

Abstract
We present here a computational, rule-based model to study the function of the SH2 domain-containing protein tyrosine phosphatase, Shp2, in intracellular signal transduction. The two SH2 domains of Shp2 differentially regulate the enzymatic activity by a well-characterized mechanism, but they also affect the targeting of Shp2 to signaling receptors in cells. Our kinetic model integrates these potentially competing effects by considering the intra- and intermolecular interactions of the Shp2 SH2 domains and catalytic site as well as the effect of Shp2 phosphorylation. Even for the isolated Shp2/receptor system, which may seem simple by certain standards, we find that the network of possible binding and phosphorylation states is composed of over 1000 members. To our knowledge, this is the first kinetic model to fully consider the modular, multifunctional structure of a signaling protein, and the computational approach should be generally applicable to other complex intermolecular interactions.
INTRODUCTION
Intracellular signal transduction pathways mediated by growth factor, cytokine, and hormone receptors control cell behavior, such as proliferation, survival, migration, and differentiation. Protein tyrosine kinases, including growth factor receptors of the receptor tyrosine kinase (RTK) class, catalyze tyrosine phosphorylation and are typically involved in the initial steps of signal transduction, but equally important in determining the magnitude and kinetics of the intracellular response are the opposing tyrosine dephosphorylation reactions, catalyzed by protein tyrosine phosphatases (PTPs) (1). In RTK signaling, the ligated receptors dimerize such that their intracellular tails may be transphosphorylated on specific tyrosine residues, which then act as a scaffold for the binding of cytosolic signaling proteins with Src homology 2 (SH2) and other phosphotyrosine-binding domains. These proteins include certain enzymes, adaptor proteins, and transcription factors, which may be activated upon binding to the receptor complex and/or by subsequent phosphorylation, dephosphorylation, or other modifications (2,3). In general, the modular design of signaling proteins, typically composed of multiple domains with specific functions, is a central paradigm in the understanding of signal transduction networks; how these domains work together in the full-length protein, and in the context of signaling mediated by certain receptors, is a problem steeped in complexity (4–6).
In this work, we analyze the function of one such signaling protein, SH2 domain-containing phosphatase (Shp) 2. Shp2 and the closely related Shp1 are intriguing because each has a classic PTP catalytic domain as well as two SH2 domains that regulate its activity and help target the enzyme to phosphorylated RTKs and other tyrosine-phosphorylated proteins (7). Whereas most PTPs are thought to simply antagonize RTK signaling, Shp2 plays a positive role in the activation of the Ras/extracellular signal-regulated kinase (Erk) signaling cascade mediated by platelet-derived growth factor (PDGF) receptors, epidermal growth factor (EGF) receptor, and other RTKs. A candidate mechanism for this effect is the dephosphorylation of the tyrosine residues in PDGF and EGF receptors that bind Ras GTPase-accelerating protein, a negative regulator of Ras activation; these residues are distinct from those that engage the SH2 domains of Shp2 (8–10). The precise dephosphorylation targets and signaling roles of Shp2 in cells have yet to be fully elucidated, however.
The structure of Shp2 and its relationship to the regulation of catalytic activity, at least in solution, are relatively well understood. The crystal structure of Shp2 clearly shows an autoinhibition of the PTP catalytic site by the N-terminal SH2 domain (N-SH2), whereas the more C-terminal SH2 domain (C-SH2) does not interact in this way (11,12). The autoinhibited, “closed” conformation is highly favored under basal conditions. Binding of the N-SH2 domain to a phosphotyrosine-containing protein stabilizes the “open” conformation of Shp2, activating the enzyme. In solution, addition of small, phosphorylated peptide sequences mimicking SH2 binding sites leads to a >10-fold increase in Shp2 enzyme activity, and a variant with the N-SH2 domain deleted exhibits an even higher activity that is not augmented further by phosphopeptide addition (13–16). A notable caveat in such studies is that, at least in certain cases, N-SH2-binding peptides can also be dephosphorylated by the PTP active site, requiring a more sophisticated biochemical analysis (17). Consistent with the crystal structure, deletion of the C-SH2 domain does not activate the enzyme, nor does it influence enzyme activation by N-SH2-binding peptides in solution; however, C-SH2 deletion does reduce the potency of synthetic peptides bearing two SH2 domain-binding phosphotyrosines, suggesting that the N-SH2 and C-SH2 domains bind such peptides and multiply phosphorylated protein complexes in a synergistic manner to better stabilize the open form of Shp2 (16,18–21).
Shp2 is also subject to tyrosine phosphorylation on two sites near its C-terminus in response to growth factor stimulation (22,23). Mutating these sites to phenylalanine leads to a modest reduction in Erk signaling (24), suggesting that Shp2 phosphorylation plays a role in enzyme activation. Selective attachment of chemical moieties that mimic the effects of phosphate addition yields higher Shp2 activity in solution and when microinjected into cells (25–27), and a conceptual model has emerged in which the N-SH2 domain, alone or in tandem with the C-SH2 domain, are engaged by the C-terminal phosphorylation sites in the same molecule. This hypothesis remains controversial because it cannot be directly tested (Shp2 dephosphorylates itself in solution) and because the phosphorylation site(s) can serve an adaptor function that might affect Ras activation (28,29).
The complexity of signal transduction has motivated the use of quantitative, mathematical modeling approaches in recent years to better understand the kinetic mechanisms involved and how they might work in concert inside the cell. Modeling can be a powerful tool for analysis, but an appropriate balance must always be struck between the inclusion of known signaling interactions and the tractability of the model (30,31). As one adds protein binding and phosphorylation states to the model, the number of species increases dramatically, an issue termed “combinatorial complexity” (32). Even in the early stages of intracellular signaling, in which the formation of receptor complexes may be governed by just a handful of simple binding rules (e.g., a cytosolic protein may associate with a receptor if a specific receptor site is phosphorylated and unoccupied), the number of different combinations of protein states can easily reach the hundreds or thousands, as seen in the recent model of FcɛRI signaling (33). Computational tools for rule-based modeling, which generate the network of rate processes and construct the governing equations automatically, are becoming increasingly available (34–37).
In this work, we have constructed a rule-based kinetic model of the interactions between Shp2 and a dimerized RTK, although the analysis may be generalized to the interactions of Shp2 or Shp1 with other multiprotein complexes. Among other effects, we have systematically analyzed the dual role of the Shp2 SH2 domains; on the one hand, they regulate the enzymatic activity, as characterized in solution, while on the other hand, they target the enzyme to receptor complexes in cells. The latter influences the activity toward substrates in the complex through an induced proximity effect, as considered in our previous model of PTP regulation (38). Accordingly, in the context of the currently accepted mechanisms of Shp2 regulation, we find regimes of receptor-Shp2 binding where N-SH2 deletion or C-terminal phosphorylation of Shp2 would either diminish or enhance receptor dephosphorylation, whereas deletion of the C-SH2 domain or both SH2 domains unilaterally impairs this function.
MATERIALS AND METHODS
Structure-based kinetic model of Shp2 regulation and function
In our models, activated receptors and Shp2 molecules are present at total concentrations of Rtot and Stot, respectively, calculated on the basis of the cytosol volume. For the sake of simplicity, we introduce activated receptors as preformed dimers (dimer concentration is Rtot/2) and do not include the processes of external receptor-ligand binding and dimer association/dissociation. We are concerned only with the steady-state behavior, and a dose response curve is implied by performing model calculations with different values of Rtot (38). The hypothetical receptor has two phosphorylation sites: Y1 is a substrate of the Shp2 catalytic (PTP) domain, and Y2 engages SH2 domains of Shp2 and is always phosphorylated in our model. In the human PDGF β-receptor, Y1 and Y2 would correspond to Tyr-771 and Tyr-1009, for example (8,9,13,39). The assumed binding/modification rules are illustrated in Fig. 1 and are summarized as follows; base-case rate constant values are given in Table 1.
FIGURE 1.

Kinetic model of Shp2 structure-function in receptor signaling. The hypothetical signaling complex contains two activated, dimerized receptors (blue), each with two tyrosine phosphorylation sites considered: phosphorylated Y1 is a substrate of the Shp2 catalytic PTP domain (red), whereas phosphorylated Y2 engages either SH2 domain (green) of Shp2. (a) Recruitment of Shp2 from the cytosol. The PTP and N-SH2 domains may only bind receptors when they are in the “open” conformation. (b) Phosphorylation and Shp2-mediated dephosphorylation of the receptor Y1 site. (c) The five types of complexes with two Shp2 domains engaged by receptor sites. (d) The two types of complexes with three Shp2 domains engaged by receptor sites. (e) Receptor-mediated phosphorylation of Shp2 favors the open PTP conformation by occupying the N-SH2 domain.
TABLE 1.
Base-case values of rate constants used in model calculations
| Rate constant | Base value(s) |
|---|---|
| kopen | 10 s−1 |
| kclose | 500 s−1 |
| kon,CSH2 = kon,NSH2 | 1 μM−1s−1 (58) |
| koff,CSH2 = koff,NSH2 | 0.1–10 s−1 (20,42) |
| kon,PTP | 1 μM−1s−1 (58) |
| koff,PTP | 10 s−1 (14,59) |
| kcat,PTP | 1 s−1 (14,59) |
| kkin,Y1 | 0.1 s−1 |
| kkin,Shp2* | 1 s−1 |
| kdePO4* | 1 s−1 |
| kon,PO4* | 100 s−1 |
| koff,PO4* | 0.1 s−1 |
Only applicable to the extended model, presented in Fig. 5.
Shp2 is autoinhibited through the reversible association of its N-SH2 and PTP domains (the closed conformation), provided that neither domain is receptor bound. From thermodynamic constraints, it follows that neither the N-SH2 nor PTP domain can engage receptor sites when Shp2 is in the closed conformation (11), whereas it is assumed that binding the C-SH2 domain to receptors is not affected by the transition between open and closed conformations (Fig. 1 a). Phosphorylation of receptor Y1 sites by the intrinsic kinase activity is modeled as a single, unimolecular step, as considered in previous models (33,40), whereas dephosphorylation of Y1 by Shp2 is modeled with explicit accounting of the enzyme-substrate interaction and catalytic step (Fig. 1 b).
Once an Shp2 molecule is recruited from the cytosol to an activated receptor complex, its free SH2 and/or PTP domains may associate with free phosphotyrosine sites in the complex. As has been suggested in the literature for PDGF β-receptor (20), we allow the two SH2 domains of Shp2 to bridge the two Y2 sites in the receptor dimer. Such ring closure transitions are unimolecular, and their rate constants are calculated by multiplying the second-order rate constants characterizing the corresponding bimolecular associations by various conversion factors χr, which have units of concentration and ensure that microscopic reversibility is satisfied (38). If one were to assume that binding partners in the same complex are confined within a volume of 100 nm3 (10−20 L), the concentration of a single molecule in that volume is ∼10 mM; this is a reasonable estimate of χr, although its value might be significantly increased or decreased subject to orientation constraints. The model here allows different values of χr for different interactions but also recognizes that not all of these values are independent. There are five types of complexes with two Shp2 domains bound (Fig. 1 c) and two types of complexes with all three Shp2 domains occupied (Fig. 1 d); the overall equilibrium constant for the formation of a complex requiring multiple steps does not depend on the order of the steps, and seven independent χr factors were identified and assigned conservative base-case values of 0.1 or 1 mM (Table 2, and Fig. S1 of the Supplementary Material). One might take as a base assumption that all such factors have the same value, as considered previously (38), but we arbitrarily assigned one of two different values for each binding mode to illustrate the flexibility of the model. Finally, for simplicity we neglect the formation of chains containing more than one dimer. We found that the large number of combinations of lateral association and ring closure interactions involving such complexes, even when limited to species containing only two dimers, makes the model intractable. Even with all of its limiting assumptions, the model described above is composed of 149 distinct species participating in 1,032 reactions.
TABLE 2.
Conversion factors for intracomplex binding of Shp2 domains
| Order of binding | χr factors (value in μM) |
|---|---|
| C-Y2; N-Y2′; P-Y1 | χr1 (103); χr6 (102) |
| C-Y2; N-Y2′; P-Y1′ | χr1 (103); χr7 (102) |
| C-Y2; P-Y1; N-Y2′ | χr2 (102); χr8 = χr1χr6/χr2 |
| C-Y2; P-Y1′; N-Y2′ | χr3 (103); χr9 = χr1χr7/χr3 |
| N-Y2′; C-Y2; P-Y1 | χr1 (103); χr6 (102) |
| N-Y2′; C-Y2; P-Y1′ | χr1 (103); χr7 (102) |
| N-Y2′; P-Y1; C-Y2 | χr4 (103); χr10 = χr1χr6/χr4 |
| N-Y2′; P-Y1′; C-Y2 | χr5 (102); χr11 = χr1χr7/χr5 |
| P-Y1; C-Y2; N-Y2′ | χr2 (102); χr8 = χr1χr6/χr2 |
| P-Y1′; C-Y2; N-Y2′ | χr3 (103); χr9 = χr1χr7/χr3 |
| P-Y1; N-Y2′; C-Y2 | χr4 (103); χr10 = χr1χr6/χr4 |
| P-Y1′; N-Y2′; C-Y2 | χr5 (102); χr11 = χr1χr7/χr5 |
Each row lists one of the 12 ways all three receptor-binding domains of Shp2 (C, C-SH2; N, N-SH2; P, PTP) may be sequentially engaged. The receptor site that ultimately engages C-SH2 is denoted Y2, and Y1 is the Shp2 substrate site on that receptor; the receptor site that ultimately engages N-SH2 is denoted Y2′, and Y1′ is the Shp2 substrate site on that receptor. The right-hand column specifies the conversion factors, χr, applied to the second and third steps of the complex formation (see also Fig. S1 of the Supplementary Material).
The base model was extended to consider the phosphorylation of Shp2 on a single tyrosine site, which can occur only when Shp2 is receptor bound. In the phosphorylated state, this site can reversibly engage the N-SH2 domain of the same molecule, provided that the N-SH2 is free and in the open conformation. This interaction maintains the PTP in the open conformation but also prevents Shp2 association with receptors via N-SH2. Dephosphorylation of the Shp2 phosphorylation site, when not occupied by N-SH2, can occur anywhere in the cell and is modeled simply as a unimolecular transition (Fig. 1 e). Adding these simple rules increased the complexity of the system by an order of magnitude, yielding a model with 1,325 species and 15,284 reactions.
Implementation of rule-based models
The binding and reaction rules and their associated rate constants were specified in the syntax of the second-generation version of BioNetGen (34), BioNetGen2, which uses graph theoretic methods to automatically generate the associated network of kinetic balances (ordinary differential equations in time). The open-source software (available through http://bionetgen.lanl.gov) uses standard numerical algorithms to solve the generated system of equations, which was deemed to be at steady state at time = 103 s. The annotated input files for the base and Shp2 phosphorylation models (Shp2_base.bngl and Shp2_extended.bngl, respectively), which specify the binding/reaction rules, and a detailed description of the BioNetGen2 syntax (TextS1.pdf) are provided as Supplementary Material.
Simplified kinetic model
The simplest model of Shp2/receptor interaction treats SH2 domain-mediated binding of cytosolic Shp2 (S) to receptor dimers as a one-step process, with effective forward and reverse rate constants k+ and k−, respectively. Shp2 binding is assumed here to be independent of the phosphorylation state of the Shp2 substrate site, which is phosphorylated by the intrinsic kinase with rate constant kkin and, when Shp2 is bound, dephosphorylated with effective rate constant kPTP. D and D* denote receptor dimers free for Shp2 binding and in the unphosphorylated and phosphorylated states, respectively, and DS and D*S denote the corresponding species with Shp2 bound. We define Dtot as the total concentration of dimers, according to
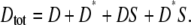 |
(1) |
The bound fraction, bD, and phosphorylated fraction, pD, are defined as
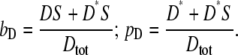 |
(2) |
At steady state, the simplified model gives the following relationship between pD and bD,
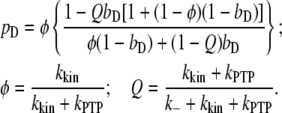 |
(3) |
As in the analysis of related models (40,41), this relationship is cast in terms of two constant parameters: φ, which compares the rates of phosphorylation and dephosphorylation when Shp2 is bound (accordingly, pD = φ when bD = 1), and Q, the exchange quotient. When Q ≈ 1 (slow exchange), all dimers not bound to Shp2 remain phosphorylated, whereas a fraction φ of the Shp2-bound dimers are phosphorylated, such that 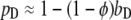 . In the limit of Q ≈ 0 (rapid exchange), the frequency of dephosphorylation on all receptors approaches bDkPTP, and thus
. In the limit of Q ≈ 0 (rapid exchange), the frequency of dephosphorylation on all receptors approaches bDkPTP, and thus 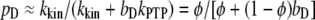 .
.
RESULTS
Shp2 targeting to receptor complexes: multivalency and serial engagement
The function of Shp2 is quantified in terms of the fraction of receptor sites Y1 that are phosphorylated (pYR/Rtot) at steady state; lower values indicate greater overall rates of Shp2-mediated dephosphorylation. The quasi-steady-state approximation is valid when there is a disparity in timescales between that of the intracellular processes (seconds) and that of the binding/trafficking processes that govern the total number of dimerized receptors (minutes), as considered previously (38,40).
Using the base model, we first systematically varied the concentrations of activated receptors and Shp2 and the binding affinities of the Shp2 SH2 domains (Fig. 2). The concentration of receptors in dimers (Rtot) was varied in the range of 0.05–0.5 μM (cytosolic volume basis, or 3 × 104 – 3 × 105 molecules/pL), whereas the concentration of Shp2 (Stot) was assigned a value of 0.05, 0.1, or 0.2 μM. These receptor and Shp2 concentrations are in the general range of values that are typical of intracellular signaling proteins. Also varied was the dissociation rate constant koff of the SH2 domain-receptor interactions, assumed to be equivalent for the C-SH2 and N-SH2 domains. The values are such that the single-site dissociation constants, KD, are in the range of 0.1–10 μM, which spans the range of highly specific and regulatory SH2-phosphotyrosine interactions (20,42). As expected, essentially all dimers are bound with Shp2 when the affinity is sufficiently high (low koff) and Shp2 outnumbers activated receptors (Rtot/2Stot < 1); this yields the minimum extent of receptor phosphorylation (Fig. 2). As the SH2 domain affinities are decreased, Shp2-receptor binding decreases dramatically, because both SH2 domains are affected.
FIGURE 2.

Steady-state analysis of Shp2-mediated receptor dephosphorylation. The fraction of activated receptors phosphorylated on the Shp2 substrate site, Y1 (pYR/Rtot), was calculated as a function of the abundance of activated receptors, mimicking a dose response or varying expression levels (Rtot), and the binding affinities of the two Shp2 SH2 domains (varied in terms of the dissociation rate constant, koff,CSH2 = koff,NSH2 = koff). The other variable is the abundance of Shp2 (Stot): (a) Stot = 0.05 μM; (b) Stot = 0.10 μM; (c) Stot = 0.20 μM. All other rate constants are as listed in Tables 1 and 2.
The fraction of phosphorylated receptors tends to increase as the number of activated receptors is increased, which saturates the binding of Shp2. When activated receptors greatly outnumber Shp2 molecules, a higher dissociation rate from receptors can actually be advantageous. This is a manifestation of the serial engagement effect, more commonly associated with T-cell receptor activation (43–45), whereby one molecule of Shp2 encounters and dephosphorylates multiple dimers before the kinase activity can restore their phosphorylation. Indeed, model calculations show that, when activated receptors are relatively abundant, there is a value of koff that maximizes the extent of receptor dephosphorylation (Fig. 2 a and b; high Rtot, koff ≈ 1–3 s−1).
The serial engagement effect can be understood in the context of a simple kinetic model in which the association and dissociation of Shp2 from receptor complexes and Shp2-mediated dephosphorylation of receptors are approximated as single steps (Fig. 3). In this model, the potential for serial engagement is encapsulated in a single dimensionless parameter, the exchange quotient Q that characterizes the number of phosphorylation and dephosphorylation reactions that occur during the lifetime of an Shp2/receptor encounter (Eq. 3, Materials and Methods). When Q ≈ 1, all activated receptors not bound to Shp2 are maintained in a fully phosphorylated state, whereas a value of Q significantly <1 is indicative of serial engagement and its degree. As a function of the fraction of dimers bound, bD, a lower value of Q always improves the overall rate of dephosphorylation. This is evident when the results from Fig. 2 are plotted in this way and compared with the predictions of the simplified kinetic model: increasing the SH2 domain dissociation constant decreases the average lifetime of the Shp2/receptor association, decreasing the apparent value of Q and enhancing serial engagement (Fig. 3).
FIGURE 3.

Serial engagement of activated receptors by Shp2. The receptor Y1 phosphorylation results shown in Fig. 2 are plotted as a function of the fraction of receptor dimers bound with at least one Shp2 (bD). Plotted in this way, the results may be compared with the predictions of a simplified, two-parameter model (Materials and Methods, Eq. 3). One parameter in this model, φ, was determined from the value of pYR/Rtot at bD ≈ 1. The value of the other, the exchange quotient Q, determines the degree of serial engagement, the ability of an Shp2 molecule to engage and dephosphorylate numerous receptor complexes for each instance of Y1 phosphorylation (solid curves). The values of Q, in descending order, are 1, 0.97, 0.81, 0.51, and 0.
Intracomplex binding, controlled by the magnitudes of the conversion factors χr, is critical for high-avidity binding of Shp2 (Fig. 4). Increasing the rates of intracomplex binding affects Shp2 function in the same manner as decreasing koff and can offset a reduction in the affinities of the individual SH2 domains. Both parameters influence the average rate of Shp2 dissociation from the receptor complex, which was taken as a lumped parameter in our simplified kinetic model (Eq. 3). Thus, when koff is low or χr are high, Shp2 binding to receptors is approximately stoichiometric (Fig. 4 a). When koff is higher and χr are modest, Shp2 binds with lower avidity, and there is also the opportunity for serial engagement when activated receptors outnumber Shp2 molecules (Fig. 4 c).
FIGURE 4.

Intracomplex binding is important for high-avidity binding of Shp2. The fraction of activated receptors phosphorylated on the Shp2 substrate site, Y1 (pYR/Rtot), was calculated as a function of the abundance of activated receptors (Rtot) and the intracomplex association rate constants (varied by multiplying the base-case values of χr, listed in Table 2, by a common factor). The value of koff,CSH2 = koff,NSH2 = koff was varied according to (a) koff = 0.1 s−1; (b) koff = 1.0 s−1; (c) koff = 5.0 s−1. In all cases, Stot = 0.10 μM, and other parameter values are as listed in Table 1.
Deletion of the Shp2 N-SH2 domain can diminish or enhance Shp2 function; C-terminal phosphorylation of Shp2 mimics N-SH2 deletion
Based on dephosphorylation of generic phosphorylated substrates in solution, it is clear that deletion of the N-SH2 domain releases the autoinhibition of the PTP catalytic site, yielding full catalytic activity of the Shp2 enzyme. Based on the cooperation of the two SH2 domains in targeting Shp2 to activated receptors and other signaling complexes at the plasma membrane, however, it is not immediately apparent how this modification would affect dephosphorylation of substrates associated with those complexes and thus modulation of signal transduction by Shp2 in cells.
Indeed, model calculations show that deletion of the N-SH2 can either diminish or enhance the dephosphorylation of a targeted substrate such as a receptor phosphorylation site (Fig. 5). The absence of the N-SH2 domain enhances the activity of Shp2 while associated with receptors through its C-SH2 domain, but at the same time it affects the avidity of the Shp2/receptor interaction. The calculations show that, under the conditions where activated receptors are predominantly associated with full-length Shp2, N-SH2 deletion tends to diminish substrate dephosphorylation; conversely, when either the activated receptors are in excess over Shp2 or the SH2 domain/receptor interactions yield a relatively low avidity, N-SH2 deletion can enhance substrate dephosphorylation (Fig. 5 a–c). Based on the concepts established in the previous section, it is clear that there are two distinct effects that contribute to the enhancement offered by N-SH2 deletion: the lack of PTP autoinhibition and the serial engagement of receptors as the lifetime of Shp2/receptor complexes is decreased.
FIGURE 5.

Conflicting roles of the N-SH2 domain in Shp2 autoinhibition and receptor targeting. The base model was modified to exclude all binding interactions of the N-SH2 domain, and the function of N-SH2-deleted Shp2 (circles) was compared with full-length Shp2 (squares) for various levels of activated receptors and an Shp2 expression level of Stot = 0.10 μM. An extended model shows that receptor-mediated phosphorylation of Shp2, which occupies the N-SH2 in an intramolecular fashion, yields an intermediate level of receptor dephosphorylation (stars). The SH2 dissociation rate constant, koff,CSH2 = koff,NSH2 = koff was varied according to (a) koff = 0.1 s−1; (b) koff = 1.0 s−1; (c and d) koff = 5.0 s−1. Other rate constant values are as listed in Tables 1 and 2. In d, the values of kkin,Y1 and kcat,PTP were increased 10-fold, and all intracomplex association rates were increased 100-fold.
To further illustrate the importance of the parameter values on the predicted effect of N-SH2 deletion in Shp2 signaling, the SH2 domain affinities were adjusted to a relatively low value (KD = 5 μM, as in Fig. 5 c) while both increasing the catalytic rates of receptor Y1 phosphorylation and dephosphorylation by 10-fold (to negate the serial engagement effect) and increasing the rates of all intracomplex associations by 100-fold (Fig. 5 d). Under these conditions, the full-length Shp2 is capable of high-avidity binding to activated receptors, even though the affinities of the individual SH2 domains are low. It is clear that N-SH2 deletion compromises Shp2 function under these conditions, particularly when Shp2 molecules outnumber activated receptors.
As illustrated in Fig. 1 e, the base model was extended to include phosphorylation of receptor-bound Shp2 on its C-terminal tail; this phosphorylation site may then engage the N-SH2 domain, which prevents N-SH2 from either autoinhibiting the PTP domain or participating in receptor binding. While engaged in this manner, Shp2 functions as it does when N-SH2 is deleted altogether. Accordingly, the output of the extended model with Shp2 phosphorylation is invariably bracketed by the results of the base model for full-length and N-SH2-deleted Shp2 (Fig. 5), depending on the relative extent of Shp2 phosphorylation and whether the Shp2 phosphorylation site or activated receptors better compete for N-SH2 binding. The model therefore predicts that, in the same fashion as N-SH2 deletion, tyrosine phosphorylation of Shp2 can either diminish or enhance Shp2-mediated dephosphorylation of receptor-associated substrates.
Deletion of the Shp2 C-SH2 domain, or of both SH2 domains, compromises Shp2 function
With its C-SH2 domain deleted, Shp2 remains autoinhibited by its N-SH2, and any receptor engagement must occur while Shp2 is in the open conformation. Even with the SH2 domain KD skewed toward the highest affinity (0.1 μM, comparable with the concentrations of Shp2 and activated receptors), the autoinhibition drastically reduces the rate of Shp2-receptor association when C-SH2 is absent. Thus, only a small fraction of activated receptors are associated with and dephosphorylated by C-SH2-deleted Shp2 (Fig. 6 a). For lower SH2 domain affinities (KD up to 10 μM) as well, it was confirmed that C-SH2 deletion always diminishes receptor dephosphorylation, despite the greater degree of serial engagement. If one swaps the frequencies of the open and close transitions, such that the open conformation is highly favored, the performance is nearly identical to the N-SH2 deletion mutant (Fig. 6 a and results not shown).
FIGURE 6.

The C-SH2 domain is essential for receptor targeting of Shp2. The base model was modified to exclude all binding interactions of the C-SH2 domain or of both the C-SH2 and N-SH2 domains. Receptor dephosphorylation was assessed for various levels of activated receptors and koff,NSH2 = 0.1 s−1. Except where indicated, all other rate constant values are as listed in Tables 1 and 2. (a) Defective Shp2 function with C-SH2 deleted (solid symbols). Shp2 concentration Stot values are as indicated. Shift of the open/close equilibrium to favor the open conformation (open symbols) mimics the N-SH2 deletion case. (b) Comparison of C-SH2 deletion (solid symbols) with deletion of both SH2 domains (open symbols) for different values of the PTP catalytic rate constant, kcat.
Like the C-SH2 deletion, truncation of both SH2 domains yields an Shp2 variant that is deficient in dephosphorylating receptors but for a different reason. Whereas the C-SH2 deletion has reduced targeting capability and full autoinhibition of the PTP, deletion of both SH2 domains yields a fully active enzyme in solution but erases all targeting capability in cells. Which of these deficiencies is more debilitating for the Shp2 function depends on the binding and catalytic properties of the enzyme (Fig. 6 b). For the set of parameter values considered here, the catalytic efficiency of the SH2-null variant is sensitive to a change in the catalytic rate constant, kcat,PTP. In contrast, for C-SH2-deleted Shp2, the cooperative binding of the N-SH2 and PTP domains gives rise to a scenario in which the lifetime of the Shp2/receptor complex is essentially limited by the PTP catalytic step, and the extent of dephosphorylation is thus determined by the rate of Shp2-receptor association.
Evaluation of restrictive binding rules for PTP-substrate binding within the complex
Our model considers all of the possible ways the two SH2 domains and PTP domain of Shp2 may participate in interactions with activated receptors. The multiplication factors for intracomplex binding, χr, were arbitrarily assigned different values to illustrate the generality of the model (Table 2), but currently there is no reason to suspect that certain modes of intracomplex binding would be favored or disfavored. One can, however, assess the possible impact of structural constraints on the dephosphorylation of receptor-associated substrates (Fig. 7).
FIGURE 7.

Potential restrictions on PTP-substrate binding within the complex. Certain of the multiplication factors, χr, were assigned either base values (Table 2; black) or a value of zero (red) to assess the corresponding modes of PTP-Y1 interaction after binding of one or both SH2 domains of Shp2: (a) χr3 = χr4 = 103 or 0 μM; (b) same as a but with N-SH2-deleted Shp2; (c) χr7 = 100 or 0 μM. In all cases, Stot = 0.10 μM, and SH2 dissociation rate constant (koff,CSH2 = koff,NSH2 = koff) values are as indicated.
For instance, if Shp2 with only one SH2 domain bound is to dephosphorylate the receptor, the PTP may be restricted to binding only one of the two receptors in the dimer. This is of little consequence for full-length Shp2, because simultaneous binding of both SH2 domains is greatly favored (Fig. 7 a). On the other hand, when one of the SH2 domains is deleted, this restriction diminishes receptor dephosphorylation significantly; under these conditions, N-SH2 deletion would be less likely to enhance Shp2 function relative to the full-length enzyme (Fig. 7 b). Conversely, when both of its SH2 domains are bound, full-length Shp2 is compromised if its PTP domain is constrained to binding only one of the receptors (e.g., whichever receptor is bound to C-SH2, Fig. 7 c), whereas this restriction is not applicable to N-SH2-deleted Shp2. Given the dynamic interactions between full-length Shp2 and a receptor dimer, it is still possible in this case for the PTP domain to interact with the substrate sites of both receptors before dissociating from the complex completely, but it does so with greatly reduced proclivity.
DISCUSSION
The function of a signaling protein is generally pieced together from structural analysis, through the identification of homologous domains and their arrangement in the protein's tertiary structure, and biochemical characterization of binding specificities and affinities in solution. In the case of Shp2, an SH2 domain-containing PTP, a consistent story has thus emerged. Full-length Shp2 tends to adopt a closed, autoinhibited conformation that blocks the active site of the PTP; stabilization of the fully active, open conformation of Shp2 requires the occupancy of its N-SH2 domain, either by tyrosine-phosphorylated proteins/peptides or by one of two tyrosine phosphorylation sites in the C-terminal tail of Shp2 itself. The function of Shp2 in cells adds another dimension of complexity, namely that the SH2 domains are also important for targeting Shp2 to signaling complexes, where the PTP enjoys an induced proximity effect. To synthesize and analyze such dynamics in signal transduction, kinetic modeling has become an increasingly valuable tool, but the numerous combinations of binding and phosphorylation states impose a significant technical challenge. The rule-based approach employed here permitted us to examine how multiple protein domains with different functionalities work in concert to affect signaling function.
The central feature of our model is the cooperative binding of the two SH2 domains of Shp2. We assume binding to symmetric phosphorylation sites on a pair of dimerized receptors, but the conclusions are expected to be sufficiently general. Experiments with individual or tandem SH2 domains of Shp2 and singly or doubly phosphorylated peptides show that the interaction of both SH2 domains, which individually possess low single-site affinities (KD ∼ 1–10 μM), yields a high overall binding avidity (effective KD ∼ 1–10 nM) that could be sufficient for near stoichiometric binding in cells (18–20). A 1,000-fold enhancement in binding translates to a value of χr that is roughly 1,000 times higher than the single-site KD, or χr ∼ 1–10 mM, in line with our conservative estimates. In the context of full-length Shp2, to the extent that N-SH2 cannot bind to phosphotyrosine while in the closed conformation, the value of χr would need to be even higher to overcome this effect (see Figs. 4 c and 5 d). When Shp2 binding avidity is not as strong, we found that the effect of serial engagement can at least partially compensate; this effect arises when encounters between Shp2 and receptors are fleeting relative to the rate of substrate phosphorylation/dephosphorylation (Fig. 3).
The disparity between affinity and avidity of the Shp2 SH2 domains is at the core of what is perhaps our most interesting finding: that deletion of the N-SH2 domain can either diminish or enhance receptor dephosphorylation (Fig. 5). Under conditions that favor cooperative binding of a high fraction of receptors, N-SH2 deletion tends to abrogate Shp2 function; otherwise, Shp2 function may be enhanced. Interestingly, the effect of N-SH2 deletion can be reversed as the number of activated receptors is modulated, as by a change in growth factor/cytokine concentration or by receptor overexpression/knock-down.
These predictions of the model can be related to published data, although it should be noted that those data do not allow for a quantitative comparison. Targeted deletion of Shp2 exon 3 in mouse yields expression of a mutant Shp2 with most of its N-SH2 domain deleted. As expected, this mutation yields greater overall PTP activity as measured in solution, but the Shp2 variant is severely defective in binding to activated receptors and other signaling complexes (46–50). The effect of this mutation on intracellular signal transduction was assessed at the level of growth factor-stimulated Erk activation, which is positively modulated by Shp2, with variable results. In one study, Erk phosphorylation stimulated by PDGF was enhanced in the mutant cells relative to wild-type, despite a lower expression level (∼25% of wild-type) of the mutant, whereas Erk signaling stimulated by fibroblast growth factor was significantly reduced in the same cells (46). In another study using the same deletion of Shp2 exon 3, Erk signaling stimulated by all growth factors tested, including PDGF, was diminished in mutant-expressing cells (48). These disparate observations are not inconsistent with our modeling results, which predict that different receptor expression levels and/or Shp2 binding avidities can affect the outcome of N-SH2 deletion. Alternatively, some receptors may activate Shp2 for dephosphorylating substrates not intimately associated with the complex, in which case targeting of Shp2 with N-SH2 deleted is not necessary.
By comparison with N-SH2 deletion, certain Shp2 mutations disrupt autoinhibition of the PTP without preventing binding of phosphorylated peptides to N-SH2; indeed, favoring the open conformation enhances this function. Interestingly, such activating Shp2 mutation sites are associated with Noonan's syndrome, a common human birth defect (51). Expression of Shp2 variants activated in this manner generally yields enhanced growth factor stimulation of Erk signaling, at least in certain cell contexts, arguing for the importance of the N-SH2 targeting function (52–56).
Our modeling approach also sheds light on the most controversial aspect of Shp2 regulation, the mechanism by which Shp2 phosphorylation modulates its activity and signaling functions. In our extended model, Shp2 phosphorylation at one site and the intramolecular binding of N-SH2 were considered. As one might have predicted, this gives a level of receptor dephosphorylation that lies between those mediated by full-length, unphosphorylated Shp2 and Shp2 with N-SH2 deleted (Fig. 5). In other words, intramolecular binding of N-SH2 mimics N-SH2 deletion in our model. What if we had included both of the Shp2 phosphorylation sites? Biochemical evidence suggests that these sites can engage both SH2 domains, favoring the open conformation of Shp2 in a cooperative manner (26). If so, it is clear that inclusion of both phosphorylation sites would mimic the deletion of both SH2 domains; such a mechanism discounts any targeting function of the SH2 domains. Alternative mechanisms for the effect(s) of Shp2 phosphorylation include binding the adaptor protein Grb2, which might help target Shp2 to Gab1 in signaling by EGF receptor and other RTKs (57), and binding of SH2 domain-containing substrates of Shp2 (24).
This modeling study underscores the complexity of interactions between signaling proteins with multiple modular domains. In the case of Shp2, it illustrates the potential tradeoffs between regulation of catalytic activity and targeting of the enzyme to substrate-containing complexes or compartments, and it shows that intracellular Shp2 and receptor expression levels must be carefully considered in the interpretation of cell signaling experiments. Conversely, these expression levels would need to be measured and varied systematically if the quantitative predictions of the model are to be validated. Rule-based kinetic modeling is a powerful computational tool for modeling the assembly of signaling complexes and signal transduction pathways, and here we have shown how it can be used to impart structure-based functionality to their molecular components.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors are grateful to Benjamin Neel (Beth Israel Deaconess Medical Center and Harvard Medical School) for helpful discussions.
This work was supported by grants from the National Science Foundation (No. 0133594) and National Institutes of Health (GM067739) to J.M.H. J.R.F. was supported by grants from the National Institutes of Health (R37GM35556) and the Department of Energy through contract DE-AC52-06NA25396.
References
- 1.Tonks, N. K., and B. G. Neel. 2001. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 13:182–195. [DOI] [PubMed] [Google Scholar]
- 2.van der Geer, P., T. Hunter, and R. A. Lindberg. 1994. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 10:251–337. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell. 103:211–225. [DOI] [PubMed] [Google Scholar]
- 4.Hunter, T. 2000. Signaling—2000 and beyond. Cell. 100:113–127. [DOI] [PubMed] [Google Scholar]
- 5.Pawson, T. 2004. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 116:191–203. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya, R. P., A. Remenyi, B. J. Yeh, and W. A. Lim. 2006. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75:655–680. [DOI] [PubMed] [Google Scholar]
- 7.Neel, B. G., H. H. Gu, and L. Pao. 2003. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28:284–293. [DOI] [PubMed] [Google Scholar]
- 8.Klinghoffer, R. A., and A. Kazlauskas. 1995. Identification of a putative Syp substrate, the PDGFβ receptor. J. Biol. Chem. 270:22208–22217. [DOI] [PubMed] [Google Scholar]
- 9.Ekman, S., A. Kallin, A. Engström, and C.-H. Heldin. 2002. SHP-2 is involved in heterodimer specific loss of phosphorylation of Tyr771 in the PDGF β-receptor. Oncogene. 21:1870–1875. [DOI] [PubMed] [Google Scholar]
- 10.Agazie, Y. M., and M. J. Hayman. 2003. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 23:7875–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hof, P., S. Pluskey, S. Dhe-Paganon, M. J. Eck, and S. E. Shoelson. 1998. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 92:441–450. [DOI] [PubMed] [Google Scholar]
- 12.Barford, D., and B. G. Neel. 1998. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 6:249–254. [DOI] [PubMed] [Google Scholar]
- 13.Lechleider, R. J., S. Sugimoto, A. M. Bennett, A. S. Kashishian, J. A. Cooper, S. E. Shoelson, C. T. Walsh, and B. G. Neel. 1993. Activation of the SH2-containing phosphotyrosine phosphatase SH-PTP2 by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor. J. Biol. Chem. 268:21478–21481. [PubMed] [Google Scholar]
- 14.Dechert, U., M. Adam, K. W. Harder, I. Clark-Lewis, and F. Jirik. 1994. Characterization of protein tyrosine phosphatase SH-PTP2: study of phosphopeptide substrates and possible regulatory role of SH2 domains. J. Biol. Chem. 269:5602–5611. [PubMed] [Google Scholar]
- 15.Sugimoto, S., T. J. Wandless, S. E. Shoelson, B. G. Neel, and C. T. Walsh. 1994. Activation of the SH2-containing protein tyrosine phosphatase, SH-PTPs by phosphotyrosine-containing peptides derived from insulin receptor substrate-1. J. Biol. Chem. 269:13614–13622. [PubMed] [Google Scholar]
- 16.Pei, D. H., J. Wang, and C. T. Walsh. 1996. Differential functions of the two Src homology 2 domains in protein tyrosine phosphatase SH-PTP1. Proc. Natl. Acad. Sci. USA. 93:1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keilhack, H., F. S. David, M. McGregor, L. C. Cantley, and B. G. Neel. 2005. Diverse biochemical properties of Shp2 mutants: implications for disease phenotypes. J. Biol. Chem. 280:30984–30993. [DOI] [PubMed] [Google Scholar]
- 18.Pluskey, S., T. J. Wandless, C. T. Walsh, and S. E. Shoelson. 1995. Potent stimulation of SHPTPs phosphatase activity by simultaneous occupancy of both SH2 domains. J. Biol. Chem. 270:2897–2900. [DOI] [PubMed] [Google Scholar]
- 19.Eck, M. J., S. Pluskey, T. Trub, S. C. Harrison, and S. E. Shoelson. 1996. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 379:277–280. [DOI] [PubMed] [Google Scholar]
- 20.Ottinger, E. A., M. C. Botfield, and S. E. Shoelson. 1998. Tandem SH2 domains confer high specificity in tyrosine kinase signaling. J. Biol. Chem. 273:729–735. [DOI] [PubMed] [Google Scholar]
- 21.Cunnick, J. M., L. Mei, C. A. Doupnik, and J. Wu. 2001. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 276:24380–24387. [DOI] [PubMed] [Google Scholar]
- 22.Feng, G., C. Hui, and T. Pawson. 1993. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. 259:1607–1610. [DOI] [PubMed] [Google Scholar]
- 23.Vogel, W., R. Lammers, J. Huang, and A. Ullrich. 1993. Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science. 259:1611–1614. [DOI] [PubMed] [Google Scholar]
- 24.Araki, T., H. Nawa, and B. G. Neel. 2003. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J. Biol. Chem. 278:41677–41684. [DOI] [PubMed] [Google Scholar]
- 25.Lu, W., D. Q. Gong, D. Bar-Sagi, and P. A. Cole. 2001. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol. Cell. 8:759–769. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, Z. S., K. Shen, W. Lu, and P. A. Cole. 2003. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J. Biol. Chem. 278:4668–4674. [DOI] [PubMed] [Google Scholar]
- 27.Lu, W., K. Shen, and P. A. Cole. 2003. Chemical dissection of the effects of tyrosine phosphorylation of SHP-2. Biochemistry. 42:5461–5468. [DOI] [PubMed] [Google Scholar]
- 28.Bennett, A. M., T. L. Tang, S. Sugimoto, C. T. Walsh, and B. G. Neel. 1994. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor β to Ras. Proc. Natl. Acad. Sci. USA. 91:7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, W., R. Nishimura, A. Kashishian, A. G. Batzer, W. J. H. Kim, J. A. Cooper, and J. Schlessinger. 1994. A new function for a phosphotyrosine phosphatase: linking Grb2-Sos to a receptor tyrosine kinase. Mol. Cell. Biol. 14:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weng, G., U. S. Bhalla, and R. Iyengar. 1999. Complexity in biological signaling systems. Science. 284:92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kholodenko, B. N. 2006. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hlavacek, W. S., J. R. Faeder, M. L. Blinov, A. S. Perelson, and B. Goldstein. 2003. The complexity of complexes in signal transduction. Biotechnol. Bioeng. 84:783–794. [DOI] [PubMed] [Google Scholar]
- 33.Faeder, J. R., W. S. Hlavacek, I. Reischl, M. L. Blinov, H. Metzger, A. Redondo, C. Wofsy, and B. Goldstein. 2003. Investigation of early events in FcɛRI-mediated signaling using a detailed mathematical model. J. Immunol. 170:3769–3781. [DOI] [PubMed] [Google Scholar]
- 34.Blinov, M. L., J. R. Faeder, B. Goldstein, and W. S. Hlavacek. 2004. BioNetGen: software for rule-based modeling of signal transduction based on the interactions of molecular domains. Bioinformatics. 20:3289–3291. [DOI] [PubMed] [Google Scholar]
- 35.Lok, L., and R. Brent. 2005. Automatic generation of cellular reaction networks with Moleculizer 1.0. Nat. Biotechnol. 23:131–136. [DOI] [PubMed] [Google Scholar]
- 36.Conzelmann, H., J. Saez-Rodriguez, T. Sauter, B. N. Kholodenko, and E. D. Gilles. 2006. A domain-oriented approach to the reduction of combinatorial complexity in signal transduction networks. BMC Bioinformatics. 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hlavacek, W. S., J. R. Faeder, M. L. Blinov, R. G. Posner, M. Hucka, and W. Fontana. 2006. Rules for modeling signal-transduction systems. Sci. STKE. 344:re6. [DOI] [PubMed] [Google Scholar]
- 38.Haugh, J. M., I. C. Schneider, and J. M. Lewis. 2004. On the cross-regulation of protein tyrosine phosphatases and receptor tyrosine kinases in intracellular signaling. J. Theor. Biol. 230:119–132. [DOI] [PubMed] [Google Scholar]
- 39.Kazlauskas, A., G. S. Feng, T. Pawson, and M. Valius. 1993. The 64-kDa protein that associates with the platelet-derived growth factor receptor β subunit via Tyr-1009 is the SH2-containing phosphotyrosine phosphatase Syp. Proc. Natl. Acad. Sci. USA. 90:6939–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haugh, J. M., and D. A. Lauffenburger. 1998. Analysis of receptor internalization as a mechanism for modulating signal transduction. J. Theor. Biol. 195:187–218. [DOI] [PubMed] [Google Scholar]
- 41.Haugh, J. M., A. C. Huang, H. S. Wiley, A. Wells, and D. A. Lauffenburger. 1999. Internalized epidermal growth factor receptors participate in the activation of p21ras in fibroblasts. J. Biol. Chem. 274:34350–34360. [DOI] [PubMed] [Google Scholar]
- 42.Ladbury, J. E., M. A. Lemmon, M. Zhou, J. Green, M. C. Botfield, and J. Schlessinger. 1995. Measurement of the binding of tyrosyl phosphopeptides to SH2 domains: a reappraisal. Proc. Natl. Acad. Sci. USA. 92:3199–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valitutti, S., S. Muller, M. Cella, E. Padovan, and A. Lanzavecchia. 1995. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 375:148–151. [DOI] [PubMed] [Google Scholar]
- 44.Valitutti, S., and A. Lanzavecchia. 1997. Serial triggering of TCRs: a basis for the sensitivity and specificity of antigen recognition. Immunol. Today. 18:299–304. [PubMed] [Google Scholar]
- 45.Coombs, D., A. M. Kalergis, S. G. Nathenson, C. Wofsy, and B. Goldstein. 2002. Activated TCRs remain marked for internalization after dissociation from pMHC. Nat. Immunol. 3:926–931. [DOI] [PubMed] [Google Scholar]
- 46.Saxton, T. M., M. Henkemeyer, S. Gasca, R. Shen, D. J. Rossi, F. Shalaby, G. Feng, and T. Pawson. 1997. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. 16:2352–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu, C., Z. Shi, R. Shen, F. Tsai, S. H. Orkin, and G. Feng. 1997. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Mol. Cell. Biol. 17:5499–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi, Z., W. Lu, and G. Feng. 1998. The Shp-2 tyrosine phosphatase has opposite effects in mediating the activation of extracellular signal-regulated and c-Jun NH2-terminal mitogen-activated protein kinases. J. Biol. Chem. 273:4904–4908. [DOI] [PubMed] [Google Scholar]
- 49.Shi, Z., D. Yu, M. Park, M. Marshall, and G. Feng. 2000. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol. Cell. Biol. 20:1526–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang, W., L. D. Klaman, B. Chen, T. Araki, H. Harada, S. M. Thomas, E. L. George, and B. G. Neel. 2006. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev. Cell. 10:317–327. [DOI] [PubMed] [Google Scholar]
- 51.Tartaglia, M., E. L. Mehler, R. Goldberg, G. Zampino, H. G. Brunner, H. Kremer, I. van der Burgt, A. H. Crosby, A. Ion, S. Jeffery, K. Kalidas, M. A. Patton, R. S. Kucherlapati, and B. D. Gelb. 2001. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29:465–468. [DOI] [PubMed] [Google Scholar]
- 52.O'Reilly, A. M., S. Pluskey, S. E. Shoelson, and B. G. Neel. 2000. Activated mutants of SHP-2 preferentially induce elongation of Xenopus animal caps. Mol. Cell. Biol. 20:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Araki, T., M. G. Mohi, F. A. Ismat, R. T. Bronson, I. R. Williams, J. L. Kutok, W. T. Yang, L. I. Pao, D. G. Gilliland, J. A. Epstein, and B. G. Neel. 2004. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutations. Nat. Med. 10:849–857. [DOI] [PubMed] [Google Scholar]
- 54.Mohi, M. G., I. R. Williams, C. R. Dearolf, G. Chan, J. L. Kutok, S. Cohen, K. Morgan, C. Boulton, H. Shigematsu, H. Keilhack, K. Akashi, D. G. Gilliland, and B. G. Neel. 2005. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 7:179–191. [DOI] [PubMed] [Google Scholar]
- 55.Chan, R. J., M. B. Leedy, V. Munugalavadla, C. S. Voorhorst, Y. J. Li, M. G. Yu, and R. Kapur. 2005. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 105:3737–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu, W. M., H. Daino, J. Chen, K. D. Bunting, and C. K. Qu. 2006. Effects of a leukemia-associated gain-of-function mutation of SHP-2 phosphatase on interleukin-3 signaling. J. Biol. Chem. 281:5426–5434. [DOI] [PubMed] [Google Scholar]
- 57.Gu, H. H., and B. G. Neel. 2003. The ‘Gab’ in signal transduction. Trends Cell Biol. 13:122–130. [DOI] [PubMed] [Google Scholar]
- 58.Northrup, S. H., and H. P. Erickson. 1992. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. USA. 89:3338–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugimoto, S., R. J. Lechleider, S. E. Shoelson, B. G. Neel, and C. T. Walsh. 1993. Expression, purification, and characterization of SH2-containing protein tyrosine phosphatase, SH-PTP2. J. Biol. Chem. 268:22771–22776. [PubMed] [Google Scholar]