

Abstract
An implicit-membrane representation based on the generalized Born theory of solvation has been developed. The method was parameterized against the water-to-cyclohexane insertion free energies of hydrophobic side-chain analogs. Subsequently, the membrane was compared with experimental data from translocon inserted polypeptides and validated by comparison with an independent dataset of six membrane-associated peptides and eight integral membrane proteins of known structure and orientation. Comparison of the insertion energy of α-helical model peptides with the experimental values from the biological hydrophobicity scale of Hessa et al. gave a correlation of 93% with a mean unsigned error of 0.64 kcal/mol, when charged residues were ignored. The membrane insertion energy was found to be dependent on residue position. This effect is particularly pronounced for charged and polar residues, which strongly prefer interfacial locations. All integral membrane proteins investigated orient and insert correctly into the implicit-membrane model. Remarkably, the membrane model correctly predicts a partially inserted configuration for the monotopic membrane protein cyclooxygenase, matching experimental and theoretical predictions. To test the applicability and usefulness of the implicit-membrane method, molecular simulations of influenza A M2 as well as the glycophorin A dimer were performed. Both systems remain structurally stable and integrated into the membrane.
INTRODUCTION
Estimates indicate that 20–30% of the human genome encodes membrane proteins (1–3). Even though the majority of current drug targets are membrane proteins such as receptors and ion channels, only 113 unique membrane protein structures are known at present (4) (August 2006, an up-to-date summary is provided by White: http://blanco.biomol.uci.edu/). The scarcity of structural data is mainly a result of substantial difficulties with overexpression and crystallization of membrane proteins (5). Due to their pharmacological role as major drug targets, there has been an increasing interest in detailed structural data as well as realistic simulation methodologies. Together with the extremely high computational cost of molecular mechanics simulations using explicit lipid-bilayer membranes (6–9), this has led to the development of a variety of implicit-membrane representations, which we briefly summarize. Knowledge-based methods include energy terms derived from a library of known protein sequences and structures (e.g., 10–12) (see also Hurwitz et al. (13) for an up-to-date review). Coarse-grained or off-lattice models have been successfully employed to study the insertion process of a variety of membrane peptides (14–16). The peptides are modeled as a linked chain of hard spheres, each representing a residue, whereas the membrane and surrounding aqueous phase are modeled by properties depending only on the membrane normal, e.g., fractional water content, polarity, and hydrophobicity. If parameterized well, this type of coarse-grained model can yield excellent results. However, these models have several limitations compared to all-atom models. 1), The potential functions used are not based on classical force fields and hence lack the extensive testing, accuracy, and versatility of the latter. 2), Peptide-peptide interactions are difficult to model correctly in the absence of side-chain atoms. 3), Complex membrane-protein functionality, like ions moving through channel pores, cannot be modeled using such simplified models.
More rigorous approaches combine standard all-atom force fields with implicit-solvent energy terms. This can be achieved by modeling both the aqueous solvent and lipid phase as a lattice of Langevin dipoles mimicking the spatial polarization of the protein environment (17). Other studies have applied distance-dependent dielectrics, as well as Gaussian screening functions to model both membrane and aqueous solvation (18). Another approach builds on atomic solvation parameter methods, which model the solvation terms as an effective surface tension multiplied by the accessible peptide surface area (19,20). More accurate is the use of the Poisson-Boltzmann equation in determining the electrostatic free energy of solvation, but its use in molecular dynamics simulations is prohibitively slow (21,22). A further overview on the large number of present and future applications of implicit-membrane models is given in recent reviews (23,24).
A much faster method, with results comparable to Poisson Boltzmann, is the generalized Born algorithm. This algorithm treats the solvent as a dielectric continuum. For spherical ions in a homogeneous isotropic dielectric, the solvation energy can be determined analytically as demonstrated by Born (25). The generalized Born solvation model extends this equation to macromolecules, which are approximated as an assortment of charged spheres (26). The immense success of this method (25,26) in globular protein and peptide folding simulations (27–31) has spurred attempts to apply the generalized Born formalism to represent the membrane environment implicitly (32–34). These models describe the membrane environment as a uniform hydrophobic slab and have been used successfully to fold and assemble small helical membrane peptides (34,35).
This study was motivated by the need for a computationally efficient implicit-membrane model, which will allow simulation of large systems and long timescales while still being accurate enough to compare reasonably well with experimental data. Statistical analyses of membrane proteins (12) as well as translocon-mediated insertion experiments of designed polypeptides (36,37) suggest that the insertion energy of a residue at a certain position along the membrane normal is a property of its local solvation environment. It therefore seems reasonable to model a membrane as a smoothly varying “solvation function” along the bilayer normal. In this study, the membrane was treated as a region that becomes increasingly apolar (i.e., increasingly inaccessible to the solvent) toward the center of the membrane. However, rather than changing the dielectric constant as a function of the membrane normal, the self-solvation energy of an atom, which accounts for the largest part of the solvation energy (38), was modified to vary smoothly between full solvation in bulk water and a limiting value for burial at the center of the membrane. At this stage of development of the method, any increased polarity at the charged bilayer interfaces was neglected. Nevertheless, most of the experimental results could be reasonably well reproduced, suggesting that the solvent exclusion properties of the membrane model account for the bulk properties of a lipid bilayer. A general problem of membrane-protein simulation lies in the correct treatment of charged residues at the interfaces. In nature, burial of a charged residue inside a membrane will almost certainly involve a change of protonation state or accompaniment by a hydration shell. Here, the charged interfaces might play an important role; however, this is beyond the means of the model described here.
The membrane model was parameterized to match the water-to-cyclohexane transfer free energies of the side-chain analogs of leucine, isoleucine, and valine (39). The model was compared to the apparent free energies of a set of translocon-inserted helices (36), the octanol and interfacial scales of Wimley and White (40,41), and cyclohexane-water transfer free energies of the remaining side-chain analogs, as well as to a recent study, that derived an implicit-membrane representation from the distributions of amino acids along the membrane normal (12). Subsequently, the membrane model was tested against a range of proteins with different membrane association and topology: 1), Transmembrane (TM) helices whose tilt and rotation angles have been determined using solid-state NMR methods in oriented lipid bilayer membranes (42); 2), a set of antimicrobial peptides known to bind to the membrane surface; 3), a set of integral membrane proteins with regular as well as irregular structures; 4), a monotopic membrane protein, which integrates only partially into the cytoplasmic leaflet of a lipid bilayer. Finally, the feasibility of the membrane model for protein dynamics simulation was confirmed by performing several simulations using both a Monte Carlo concerted backbone rotation method (27) and molecular dynamics.
METHODS
The generalized Born method
The present generalized Born membrane is based on a method developed by Spassov et al. (33), in which the membrane is modeled as a planar dielectric slab with thickness 2L, surrounded by a uniform polar solvent with a dielectric constant ɛw = 80. Both the protein interior and the slab are assumed to have the same interior dielectric constant of ɛm = 2. The generalized Born method has been described in detail in a recent review (43). The Born radii are calculated using the fast asymptotic pairwise summation of Qiu and Still (44), which has been demonstrated to yield excellent results in predicting experimental free energies of solvation as well as hydration effects on conformational equilibria (45). Spassov et al. (33) modeled the membrane by restricting the pairwise summation to solute atoms outside of the membrane. This was done by switching the volumes Vi(zi) of atoms i inside the membrane to zero at the interfaces z = ±L. The contribution due to the membrane was modeled using an exponential switching function G(zi, L) that changes the self-solvation term of each atom between a buried and a solvated state at the interfaces. Thus, the solvation energy for each atom is given by
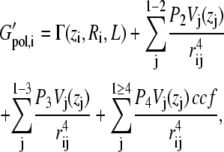 |
(1) |
where P1–P4 are the parameters determined by Qiu et al. (44), and ccf is a close contact function. G(zi, L) was obtained by fitting a smooth function to the polarization energy of a singly charged ion, determined by a Poisson-Boltzmann solver, as it is moved through the dielectric slab.
A real membrane, however, is not a hydrophobic slab with a uniform dielectric constant, but rather a heterogeneous medium with a highly nonuniform distribution of charge, density, and polarizable solvent. In this study, the membrane was therefore treated as a region that becomes increasingly apolar (i.e., increasingly inaccessible to the solvent) toward the center of the membrane. The self-solvation terms G(zi, L), as well as the atomic volumes V(zi), were modified to vary smoothly between full solvation and a limiting value for burial at the center of the membrane. In this work, we propose a Gaussian shape
 |
(2) |
where gbulk is the limiting value of Γ at a large distance from the membrane (i.e., z ≫ L) corresponding to the self-solvation term of the unmodified generalized Born method 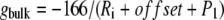 , while gcenter is the value of Γ at the membrane center.
, while gcenter is the value of Γ at the membrane center.
The nonpolar part of the solvation free energy is modeled using an effective surface tension associated with the solvent-accessible surface area (SASA) (44). Instead of a costly calculation of the accurate surface area, a mimic based on the Born radii is used, which has been shown to be very accurate, but much faster (46). As it is moved toward the center of the membrane, the surface energy contribution of each atom is varied using a Gaussian function: For distances far from the membrane (i.e., z ≫ L), the nonpolar contribution is included with the positive surface tension of solvation in water, whereas in the center of the membrane the surface tension is negative (i.e., energy is gained by moving into this phase from the gas phase), as determined experimentally (39). Gaussians were chosen in good agreement with experimental evidence from lipid distortion (47,48), x-ray and neutron diffraction experiments on fluid liquid-crystalline bilayers (49–51), and partitioning experiments on hexane (52) in lipid bilayers.
It should be noted that the present membrane model neglects any effects due to differences in lipid composition and charge distribution of the two bilayer leaflets, as well as effects due to the transmembrane voltage. However, it is in principle possible to include these properties by replacing the Gaussians with an equivalent nonsymmetric function.
Parameterization of the Born membrane
There are several adjustable parameters in the present generalized Born implicit-membrane representation. The first is the width of the membrane, given by the width of the Gaussian curve describing the self-solvation energy term in the Qiu and Still formula (44) (Eq. 1). We assumed a Gaussian with γ = −3.0 (also used for the volume term V(z)) and a membrane half-width of L = 15 Å (Fig. 1 A). The second adjustable parameter is the energy of transferring an isolated unit charge from vacuum to the center of the membrane, corresponding to the height of the Gaussian self-term gcenter. This value was set to gcenter = −7.67 kcal/mol, as reported in other studies (33,53). The surface tension contribution of each atom was varied as described above using a Gaussian function with γ = −1.5, interpolating between the limiting values of 12 cal/mol·Å2 in the surrounding medium and −19 cal/mol·Å2 in the membrane. These values were determined from a fit of the membrane insertion energy of leucine (−4.85 kcal/mol), isoleucine (−4.99 kcal/mol), and valine (−4.63 kcal/mol) side-chain analogs to the experimental water-to-cyclohexane transfer free energies (39) (see Table 2). Since these analogs are virtually neutral their experimental cyclohexane-water transfer free energies can be assumed to be only dependent on nonpolar interactions, thus providing an estimate of the effective surface tension of the membrane. The mean error for the optimal fit with respect to the experimental values was 0.24 ± 0.30 kcal/mol. Since the OPLS all-atom force field partial charges of these analogs are virtually zero the polar contribution of the implicit membrane was <0.2 kcal/mol for these analogs.
FIGURE 1.

(A) Exponential (solid) and Gaussian functions with γ = −1.5 (dashed) indicating the attractive nonpolar term, and γ = −3 (dotted) indicating the repulsive generalized Born term. (B) Calculated insertion energy of the LnA(19−n) set of designed peptides together with the experimental best-fit line (EInsertion = −0.66nLeu + 2.14). The experimental error is estimated at ±0.2–0.3 kcal/mol (36). Sequences of the symmetrical peptides used for residue-pair scans are shown below the graph. Results for the positional dependency of the insertion energy as determined by symmetrical scans of leucine pairs are shown as points in the graph. Results for the remaining scans are given in Fig. 4.
TABLE 2.
Bulk-solvent-to-membrane-center transfer free energies of the 20 natural amino acids integrated into an α-helical conformation compared with the biological scale and a statistical membrane model
| Residue transfer energies (kcal/mol)
| ||||
|---|---|---|---|---|
| Residue | Generalized Born membrane | Biological scale | Statistical membrane | Water to cyclohexane |
| ALA | 0.11 | 0.11 | −0.42 | −1.81 |
| ASN | 3.55 | 2.05 | 0.90 | 6.64 |
| CYS | 0.25 | −0.13 | — | −1.28 |
| GLN | 3.39 | 2.36 | 1.21 | 5.54 |
| GLY | 0.58 | 0.74 | −0.30 | −0.94 |
| HIS | 3.33 | 2.06 | −0.06 | 4.66 |
| ILE | −0.87 | −0.60 | −0.68 | −4.92 |
| LEU | −1.21 | −0.55 | −0.64 | −4.92 |
| MET | 0.38 | −0.10 | −0.55 | −2.35 |
| PHE | −0.81 | −0.32 | −0.83 | −2.98 |
| PRO | 1.93 | 2.23 | 0.65 | — |
| SER | 2.08 | 0.84 | 0.00 | 3.40 |
| THR | 1.71 | 0.52 | 0.00 | 2.57 |
| TRP | 1.12 | 0.30 | −0.18 | −2.33 |
| TYR | 0.84 | 0.68 | −0.05 | 0.14 |
| VAL | −0.59 | −0.31 | −0.58 | −4.04 |
| ARG | 11.42 | 2.58 | 2.02 | 14.92 |
| ASP | 23.61 | 3.49 | 2.19 | 8.72 |
| GLU | 27.23 | 2.68 | 2.11 | 6.81 |
| LYS | 20.85 | 2.71 | 1.83 | 5.55 |
Experimental water to cyclohexane transfer free energies of side chain analogs are also shown.
Translocon-inserted polypeptides
Hessa et al. (36) challenged the endoplasmic reticulum translocon Sec61 with an extensive set of designed polypeptide segments using an in vitro assay to measure the efficiency of membrane integration (54). The peptides have the general design GGPG-X19-GPGG, where the GGPG flanks serve to insulate the central 19-residue stretch from the surrounding sequence by having a low probability of secondary structure formation. In this study, all peptides were modeled as perfect α-helices with extended GGPG flanking segments. Preequilibrated side-chain conformations were used and the segment termini were acetylated (C-terminus) and methylated (N-terminus).
Calculating the minimal energy conformation
The minimal energy conformation was calculated by exploring the entire translational and rotational space of the peptide in the membrane. The principal axis of the protein was determined through diagonalization of the inertia tensor using only the heavy backbone atoms. The tilt angle was defined as the angle of the principal axis with respect to the membrane normal, whereas the rotation angle was defined as the angle of rotation around the principal axis.
The helix was translated from −50 Å to +50 Å along the membrane normal (membrane center = 0 Å) in 0.5 Å steps. At each step, the protein was rotated through all space to find the orientation of minimum energy by first tilting it with respect to the membrane normal and subsequently rotating it around its principal axis until all tilt and rotational states had been sampled with a step size of 1°. The lowest energy conformation encountered was then subjected to a rigid-body minimization to ascertain the precise location of the global energy minimum.
Experimental structures
The implicit-membrane model was tested on a range of experimental structures with different membrane topologies (see Table 1): The M2 channel segment of the δ-subunit of the nicotinic acetylcholine receptor (AchR) (1cek) (55), influenza A M2 (1mp6) (56), and the membrane conformation of FD coat protein (1mzt) (57) are single membrane-spanning α-helices. Their tilt angles are known from solid-state NMR measurements in aligned lipid bilayers (42). Since the solid-state NMR structure of the AchR M2 (1cek) lacks the charged residues at the termini, the equivalent solution NMR structures (55) (1a11) were used, which have the complete sequence. The structures of the antimicrobial peptides magainin (2mag) (58), ovispirin (1hu5) (59), and a cecropin-magainin hybrid (1f0d) (60) were determined by solution NMR in micelles. These peptides are generally believed to form amphipathic α-helices oriented parallel to the membrane in a surface-bound fashion (61–64). The transmembrane helix dimer glycophorin A (1afo) (65) has been determined by solution NMR. The relatively regular integral membrane proteins bacteriorhodopsin (1cwq) (66) and sensory rhodopsin (1h68) (67), as well as the more irregular aquaporin (1j4n, 1rc2) (68,69) and chloride channel structures (1kpk, 1kpl) (70) were all determined by x-ray crystallography. The membrane model was also challenged with the monotopic membrane protein cyclooxygenase 2 (1cx2) (71), which is known to insert only partially into one monolayer of a membrane.
TABLE 1.
Minimum energy orientations for membrane proteins of known conformation
| Calculated minimum energy orientation
| |||||
|---|---|---|---|---|---|
| PDB | Protein | Organism | Tilt angle (°) | z (Å) | ΔE (kcal/mol) |
| 1afo | Glycophorin A | Human red blood cell | 14 | 3.7 | −70.7 |
| 1cwq | Bacteriorhodopsin | Halobacterium salinarium | 30 | 1 | −29.9 |
| 1h68 | Sensory rhodopsin II | Natronomonas pharaonis | 3 | 0.7 | −86.6 |
| 1j4n | Aquaporin | Bovine red blood cell | 14 | 0.3 | −83.2 |
| 1rc2 | Aquaporin | Escherichia coli | 8 | 0.7 | −108.1 |
| 1kpk | Chloride channel homologue | E. coli | 3 | 3 | −90.5 |
| 1kpl | Chloride channel homologue | Salmonella typhimurium | 3 | 3.1 | −82.6 |
| 1cx2 | Cyclooxygenase-2 | Mouse | 0 | 36 | −95.3 |
The protein tilt angle, displacement of the center of mass with respect to the membrane center (z), and insertion energy (ΔE) are listed for all systems. The membrane center is located at z = 0 and negative z values correspond to shifts toward the extracellular side, whereas positive z values correspond to shifts toward the cytoplasm. For 1afo, 1j4n, 1rc2, 1kpk, 1kpl, and 1cx2, the tilt angle is given with respect to the multimer.
Molecular simulations
The simulations were run with a Monte Carlo program developed by the authors specifically for the simulation of membrane proteins in a GB/SA continuum solvent. An efficient concerted rotation sampling technique (72) is used to move the protein backbone; in addition, there are single rapid side-chain moves, with a side-chain move/backbone move ratio of 3. This method has been demonstrated to be at least as efficient as molecular dynamics sampling (73). The potential energy was evaluated with the OPLS-AA force field (74), and the Monte Carlo simulations used Metropolis sampling at a temperature of 300 K. All nonbonded interactions as well as the generalized Born energy were truncated using a cutoff of 14 Å, and the Born radii were recomputed for every configuration. The described setup has been shown to perform well in sampling DNA (75) and protein-folding simulations (27), where the native state of several polypeptides was rapidly determined starting from extended conformations. Here we report the first application of this method to the simulation of membrane proteins in implicit solvation.
Molecular dynamics simulations were performed with the GROMACS software package (76), modified to include the implicit-membrane model described above. Fixed bond lengths (77) and a time step of 2 fs for numerical integration were used. The simulation conditions and parameters were the same as for the Monte Carlo simulations and comparison of a test trajectory showed both methods to sample the same potential function.
RESULTS AND DISCUSSION
Correlation with experimental scales
The biological hydrophobicity scale was derived by calculating the insertion energies for the 19-residue helical model peptides and replacing the central residue with all 20 natural amino acids. For each peptide, the insertion energy was calculated by performing transrotational minimum-energy search in the membrane. All peptides except those with charged amino acids were found to have local minima for fully inserted configurations (z = ±2.5 Å) with small tilt angles (<10°). In their experimental derivation, Hessa et al. (36) varied the number of Leu residues in the 19-residue segment to obtain apparent free energies of insertion in the range −1.2 to 1.2 kcal/mol, since the experimental measurement is maximally sensitive at this range. Using the insertion energies of these peptides, the biological scale can subsequently be computed in a stepwise procedure (see Supplementary Material). In principle, this derivation is equivalent to replacing the central residue X of the GGPG-AAAALAAAAXAAAALAAAA-GPGG peptides with all 20 natural amino acids and subtracting the energy for X = A. Indeed, for this membrane model both methods give almost identical values and the final scale was calculated as their average (see Supplementary Material). Both the experimental and computed scales were made to coincide for alanine (see Positional dependency below).
Fig. 2 compares the computationally derived scale with the experimental apparent free energies of the biological hydrophobicity scale (36). Considering the simplicity of the membrane model, both scales correlate remarkably well (C = 93%). The linear fit has a slope of 0.6 and the scales have identical origins (rsquare = 0.86). Comparison with a statistical scale derived from membrane protein structures (12) gave a correlation of 87%, an offset of −0.41 kcal/mol and a slope of 0.32 (rsquare = 0.73). The calculated scale also correlates remarkably well (C = 97%) with experimental transfer free energies of side-chain analogs into cyclohexane (39). On the other hand, Fig. 3 shows that there is little to no correlation with both the octanol (40) and interface scales (41). The individual energies are reported in Table 2.
FIGURE 2.

(A) Calculated insertion energy of the designed peptides versus experimental transfer free energies of side-chain analogs from water into cyclohexane. (B) Insertion energy of the designed peptides: generalized Born membrane versus the biological hydrophobicity scale (36). (C) Calculated insertion energy of the designed peptides against insertion potentials from a statistical membrane potential (12).
FIGURE 3.

(A) Calculated insertion energy of the designed peptides against the Wimley and White interfacial scale (41). (B) Calculated insertion energy of the designed peptides against the Wimley and White octanol scale (40).
Charged residues were excluded from the scale since their insertion energies are considerably overestimated by the generalized Born membrane. The energy penalties for the burial of charged residues at the center of the membrane are much higher than the corresponding values from the biological scale (23.6 kcal/mol for Asp, 11.4 kcal/mol for Arg, 27.2 kcal/mol for Glu, and 20.9 kcal/mol for Lys). Upon burial in the hydrophobic membrane core, a charged residue is likely to be either neutralized or accompanied by a shell of water molecules, which will lower its insertion energy significantly. In fact, there is no energy penalty for burial of charged amino acids with neutralized side chains (see Supplementary Material). However, simulation of ionization-state changes or the inclusion of a hydration shell, although both possible in principle, is beyond the means of the simple membrane model proposed here.
Comparison of the insertion free energies for peptides in which either one or two Ala residues have been changed to Gly, Ser, Trp, or Tyr demonstrated that the biological scale was approximately additive (36). This property was also found for insertion of the same peptides into the generalized Born membrane (see Supplementary Material).
In the experiment, lengthening of the flanking stretches from GGPG-X19-GPGG to GGGGGGPG-X19-GPGGGGGG through the stepwise addition of Gly residues resulted in variations to the apparent free energy of no more than ±0.2 kcal/mol, demonstrating that residues outside the H-segment have little influence on the results. The corresponding results for the generalized Born membrane found no measurable change to the insertion energy (see Supplementary Material). However, replacing the GGPG-X19-GPGG flanks with NNPN-X19-NPNN resulted in a 1.1-kcal/mol increase in insertion energy. The experiment found a more measured increase of 0.5 kcal/mol.
Positional dependency
To study the effect of amino acid position on the insertion energy of the designed peptides, Hessa et al. (36) performed symmetrical scans in which a pair of identical amino acids was moved from the center of the peptide toward the carboxy and amino terminus, respectively. These scans were repeated in the generalized Born membrane by building α-helical models of all peptide segments synthesized in the experiments and performing an orientational minima search in the membrane (see Methods). For each system, a Monte Carlo simulation was performed to yield the average conformational energies both in bulk solvent (z = ∞) and for the completely inserted transmembrane configuration (z = 0). The backbone was kept fixed while the side chains were sampled with flexible bonds, angles, and dihedrals. The results generally converge after 20 million steps, with a 0.5-million-step equilibration period. In general, the insertion energy is fairly insensitive to the actual side chain conformation. The maximum fluctuation of the solvation energy for flexible side chains was found to be ∼1 kcal/mol.
Fig. 1 shows the results of symmetrical scans for pairs of Leu residues together with the experimental best fit. Since we assume helical structures for both the membrane-bound state as well as for a larger separation from the membrane, the theoretical model neglects the free energy of folding the hydrophobic helix in the solvent phase. Experimentally, the structure in the solvent phase is unknown, but it is almost certainly not a helix, given the strong hydrophobicity of that structure. We found a constant ΔG = 0.6 kcal/mol per residue, which includes the free energy of folding the system in water as well as the free energy cost of immobilizing the helix in the membrane, as determined from the best fit of the experimental and computational data. Despite the uncertainty of the experimental structures, which in principle limits the accuracy of this study, both computational and experimental results can be seen to correlate remarkably well. However, although the experiment shows little to no positional dependency for Leucine the computational results found them to be slightly more favorable at the center of the membrane. This is a reflection of the fact that for hydrophobic residues the insertion energy is dominated by the nonpolar SASA term, which has the shape of a Gaussian centered on the membrane core (see The generalized Born method). Phe residue scans show a very similar behavior (Fig. 4).
FIGURE 4.

Comparison of the calculated (squares) and experimental (diamonds) positional dependency on the insertion energy for residues Asn, Lys, Phe, Ser, Trp, and Tyr. Note that the graph for Lys has two different scales for the calculated (left) and experimental (right) results due to the overestimation of burial of charged residues in the membrane by the generalized Born method (see text). The zero potential lines are indicated. The upper left panel (SER) shows the average error bar of the calculations, which fluctuate around a mean of ∼1 kcal/mol.
As expected from theory and experimental data, the polar residues Asn and Ser both show a preference for positions near the helix termini (see Fig. 4), with a larger slope encountered than the experimental data. This is due to the uncertainty of the experimental structures, which we assume to be helices for computational feasibility.
Aromatic residues are known to prefer the interface transmembrane segments (78). In this analysis, Tyr and Trp do indeed show a slight preference to locate near the interfaces. However, the penalty for inserting Trp is slightly lower in the experiments. Trp is the largest amino acid side chain and its desolvation in the membrane is likely to cause some disruption of the bilayer core, probably resulting in effects beyond those of an implicit-membrane model.
Fig. 4 shows that burial of Lys residues at the center of the membrane generally entails a large energy penalty, whereas they are favorable at the interfaces. This property matches the experimental and theoretical expectations. However, both the penalty of burial and the solvation energy at the interfaces is much larger compared to the experimental values. This is due to the general problem relating to the simulation of ionizable residues (see above).
Transmembrane and surface-bound helices
As a first test of this model, the minimum energy conformation of a set of six TM helices and antimicrobial peptides was calculated. All TM helices inserted correctly, with insertion energies in the range 1–13 kcal/mol, whereas all antimicrobial peptides were found to occupy surface-bound conformations, in agreement with experimental observations (58,62,64,79). The results closely match those from a previously reported statistical membrane potential (80) and are summarized in Table 3.
TABLE 3.
Comparison of the experimental and computational results for membrane-associated helices and membrane proteins of known conformation
| Experimental and computed peptide orientations
| ||||||
|---|---|---|---|---|---|---|
| Experiment
|
Calculated
|
|||||
| PDB | Name | Helix residues | Tilt angle (°) | Tilt angle (°) | z (Å) | ΔE (kcal/mol) |
| 1a11 | Acetylcholine M2 (1cek) | 2–24 | 11 | 11 ± 5 | −1.0 ± 0.9 | −5.0 ± 0.9 |
| 1mp6 | Influenza A M2 | 23–45 | 37 | 25 | −1.1 | −12.8 |
| 1mzt | FD coat protein | 21–44 | 19 | 15 | −1.5 | −0.83 |
| 2mag | Magainin | 4–21 | ∼90 | 85 ± 4 | 17.6 ± 0.9 | −4.6 ± 0.8 |
| 1hu5 | Ovispirin 1 | 4–16 | ∼90 | 89 ± 12 | 18.1 ± 0.6 | −3.3 ± 0.4 |
| 1f0d | Magainin-cecropin hybrid | 11–17 | ∼90 | 89 ± 27 | 21.3 ± 2.0 | −2.3 ± 0.6 |
The residues defining the helical segment, as well as the experimental tilt angle, are listed. The helix tilt angle, displacement of the center of mass of the helix with respect to the membrane center (z), and insertion energy (ΔE) are listed for all systems. The experimental tilt angles were calculated from the aligned solid-state NMR structures deposited in the Protein Databank.
Integral membrane proteins
Fig. 5 shows the insertion profiles for the transmembrane section of the glycophorin A homodimer (1afo, model 12) (65), the integral membrane protein sensory rhodopsin (1h68) (67), and the more irregular structures of aquaporin from bovine red blood cell (1j4n) (68) and a chloride channel homolog from Escherichia coli (1kpk) (70). All proteins investigated have an energy minimum for inserted configurations with their center of geometry close to the core of the membrane and low tilt angles (see Table 1). Interestingly, all proteins exhibit similar energy profiles with an energy penalty for insertion from both membrane interfaces. In all cases, insertion from the extracellular side involves traversing a higher energy barrier than insertion from the cytoplasmic side. This is a reflection of the different topology of extra- and intracellular membrane protein interfaces. Generally, membrane proteins have a much larger number of charged residues at their intracellular interface (12,81), resulting in higher energy penalties for burial compared to the cytoplasmic side.
FIGURE 5.

(A) Global minimum energy conformation of cyclooxygenase 2 in the membrane. (B) Energy profiles obtained by pushing aligned proteins through the membrane. AQP, aquaporin from bovine red blood cell (1j4n); ClC, chloride channel homolog from E. coli (1kpk); SR, sensory rhodopsin (1h68); GpA, glycophorin A dimer (1afo, model 12); Cox 2, cyclooxygenase 2 (1cx2). The extracellular side is to the left. Except for Cox 2, all proteins can be seen to have a larger insertion penalty from the extracellular side, as well as a global insertion energy minimum at the membrane center. The insertion energy of Cox 2 is displayed on the y axis to the right, whereas all the other insertion energies are given on the left y axis.
Monotopic membrane protein
The monotopic membrane protein cyclooxygenase 2 (1cx2) is a homodimer that inserts partially into the cytoplasmic leaflet of the membrane and is a major target for anti-inflammatory drugs (e.g., Ibuprofen) (71). Fig. 5 shows that a fully inserted configuration carries an extremely high energy penalty, whereas the global energy minimum corresponds to a partially inserted conformation 36.0 Å from the membrane center. Interestingly, the statistical membrane potential found the same global insertion minimum orientation with the protein only slightly closer to the membrane center (33.5 Å). The tilt angle of the dimer with respect to the membrane normal was found to be 0°. Generally the orientation and partially inserted configuration closely match experimental and theoretical expectations. However, the relatively thin 30-Å membrane might overestimate the depth of insertion. Nevertheless, it is remarkable that both the statistical and generalized Born membrane models can predict the correct membrane orientation of such a complex system.
Insertion-energy landscape
To investigate the insertion-energy landscape for other, local minimum-energy orientations, the potential was plotted as a function of position along the membrane normal and tilt angle with the rotation angle optimized (i.e., the rotation angle for each position and tilt angle is such that the energy is minimal). Fig. 6 shows the resulting insertion energy landscapes for AchR M2 (1a11, model 1) and magainin (2mag, model 1). The zero point of the potential was chosen at an infinite distance from the membrane. AchR has four distinct minima, the two deepest corresponding to inserted configurations with the helices approximately parallel to the membrane normal. The other two minima are surface bound configurations with the helix axis parallel to the plane of the membrane. It should be noted that due to the symmetry of the membrane model, the cytoplasmic and intracellular minima have identical insertion energies, as do the two inserted minima. Repeating the analysis for all 10 AchR solution NMR structures gave topologically identical energy landscapes.
FIGURE 6.

Insertion energy landscape of a helix as a function of the tilt angle and center of mass position along the membrane normal for optimized rotation angle (around the long axis of the helix). The profile for the acetylcholine receptor helix M2 (1a11, model 1) is representative of all transmembrane helices investigated, displaying four distinct minima, whereas the profile for magainin (2mag, model 1) is typical of all the other antimicrobial peptides analyzed in this study with two surface-bound energy minima. The potential zero was chosen to be at an infinite distance from the membrane.
Generally, inserted TM configurations correspond to the global energy minima. The mean insertion energy (−5.0 ± 0.9 kcal/mol), tilt angle (12° ± 5°), and position close to the center of the membrane (−1.0 ± 0.9 Å) compare well to the values previously calculated for a statistical membrane potential (−4.7 ± 0.1 kcal/mol, 20° ± 5°, and 0.5 ± 0.2 Å) (80). Adsorption of the peptide onto the membrane surface is also favorable but to a lesser extent, with energy minima of −3.2 ± 0.3 kcal/mol. The minima correspond to helices bound to the interfacial membrane surface in a parallel orientation with tilt angles of 92° ± 10°. The equivalent values for the statistical membrane potential are −3.5 ± 0.4 kcal/mol and 92°±2°. At a distance of 18.5 ± 2.5 Å, the helix is farther away than the 9.3 ± 0.8 Å found previously. Nevertheless, the overall agreement between the two membrane models is remarkable. Both membranes support an insertion model where a helix will first bind to the membrane surface in a parallel orientation and subsequently change to a fully inserted transmembrane configuration. This is in accordance with general theoretical considerations (82) as well the two-stage folding model for α-helical membrane proteins (83,84). There is strong experimental evidence for independent helix formation and insertion in the first stage.
The above results are also in very good agreement with a theoretical study of the same structures (85,86), which found average energies of −4.7 ± 2.1 kcal/mol and −2.6 ± 2.4 kcal/mol for inserted and surface-bound configurations, respectively. The study used a theoretical continuum-solvent method developed by Ben-Tal (87) that has been successfully applied to estimate the insertion energies of TM peptides and proteins (88). To compare the results, the helix-coil transition free energy (ΔGcon = −2.4 kcal/mol) was subtracted, since the present data estimates the insertion energy of a folded helix.
For the antibiotic peptide magainin the free-energy landscape can be seen to differ substantially in topology. There are only two interfacial minima and the membrane region forms a large barrier spanning the entire tilt range of the helix with a 50 to 100 kcal/mol energy penalty for insertion into the membrane. The association depth (17.6 ± 0.9 Å), tilt angle (85° ± 4°), and insertion energy (−4.6 ± 0.8 kcal/mol) compare well with the statistical membrane model, which found values of 13 ± 1 Å, 87° ± 7°, and −3.5 ± 0.1 kcal/mol, respectively.
Conformational sensitivity
The purpose of an implicit-membrane representation is to enable significantly longer simulation times for the study of membrane proteins than are possible with an explicit lipid bilayer and solvent molecules. The present generalized Born membrane model is only a factor of ∼2 slower than a corresponding simulation in vacuum, whereas simulations using explicit lipid and solvent molecules are several orders of magnitude slower. To test the applicability of the method for modeling membrane proteins, 350 million Monte Carlo step simulations of the influenza A M2 helix (1mp6), as well as a 2.5 ns molecular dynamics simulation of the glycophorin A dimer (1afo), were performed. The simulations started from the experimental structures, with the helices inserted into the center of the membrane. The backbone root mean-square deviation (RMSD) with respect to the experimental structure over the course of the simulation was found to have an average of ∼1.8 ± 0.6 Å for influenza A and ∼1.8 ± 0.3 Å for glycophorin A. All helices remained firmly integrated near the center of the membrane in a transmembrane configuration (Fig. 7). Tilt angles of the influenza A helix with respect to the membrane normal fluctuate in the range 0–30° (average of 9° ± 5°) throughout the simulation and there is frequent bending and kinking. Fig. 7 shows the overlay of the glycophorin A dimer simulation at 0 and 2.5 ns, with an RMSD of 1.6 Å when the dimer is fitted as a whole. The system remains stable and the relative orientation of the helices is constant throughout the simulation. Both runs demonstrate that membrane systems can be reliably studied in an implicit membrane. Future work will concentrate on the role of the parameters on experimentally determined properties, such as tilt and kinking angles as well as helix-helix fluctuations.
FIGURE 7.

(A) Tilt angles of helices A and B, and center of mass position of the glycophorin A dimer simulation. (B) Initial global minimum energy configuration of the glycophorin A dimer TM domain (1afo, model 12) in black. The final structure at the end of the simulation (2.5 ns) is shown in gray. This model is representative of the 11 of 20 structures that were found to insert.
CONCLUSION
Translocon-mediated insertion experiments of designed polypeptides (36), as well as statistical analyses of membrane proteins (12), seem to indicate that the insertion properties of a transmembrane helix are determined chiefly by its solvation energy in the membrane environment. This study used the generalized Born method to derive an implicit-membrane representation based on the assumption that the bulk properties of a lipid bilayer can be captured by treating it as a region that becomes increasingly apolar (i.e., increasingly inaccessible to the solvent) toward the center of the membrane. The polar part of the limiting value of maximum desolvation at the center was taken from theoretical calculations (33,53), whereas the nonpolar part was determined by fitting the calculated insertion energies of hydrophobic side-chain analogs against those measured experimentally (39).
Subsequent comparison of the insertion energy of α-helical model peptides, containing the remaining 20 natural amino acids, with the experimental values from the biological hydrophobicity scale of Hessa et al. (36) gave a correlation of 93% with a mean unsigned error of 0.64 kcal/mol. The calculated scale also correlates remarkably well (C = 97%) with experimental transfer free energies of side-chain analogs into cyclohexane (39), though it displays little to no correlation with either the octanol (40) or the interface scale (41).
TM peptides of known orientation were found to insert and orient correctly, with an average tilt deviation of 6° from the NMR values. Antimicrobial peptides were found to orient parallel to the membrane surface, pointing their hydrophobic residues toward the membrane center while exposing their polar residues to the aqueous environment, in excellent agreement with experimental data. The free-energy landscapes showed that there is a large penalty for insertion into the membrane regardless of the tilt angle.
In general, the translational and rotational energy landscapes described represent a detailed search of the orientation space of the peptides considered. The insertion-energy surfaces are surprisingly smooth and the good overall agreement with theoretical and experimental data is encouraging. It should be noted that the insertion-energy landscapes are similar to those found for previously derived membrane potential based on 46 α-helical membrane protein structures (80).
All integral membrane proteins investigated orient and insert correctly into the implicit-membrane model, regardless of whether their structure is composed of a fairly regular arrangement of helices or not. One of the most remarkable findings of the study was that the membrane model predicts a partially inserted configuration for the monotopic membrane protein cyclooxygenase 2. The part inserted into the membrane agrees well with experimental and theoretical predictions.
Simulations of influenza A as well the glycophorin A dimer proved that both systems remain stable over the timescale of the simulations. These simulations run a factor of only ∼2 slower than the equivalent systems in vacuum, demonstrating the feasibility and applicability of the current method to large-scale membrane protein simulations.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Profs. Stephen White and Gunnar von Heijne for providing us with the experimental insertion data of the designed polypeptide segments.
M.B.U. is funded by an international fellowship of the Wellcome Trust. J.P.U. is funded by an Emmy Noether fellowship of the Deutsche Forschungs-gemeinschaft. Research in M.S.P.S.'s group is supported by the Wellcome Trust, the Medical Research Council, the Biotechnology and Biological Sciences Research Council, and the Engineering and Physical Sciences Research Council.
References
- 1.Krogh, A., B. Larsson, G. von Heijne, and E. L. Sonnhammer. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580. [DOI] [PubMed] [Google Scholar]
- 2.Wallin, E., and G. von Heijne. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7:1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones, D. T. 1998. Do transmembrane protein superfolds exist? FEBS Lett. 423:281–285. [DOI] [PubMed] [Google Scholar]
- 4.Terstappen, G. C., and A. Reggiani. 2001. In silico research in drug discovery. Trends Pharmacol. Sci. 22:23–26. [DOI] [PubMed] [Google Scholar]
- 5.Grisshammer, R., and C. G. Tate. 1995. Overexpression of integral membrane-proteins for structural studies. Q. Rev. Biophys. 28:315–422. [DOI] [PubMed] [Google Scholar]
- 6.Biggin, P. C., and M. S. P. Sansom. 1999. Interactions of α-helices with lipid bilayers: a review of simulation studies. Biophys. Chem. 76:161–183. [DOI] [PubMed] [Google Scholar]
- 7.Forrest, L. R., and M. S. P. Sansom. 2000. Membrane simulations: bigger and better? Curr. Opin. Struct. Biol. 10:174–181. [DOI] [PubMed] [Google Scholar]
- 8.Domene, C., P. Bond, and M. S. P. Sansom. 2003. Membrane protein simulation: ion channels and bacterial outer membrane proteins. Adv. Prot. Chem. 66:159–193. [DOI] [PubMed] [Google Scholar]
- 9.Nymeyer, H., T. B. Woolf, and A. E. Garcia. 2005. Folding is not required for bilayer insertion: replica exchange simulations of an α-helical peptide with an explicit lipid bilayer. Proteins. 59:783–790. [DOI] [PubMed] [Google Scholar]
- 10.Pellegrini-Calace, M., A. Carotti, and D. T. Jones. 2003. Folding in lipid membranes (FILM): a novel method for the prediction of small membrane protein 3D structures. Proteins. 50:537–545. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, Y., M. E. Devries, and J. Skolnick. 2006. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput. Biol. 2:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulmschneider, M. B., M. S. Sansom, and A. Di Nola. 2005. Properties of integral membrane protein structures: derivation of an implicit membrane potential. Proteins. 59:252–265. [DOI] [PubMed] [Google Scholar]
- 13.Hurwitz, N., M. Pellegrini-Calace, and D. T. Jones. 2006. Towards genome-scale structure prediction for transmembrane proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milik, M., and J. Skolnick. 1995. A Monte Carlo model of fd and Pf1 coat proteins in lipid membranes. Biophys. J. 69:1382–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maddox, M. W., and M. L. Longo. 2002. A Monte Carlo study of peptide insertion into lipid bilayers: equilibrium conformations and insertion mechanisms. Biophys. J. 82:244–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milik, M., and J. Skolnick. 1993. Insertion of peptide chains into lipid membranes: an off-lattice Monte Carlo dynamics model. Proteins. 15:10–25. [DOI] [PubMed] [Google Scholar]
- 17.Grossfield, A., J. Sachs, and T. B. Woolf. 2000. Dipole lattice membrane model for protein calculations. Proteins. 41:211–223. [DOI] [PubMed] [Google Scholar]
- 18.Lazaridis, T. 2003. Effective energy function for proteins in lipid membranes. Proteins. 52:176–192. [DOI] [PubMed] [Google Scholar]
- 19.Efremov, R. G., D. E. Nolde, G. Vergoten, and A. S. Arseniev. 1999. A solvent model for simulations of peptides in bilayers. I. Membrane-promoting α-helix formation. Biophys. J. 76:2448–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efremov, R. G., D. E. Nolde, G. Vergoten, and A. S. Arseniev. 1999. A solvent model for simulations of peptides in bilayers. II. Membrane-spanning α-helices. Biophys. J. 76:2460–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengupta, D., L. Meinhold, D. Langosch, G. M. Ullmann, and J. C. Smith. 2005. Understanding the energetics of helical peptide orientation in membranes. Proteins. 58:913–922. [DOI] [PubMed] [Google Scholar]
- 22.Ben-Tal, N., B. Honig, C. K. Bagdassarian, and A. Ben-Shaul. 2000. Association entropy in adsorption processes. Biophys. J. 79:1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woolf, T. B., D. M. Zuckerman, N. D. Lu, and H. B. Jang. 2004. Tools for channels: moving towards molecular calculations of gating and permeation in ion channel biophysics. J. Mol. Graph. 22:359–368. [DOI] [PubMed] [Google Scholar]
- 24.Efremov, R. G., D. E. Nolde, A. G. Konshina, N. P. Syrtcev, and A. S. Arseniev. 2004. Peptides and proteins in membranes: What can we learn via computer simulations? Curr. Med. Chem. 11:2421–2442. [DOI] [PubMed] [Google Scholar]
- 25.Born. 1920. Volumen und Hydratationswärme der Ionen. Z. Phys. 1:45–48. [Google Scholar]
- 26.Still, W. C., A. Tempczyk, R. C. Hawley, and T. Hendrickson. 1990. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112:6127–6129.α [Google Scholar]
- 27.Ulmschneider, J. P., and W. L. Jorgensen. 2004. Polypeptide folding using Monte Carlo sampling, concerted rotation, and continuum solvation. J. Am. Chem. Soc. 126:1849–1857. [DOI] [PubMed] [Google Scholar]
- 28.Chowdhury, S., W. Zhang, C. Wu, G. M. Xiong, and Y. Duan. 2003. Breaking non-native hydrophobic clusters is the rate-limiting step in the folding of an alanine-based peptide. Biopolymers. 68:63–75. [DOI] [PubMed] [Google Scholar]
- 29.Jang, S., S. Shin, and Y. Pak. 2002. Molecular dynamics study of peptides in implicit water: ab initio folding of β-hairpin, β-sheet, and ββα-motif. J. Am. Chem. Soc. 124:4976–4977. [DOI] [PubMed] [Google Scholar]
- 30.Simmerling, C., B. Strockbine, and A. E. Roitberg. 2002. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 124:11258–11259. [DOI] [PubMed] [Google Scholar]
- 31.Snow, C. D., N. Nguyen, V. S. Pande, and M. Gruebele. 2002. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 420:102–106. [DOI] [PubMed] [Google Scholar]
- 32.Tanizaki, S., and M. Feig. 2005. A generalized Born formalism for heterogeneous dielectric environments: application to the implicit modeling of biological membranes. J. Chem. Phys. 122:124706. [DOI] [PubMed] [Google Scholar]
- 33.Spassov, V. Z., L. Yan, and S. Szalma. 2002. Introducing an implicit membrane in generalized Born/solvent accessibility continuum solvent models. J. Phys. Chem. B. 106:8726–8738. [Google Scholar]
- 34.Im, W., M. Feig, and C. L. Brooks III. 2003. An implicit membrane generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 85:2900–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Im, W., and C. L. Brooks III. 2004. De novo folding of membrane proteins: an exploration of the structure and NMR properties of the fd coat protein. J. Mol. Biol. 337:513–519. [DOI] [PubMed] [Google Scholar]
- 36.Hessa, T., H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White, and G. von Heijne. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 433:377–381. [DOI] [PubMed] [Google Scholar]
- 37.Hessa, T., S. H. White, and G. von Heijne. 2005. Membrane insertion of a potassium-channel voltage sensor. Science. 307:1427. [DOI] [PubMed] [Google Scholar]
- 38.Totrov, M. 2004. Accurate and efficient generalized Born model based on solvent accessibility: derivation and application for LogP octanol/water prediction and flexible peptide docking. J. Comput. Chem. 25:609–619. [DOI] [PubMed] [Google Scholar]
- 39.Radzicka, A., and R. Wolfenden. 1988. Comparing the polarities of the amino-acids - side-chain distribution coefficients between the vapor-phase, cyclohexane, 1-octanol, and neutral aqueous-solution. Biochemistry. 27:1664–1670. [Google Scholar]
- 40.Wimley, W. C., T. P. Creamer, and S. H. White. 1996. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 35:5109–5124. [DOI] [PubMed] [Google Scholar]
- 41.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- 42.Opella, S. J., and F. M. Marassi. 2004. Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 104:3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bashford, D., and D. A. Case. 2000. Generalized Born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 51:129–152. [DOI] [PubMed] [Google Scholar]
- 44.Qiu, D., P. S. Shenkin, F. P. Hollinger, and W. C. Still. 1997. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J. Phys. Chem. A. 101:3005–3014. [Google Scholar]
- 45.Jorgensen, W. L., J. P. Ulmschneider, and J. Tirado-Rives. 2004. Free energies of hydration from a generalized Born model and an ALL-atom force field. J. Phys. Chem. B. 108:16264–16270. [Google Scholar]
- 46.Schaefer, M., C. Bartels, and M. Karplus. 1998. Solution conformations and thermodynamics of structured peptides: molecular dynamics simulation with an implicit solvation model. J. Mol. Biol. 284:835–848. [DOI] [PubMed] [Google Scholar]
- 47.Killian, J. A. 2003. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 555:134–138. [DOI] [PubMed] [Google Scholar]
- 48.de Planque, M. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- 49.Wiener, M. C., and S. H. White. 1991. Fluid bilayer structure determination by the combined use of x-ray and neutron diffraction. I. Fluid bilayer models and the limits of resolution. Biophys. J. 59:162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiener, M. C., and S. H. White. 1991. Fluid bilayer structure determination by the combined use of x-ray and neutron diffraction. II. “Composition-space” refinement method. Biophys. J. 59:174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White, S. H., G. I. King, and J. I. Cain. 1981. Location of hexane in lipid bilayers determined by neutron diffraction. Nature. 290:161–163. [Google Scholar]
- 53.Parsegian, A. 1969. Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature. 221:844–846. [DOI] [PubMed] [Google Scholar]
- 54.Saaf, A., E. Wallin, and G. von Heijne. 1998. Stop-transfer function of pseudo-random amino acid segments during translocation across prokaryotic and eukaryotic membranes. Eur. J. Biochem. 251:821–829. [DOI] [PubMed] [Google Scholar]
- 55.Opella, S. J., F. M. Marassi, J. J. Gesell, A. P. Valente, Y. Kim, M. Oblatt-Montal, and M. Montal. 1999. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat. Struct. Biol. 6:374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang, J., S. Kim, F. Kovacs, and T. A. Cross. 2001. Structure of the transmembrane region of the M2 protein H(+) channel. Protein Sci. 10:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marassi, F. M., and S. J. Opella. 2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 12:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bechinger, B., M. Zasloff, and S. J. Opella. 1993. Structure and orientation of the antibiotic peptide magainin in membranes by solid-state nuclear magnetic resonance spectroscopy. Protein Sci. 2:2077–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sawai, M. V., A. J. Waring, W. R. Kearney, P. B. McCray, Jr., W. R. Forsyth, R. I. Lehrer, and B. F. Tack. 2002. Impact of single-residue mutations on the structure and function of ovispirin/novispirin antimicrobial peptides. Protein Eng. 15:225–232. [DOI] [PubMed] [Google Scholar]
- 60.Oh, D., S. Y. Shin, S. Lee, J. H. Kang, S. D. Kim, P. D. Ryu, K. S. Hahm, and Y. Kim. 2000. Role of the hinge region and the tryptophan residue in the synthetic antimicrobial peptides, cecropin A(1–8)-magainin 2(1–12) and its analogues, on their antibiotic activities and structures. Biochemistry. 39:11855–11864. [DOI] [PubMed] [Google Scholar]
- 61.Steiner, H., D. Andreu, and R. B. Merrifield. 1988. Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim. Biophys. Acta. 939:260–266. [DOI] [PubMed] [Google Scholar]
- 62.Ramamoorthy, A., F. M. Marassi, M. Zasloff, and S. J. Opella. 1995. Three-dimensional solid-state NMR spectroscopy of a peptide oriented in membrane bilayers. J. Biomol. NMR. 6:329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gazit, E., I. R. Miller, P. C. Biggin, M. S. P. Sansom, and Y. Shai. 1996. Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes. J. Mol. Biol. 258:860–870. [DOI] [PubMed] [Google Scholar]
- 64.Yamaguchi, S., D. Huster, A. Waring, R. I. Lehrer, W. Kearney, B. F. Tack, and M. Hong. 2001. Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys. J. 81:2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacKenzie, K. R., J. H. Prestegard, and D. M. Engelman. 1997. A transmembrane helix dimer: structure and implications. Science. 276:131–133. [DOI] [PubMed] [Google Scholar]
- 66.Sass, H. J., G. Buldt, R. Gessenich, D. Hehn, D. Neff, R. Schlesinger, J. Berendzen, and P. Ormos. 2000. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 406:649–653. [DOI] [PubMed] [Google Scholar]
- 67.Royant, A., P. Nollert, K. Edman, R. Neutze, E. M. Landau, E. Pebay-Peyroula, and J. Navarro. 2001. X-ray structure of sensory rhodopsin II at 2.1-A resolution. Proc. Natl. Acad. Sci. USA. 98:10131–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sui, H., B. G. Han, J. K. Lee, P. Walian, and B. K. Jap. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature. 414:872–878. [DOI] [PubMed] [Google Scholar]
- 69.Savage, D. F., P. F. Egea, Y. Robles-Colmenares, J. D. O'Connell 3rd, and R. M. Stroud. 2003. Architecture and selectivity in aquaporins: 2.5 a X-ray structure of aquaporin Z. PLoS Biol. 1:E72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a CIC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- 71.Kurumbail, R. G., A. M. Stevens, J. K. Gierse, J. J. McDonald, R. A. Stegeman, J. Y. Pak, D. Gildehaus, J. M. Miyashiro, T. D. Penning, K. Seibert, P. C. Isakson, and W. C. Stallings. 1996. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 384:644–648. [DOI] [PubMed] [Google Scholar]
- 72.Ulmschneider, J. P., and W. L. Jorgensen. 2003. Monte Carlo backbone sampling for polypeptides with variable bond angles and dihedral angles using concerted rotations and a Gaussian bias. J. Chem. Phys. 118:4261–4271. [Google Scholar]
- 73.Ulmschneider, J. P., M. B. Ulmschneider, and A. Di Nola. 2006. Monte Carlo vs molecular dynamics for all-atom polypeptide folding simulations. J. Phys. Chem. B. 110:16733–16742. [DOI] [PubMed] [Google Scholar]
- 74.Jorgensen, W. L., D. S. Maxwell, and J. Tirado-Rives. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236. [Google Scholar]
- 75.Ulmschneider, J. P., and W. L. Jorgensen. 2004. Monte Carlo backbone sampling for nucleic acids using concerted rotations including variable bond angles. J. Phys. Chem. B. 108:16883–16892. [Google Scholar]
- 76.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 95:43–56. [Google Scholar]
- 77.Hess, B., J. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 18:1463–1472. [Google Scholar]
- 78.Yau, W. M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- 79.Marassi, F. M., S. J. Opella, P. Juvvadi, and R. B. Merrifield. 1999. Orientation of cecropin A helices in phospholipid bilayers determined by solid-state NMR spectroscopy. Biophys. J. 77:3152–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ulmschneider, M. B., M. S. P. Sansom, and A. Di Nola. 2006. Evaluating tilt angles of membrane-associated helices: comparison of computational and NMR techniques. Biophys. J. 90:1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.von Heijne, G. 1992. Membrane-protein structure prediction: hydrophobicity analysis and the Positive-inside Rule. J. Mol. Biol. 225:487–494. [DOI] [PubMed] [Google Scholar]
- 82.Biggin, P. C., and M. S. Sansom. 1999. Interactions of α-helices with lipid bilayers: a review of simulation studies. Biophys. Chem. 76:161–183. [DOI] [PubMed] [Google Scholar]
- 83.Popot, J. L., and D. M. Engelman. 1990. Membrane-protein folding and oligomerization: the 2-stage model. Biochemistry. 29:4031–4037. [DOI] [PubMed] [Google Scholar]
- 84.Engelman, D. M., Y. Chen, C. N. Chin, A. R. Curran, A. M. Dixon, A. D. Dupuy, A. S. Lee, U. Lehnert, E. E. Matthews, Y. K. Reshetnyak, A. Senes, and J. L. Popot. 2003. Membrane protein folding: beyond the two stage model. FEBS Lett. 555:122–125. [DOI] [PubMed] [Google Scholar]
- 85.Kessel, A., D. Shental-Bechor, T. Haliloglu, and N. Ben-Tal. 2003. Interactions of hydrophobic peptides with lipid bilayers: Monte Carlo simulations with M2δ. Biophys. J. 85:3431–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kessel, A., T. Haliloglu, and N. Ben-Tal. 2003. Interactions of the M2δ segment of the acetylcholine receptor with lipid bilayers: a continuum-solvent model study. Biophys. J. 85:3687–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ben-Tal, N., A. Ben-Shaul, A. Nicholls, and B. Honig. 1996. Free-energy determinants of α-helix insertion into lipid bilayers. Biophys. J. 70:1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kessel, A., D. P. Tieleman, and N. Ben-Tal. 2004. Implicit solvent model estimates of the stability of model structures of the alamethicin channel. Eur. Biophys. J. 33:16–28. [DOI] [PubMed] [Google Scholar]