows ∼50% of common residues. Significantly, the common ones predominantly belong to the secondary structures α1, α5, α9, and α10, which were found to be part of the strongly interacting largest cluster.
The hub composition at 400 K is very similar to that from the 300 K simulation at Imin = 0% and 3%. Further, the number of hubs is drastically reduced in both parts of the 500 K simulation. During the first part of the 500 K simulation, many of the hub residues at Imin = 0% are common with those of the 300 K simulation, which is not the case during the second part of the 500 K simulation. Furthermore, the number of 3% hubs from the 500 K simulation is too small. This is also consistent with results from the top-largest-clusters analysis, where the size of the cluster had reduced and only two clusters of significant size were seen.
CORRELATION WITH EXPERIMENTS
Mutational studies
A large number of mutations have been carried out on T4 lysozyme and the effects of mutation on the activity/stability of the enzyme have been assessed (49–58). Here we present the hub nature of the mutated residues based on our PSN network analysis. Earlier, we had demonstrated the importance of hubs in the thermal stability of thermophilic proteins (27) and in the stability of interfaces in multimeric proteins (47). Here we examine the effect of mutation on the stability/activity of the enzyme in terms of residues that form hubs. The effect of mutation on some of the hub residues (Imin = 0%, Table 2), as observed in experiments, has been listed in supplementary Table S3. A few mutations of residues that are not hubs (nonhubs) have also been listed for comparison. The mutation of the hub residues to alanine has considerably decreased the stability of the protein, whereas such a destabilization is not clear in the case of the nonhub mutations. It should be noted that many of the mutated hubs causing destabilization are hubs not only at 300 K but also remain hubs at 400 K, emphasizing the importance of these residues in maintaining the integrity of the tertiary structure. Furthermore, the hubs V111, W138, and F153 are also hubs at 3% in the 300 K simulation, indicating a strong interaction of these residues with several other residues. Finally, some of the residues, such as F104, R145, and R148, are hubs even at 400 K and at 3%, for which mutation results are not available. Here we predict that the mutation of these residues to alanine would cause further destabilization of the protein.
Stages of domain formation during folding
Folding experiments on fragments of T4 lysozyme have shown (40) that only the C-terminal subdomain (domain D2) is capable of autonomous folding. Also, experiments have shown (40) that the intermediate state of T4 lysozyme is comprised of predominantly unfolded D1 subdomain with loosely packed D2 sub-domain. Thus, it is clear that domain D2 is formed earlier than domain D1. From our cluster analysis at Imin = 5%, three clusters of significant size have been identified from the native structure (Fig. 8, second panel). Among them, two top clusters are composed of residues from domain D2 and the third top cluster is made of residues from domain D1. During the collapse state (2.2-ns snapshot of 500 K simulation; Fig. 8, fifth panel), only two clusters of significant size are present and the top one is now composed of residues from both the domains, with the major contribution from domain D2. Further, the second top cluster is also made up of only the residues of domain D2. Thus, domain D1 loses its structural identity long before domain D2 does. Conversely, we can conclude that domain D2 is formed at an early stage, which reinforces the experimental findings.
Folding free energies
The proton exchange capacity of backbone amide protons of T4 lysozyme has been extensively investigated from NMR experiments as a function of unfolding reagent (48). Based on these experiments, the residue-wise free energy of folding has been calculated. From these studies, the helices α1(A), α5(E), and α10(H) have been identified as the most stable portion of the protein. Our investigation of the largest cluster is in agreement with this conclusion, as the largest cluster at interaction strength as high as 5% encompasses this region not only in 300 K and 400 K simulations, but also in the early part of 500 K simulation. Specifically, the residues D10, I100, N101, F104, Q105, K147, and F153, which have high free energy of folding, are part of the largest cluster at Imin = 5% in both 300 K and 400 K simulations. Furthermore, the helix α9 is also a part of the largest cluster, even at high interaction strength of Imin = 5%. However, experimental free energy values have not been reported for the residues in this helix. Our results suggest that residues R137, W138, N140, and Q141 are also part of the most stable portion of the protein. Particularly, W138 is not only a part of the cluster at Imin = 5%, but also appears as a hub at Imin = 3% in 300 K and 400 K simulations. We predict that residues such as D10, N101, M102, F104, W138, R145, R148, and F153, which are part of the largest cluster at Imin = 5% and also hubs at Imin = 3% are important for the stability of both the native state and the folding intermediates.
SUMMARY
The concept of the protein structure network was integrated with molecular dynamics simulations, using the example of T4 lysozyme. The PSNs were constructed as a function of interaction strength (Imin) between noncovalently interacting residues. The equilibrium dynamics of the PSN was investigated in the 300 K simulation and the dynamics of unfolding was probed in the 500 K simulations. Mapping the structures onto graphs provides a global view of the structure and associated changes. This information, obtained from the global view, is more significant when compared with what can be obtained from pairwise interresidue contact analysis. Important results from the general network perspective, as well as from the unfolding events of T4 lysozyme, are summarized below.
The degree distribution profiles at 300 K exhibit similar complex behavior, as was observed in the case of a large number of protein structures. Specifically, the profile of the distribution of nodes with links 0–10 undergoes a transition from a bell-shaped to a decay-like curve at a critical interaction strength. The snapshots from 500 K simulations also exhibit a similar behavior, but the transition occurs at a lower Imin. Further, the size of the largest cluster undergoes a transition at a critical Imin (Icritical) in both the 300 K and 500 K simulations. Here, again, the Icritical shifts to a lower value in the 500 K simulations.
The folding transition has been identified, from the 500 K simulation, as the point at which the ratio of the native/nonnative contacts is 1, when evaluated at an interaction strength close to Icritical. This has also been supported by conformational cluster analysis.
The composition of the largest cluster was deduced from the simulation snapshots. At 300 K, residues from both the domains of T4 lysozyme are part of the largest cluster at Imin = Icritical. However, a clear separation of domains becomes apparent as separate clusters at Imin > Icritical. Hydrophobic residues dominate the largest cluster at Icritical and their contribution drastically reduces at Imin > Icritical.
The largest cluster size is reduced significantly in the 500 K simulation. The composition of the largest cluster, evaluated even at higher Imin compares well with that of the native structure (from the 300 K simulation) until some point (∼2 ns) after the transition from folding to unfolding. The clusters indicate that domain D2 is intact for a longer time than domain D1. Furthermore, incorrectly folded structures can be detected by examining the composition of the largest clusters.
Hub residues (defined as residues that are connected to more than three other residues) have been identified. It is suggested that those residues that are hubs around Icritical in both the 300 K and 400 K simulations influence the stability of the protein structure, and these observations have been correlated with mutation experiments.
The residues in the largest cluster at Imin > Icritical play an important role in the folding process. This has been confirmed by comparison with experimental results on unfolding. Some of the residues for which experimental data is not available are predicted to be important in stabilizing the transition-state intermediate.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We acknowledge support from the Computational Genomics Initiative at the Indian Institute of Science, funded by the Department of Biotechnology (DBT), India, and the computational facilities at the Supercomputer Education and Research Centre (SERC), Indian Institute of Science, Bangalore.
K. V. Brinda's present address is Dept. of Computer Science, Cornell University, Ithaca, NY.
References
- 1.Snow, C. D., E. J. Sorin, Y. M. Rhee, and V. S. Pande. 2005. How well can simulation predict protein folding kinetics and thermodynamics? Annu. Rev. Biophys. Biomol. Struct. 34:43–69. [DOI] [PubMed] [Google Scholar]
- 2.Daggett, V. 2000. Long timescale simulation. Curr. Opin. Struct. Biol. 10:160–164. [DOI] [PubMed] [Google Scholar]
- 3.Duan, Y., and P. A. Kollman. 1998. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 282:740–744. [DOI] [PubMed] [Google Scholar]
- 4.Snow, C. D., H. Nguyen, V. S. Pande, and G. Martin. 2002. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 420:102–106. [DOI] [PubMed] [Google Scholar]
- 5.Day, R., B. J. Bennion, S. Ham, and V. Daggett. 2002. Increasing temperature accelerates protein unfolding without changing the pathway of unfolding. J. Mol. Biol. 322:189–203. [DOI] [PubMed] [Google Scholar]
- 6.Daura, X., B. Jaun, D. Seebach, W. F. van Gunstreen, and A. E. Mark. 1998. Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 280:925–932. [DOI] [PubMed] [Google Scholar]
- 7.Kazmirski, S. L., and V. Daggett. 1998. Non-native interactions in protein folding intermediates: molecular dynamics simulation of hen lysozyme. J. Mol. Biol. 284:793–806. [DOI] [PubMed] [Google Scholar]
- 8.Mark, A. E., and W. F. van Gunsteren. 1992. Simulation of the thermal denaturation of hen egg white lysozyme: trapping the molten globule state. Biochemistry. 31:7745–7748. [DOI] [PubMed] [Google Scholar]
- 9.Hunenberger, P. H., A. E. Mark, and W. F. van Gunsteren. 1995. Computational approaches to study protein unfolding: hen egg white lysozyme as a case study. Proteins. 21:196–213. [DOI] [PubMed] [Google Scholar]
- 10.Radkiewicz, J. L., and C. L. Brooks III. 2000. Protein dynamics in enzymatic catalysis exploration of dihydrofolate reductase. J. Am. Chem. Soc. 122:225–231. [Google Scholar]
- 11.Vijayakumar, S., S. Vishveshwara, G. Ravishanker, and D. L. Beveridge. 1993. Differential stability of β-sheets and α-helices in β-lactamase: a high temperature molecular dynamics study of unfolding intermediates. Biophys. J. 65:2304–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daggett, V., and M. Levitt. 1992. A model of the molten globule state from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 89:5142–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daggett, V., and M. Levitt. 1993. Protein unfolding pathways explored through molecular dynamics simulations. J. Mol. Biol. 232:600–619. [DOI] [PubMed] [Google Scholar]
- 14.Daggett, V. 1993. A model for the molten globule state of CTF generated using molecular dynamics. In Techniques in Protein Chemistry IV. R. H. Angeletti, editor. Academic Press, San Diego. 525–532.
- 15.Caflisch, A., and M. Karplus. 1994. Molecular dynamics simulation of protein denaturation: solvation of the hydrophobic cores and secondary structure of barnase. Proc. Natl. Acad. Sci. USA. 91:1746–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caflisch, A., and M. Karplus. 1995. Acid and thermal denaturation of barnase investigated by molecular dynamics simulations. J. Mol. Biol. 252:672–708. [DOI] [PubMed] [Google Scholar]
- 17.Dastidar, S. G., and C. Mukhopadhyay. 2005. Unfolding dynamics of the protein ubiquitin: insight from simulation. Phys. Rev. E. 72:51928. [DOI] [PubMed] [Google Scholar]
- 18.Barabasi, A. L. 2002. Linked: The New Science of Networks. Perseus Publishing, Cambridge, MA.
- 19.Dokholyan, N. V., B. Shakhnovich, and E. I. Shakhnovich. 2002. Expanding protein universe and its origin from the biological Big Bang. Proc. Natl. Acad. Sci. USA. 99:14132–14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dokholyan, N. V., L. Li, F. Ding, and E. I. Shakhnovich. 2002. Topological determinants of protein folding. Proc. Natl. Acad. Sci. USA. 99:8637–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vendruscolo, M., N. V. Dokholyan, E. Paci, and M. Karplus. 2002. Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E. 65:061910. [DOI] [PubMed] [Google Scholar]
- 22.Vendruscolo, M., E. Paci, C. M. Dobson, and M. Karplus. 2001. Three key residues form a critical contact network in a protein folding transition state. Nature. 409:641–645. [DOI] [PubMed] [Google Scholar]
- 23.Atilgan, A. R., P. Akan, and C. Baysal. 2004. Small-world communication of residues and significance for protein dynamics. Biophys. J. 86:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene, L. H., and V. A. Higman. 2003. Uncovering network systems with protein structures. J. Mol. Biol. 334:781–791. [DOI] [PubMed] [Google Scholar]
- 25.Rao, F., and A. Caflisch. 2004. The protein folding network. . J. Mol. Biol. 342:299–306. [DOI] [PubMed] [Google Scholar]
- 26.Hubner, I. A., E. J. Deeds, and E. I. Shakhnovich. 2005. High-resolution protein folding with a transferable potential. Proc. Natl. Acad. Sci. USA. 102:18914–18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brinda, K. V., and S. Vishveshwara. 2005. A network representation of protein structures: implications for protein stability. Biophys. J. 89:4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Case, D. A., T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, B. Wang, D. A. Pearlman, M. Crowley, S. Brozell, V. Tsui, H. Gohlke, J. Mongan, V. Hornak, G. Cui, P. Beroza, C. Schafmeister, J. W. Caldwell, W. S. Ross, and P. Kollman. 2004. AMBER 8. University of California, San Francisco.
- 29.Cheatham, T. E. III, P. Cieplak, and P. A. Kollman. 2002. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16:845–861. [DOI] [PubMed] [Google Scholar]
- 30.Weaver, L. H., and B. W. Matthews. 1987. Structure of bacteriophage T4 lysozyme refined at 1.7 Å resolution. . J. Mol. Biol. 193:189–199. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 32.Darden, T. A., D. M. York, and L. G. Pedersen. 1993. Particle mesh Ewald: an N log (N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 33.Creighton, T. E. 1996. Proteins: Structures and Molecular Properties, 2nd ed. W. H. Freeman, New York.
- 34.Kannan, N., and S. Vishveshwara. 1999. Identification of side-chain clusters in protein structures by a graph spectral method. J. Mol. Biol. 292:441–464. [DOI] [PubMed] [Google Scholar]
- 35.West, D. B. 2000. Introduction to Graph Theory, 2nd ed. Prentice Hall, Englewood Cliffs, NJ.
- 36.Stauffer, D. 1985. Introduction to Percolation Theory. Taylor and Francis, London.
- 37.Clementi, C., A. E. Garcia, and J. N. Onuchic. 2003. Interplay among tertiary contacts, secondary structure formation and side-chain packing in the protein folding mechanism: all-atom representation study of protein L. J. Mol. Biol. 326:933–954. [DOI] [PubMed] [Google Scholar]
- 38.Li, A., and V. Daggett. 1996. Identification and characterization of the unfolding transition state of chymotrypsin inhibitor 2 by molecular dynamics simulations. J. Mol. Biol. 257:412–429. [DOI] [PubMed] [Google Scholar]
- 39.DeLano, W. L. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA. http://www.pymol.org
- 40.Llinas, M., and S. Marqusee. 1998. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 7:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobson, C. M., P. A. Evans, and S. E. Radford. 1994. Understanding how proteins fold: the lysozyme story so far. Trends Biochem. Sci. 19:31–37. [DOI] [PubMed] [Google Scholar]
- 42.Bryngelson, J. D., J. N. Onuchic, N. D. Socci, and P. G. Wolynes. 1995. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 21:167–195. [DOI] [PubMed] [Google Scholar]
- 43.Rose, G. D., P. J. Fleming, J. R. Banavar, and A. Maritan. 2006. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA. 103:16623–16633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karplus, M., and D. L. Weaver. 1994. Protein folding dynamics: the diffusion-collision model and experimental data. Protein Sci. 3:650–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fersht, A. R. 1995. Optimization of rates of protein folding: the nucleation-condensation mechanism and its implications. Proc. Natl. Acad. Sci. USA. 92:10869–10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White, G. W., S. Gianni, J. G. Grossmann, P. Jemth, A. R. Fersht, and V. Daggett. 2005. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation condensation to framework mechanism of folding. J. Mol. Biol. 350:757–775. [DOI] [PubMed] [Google Scholar]
- 47.Brinda, K. V., and S. Vishveshwara. 2005. Oligomeric protein structure networks: insights into protein-protein interactions. BMC Bioinformatics. 6:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Llinas, M., B. Gillespie, F. W. Dahlquist, and S. Marqusee. 1999. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Biol. 6:1072–1078. [DOI] [PubMed] [Google Scholar]
- 49.Eriksson, A. E., W. A. Baase, X. J. Zhang, D. W. Heinz, M. Blaber, E. P. Baldwin, and B. W. Matthews. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 255:178–183. [DOI] [PubMed] [Google Scholar]
- 50.Heinz, D. W., W. A. Baase, and B. W. Matthews. 1992. Folding and function of a T4 lysozyme containing 10 consecutive alanines illustrate the redundancy of information in an amino acid sequence. Proc. Natl. Acad. Sci. USA. 89:3751–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hurley, J. H., W. A. Baase, and B. W. Matthews. 1992. Design and structural analysis of alternative hydrophobic core packing arrangements in bacteriophage T4 lysozyme. J. Mol. Biol. 224:1143–1159. [DOI] [PubMed] [Google Scholar]
- 52.Baldwin, E., J. Xu, O. Hajiseyedjavadi, W. A. Baase, and B. W. Matthews. 1996. Thermodynamic and structural compensation in “size-switch” core repacking variants of bacteriophage T4 lysozyme. J. Mol. Biol. 259:542–559. [DOI] [PubMed] [Google Scholar]
- 53.Zhang, X. J., W. A. Baase, and B. W. Matthews. 1992. Multiple alanine replacements within α-helix 126–134 of T4 lysozyme have independent, additive effects on both structure and stability. Protein Sci. 1:761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dao-pin, S., D. E. Anderson, W. A. Baase, F. W. Dahlquist, and B. W. Matthews. 1991. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry. 30:11521–11529. [DOI] [PubMed] [Google Scholar]
- 55.Blaber, M., J. D. Lindstrom, J. Xu, N. Gassner, D. W. Heinz, and B. W. Matthews. 1993. Energetic cost and structural consequences of burying a hydroxyl group within the core of a protein determined from Ala → Ser and Val → Thr substitutions in T4 lysozyme. Biochemistry. 32:11363–11373. [DOI] [PubMed] [Google Scholar]
- 56.Balber, M., W. A. Baase, N. Gassner, and B. W. Matthews. 1995. Alanine scanning mutagenesis of the α-helix 115–123 of phage T4 lysozyme: effects on structure, stability and the binding of solvent. J. Mol. Biol. 246:317–330. [DOI] [PubMed] [Google Scholar]
- 57.Gassner, N. C., W. A. Baase, and B. W. Matthews. 1996. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc. Natl. Acad. Sci. USA. 93:12155–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elwell, M. L., and J. A. Schellman. 1977. Stability of phage T4 lysozyme. I. Native properties and thermal stability of wild type and two mutant lysozymes. Biochim. Biophys. Acta. 494:367–383. [DOI] [PubMed] [Google Scholar]