

Abstract
Identifying the molecules that regulate both the recycling of synaptic vesicles and the SNARE components required for fusion is critical for elucidating the molecular mechanisms underlying synaptic plasticity. SNAP-29 was initially isolated as a syntaxin-binding and ubiquitously expressed protein. Previous studies have suggested that SNAP-29 inhibits SNARE complex disassembly, thereby reducing synaptic transmission in cultured superior cervical ganglion neurons in an activity-dependent manner. However, the role of SNAP-29 in regulating synaptic vesicle recycling and short-term plasticity in the central nervous system remains unclear. In the present study, we examined the effect of SNAP-29 on synaptic transmission in cultured hippocampal neurons by dual patch clamp whole-cell recording, FM dye imaging, and immunocytochemistry. Our results demonstrated that exogenous expression of SNAP-29 in presynaptic neurons significantly decreased the efficiency of synaptic transmission after repetitive firing within a few minutes under low and moderate frequency stimulations (0.1 and 1 Hz). In contrast, SNAP-29 did not affect the density of synapses and basal synaptic transmission. Whereas neurotransmitter release was unaffected during intensive stimulation, recovery after synaptic depression was impaired by SNAP-29. Furthermore, knockdown of SNAP-29 expression in neurons by small interfering RNA increased the efficiency of synaptic transmission during repetitive firing. These findings suggest that SNAP-29 acts as a negative modulator for neurotransmitter release, probably by slowing recycling of the SNARE-based fusion machinery and synaptic vesicle turnover.
Synaptic vesicle cycling, the process of exocytosis, membrane retrieval, and formation and recruitment of new vesicles at the release site, is extremely important in maintaining effective transmission during sustained synaptic activity. Repetitive action potential activity in the presynaptic neuron causes changes in the strength of the postsynaptic response in a time-and activity-dependent manner; this can lead to facilitation, depression, or a mixture of both forms of short-term plasticity (1). Synaptic strength depends on both the size of the readily releasable pool of synaptic vesicles and the release probability of individual vesicles following an action potential. In addition to the readily releasable pool, there is a large reserve pool of vesicles that have a lower release probability and are not thought to be primed for immediate release (2). In cultured mammalian central nervous system synapses, only a small number of readily releasable vesicles that are refilled via the fast endocytic pathway are reused during rapid stimulation (“tetanic rundown”) (3, 4). This recycling pool is a small fraction of the total and exchanges slowly with the reserve pool at low and moderate frequencies of stimulation. Synaptic vesicle replenishment is essential for ongoing synaptic transmission. Under repetitive release conditions such as high frequency stimulation, vesicles in the reserve pool are rapidly recruited to the release sites for continued transmitter release.
The maintenance of efficient neurotransmitter release depends upon the interaction of numerous protein complexes that determine the life cycle of synaptic vesicles and the recycling of the SNARE-based release machinery (5–7). The synaptic vesicle-associated protein synaptobrevin (VAMP)1 interacts with two membrane proteins, SNAP-25 and syntaxin, to form a stable SNARE complex, bringing the synaptic vesicle and plasma membranes into close apposition (8–13). During an action potential, calcium enters the presynaptic terminal and triggers synaptic vesicle fusion by activating a calcium sensor. Synaptotagmins, integral Ca2+-binding proteins of the synaptic vesicle membrane, provide Ca2+-dependent regulation of the fusion machinery (14, 15). The SNARE complex converts from a trans- to a cis-state, resulting in complete fusion of the two membranes and the release of neurotransmitter. The stable cis-SNARE core complex is subsequently dissociated by the action of α-SNAP and the ATPase N-ethylmaleimide-sensitive factor (16–21). The majority of SNAP-25 and syntaxin molecules remain associated with the plasma membrane, whereas VAMP recycles with internalized vesicles, allowing the individual SNARE components to take part in another round of membrane fusion. Efficient SNARE complex formation and dissociation are particularly important in neurons, which experience high frequency action potentials (22–24). Despite the simplicity of these individual protein interactions, exocytosis is a complex event. It is characterized by a fast response with a very short delay between excitation and secretion and by limited release of a small percentage of docked vesicles upon Ca2+influx. Considerable evidence indicates that mechanisms that modulate presynaptic activity, specifically those that change release probability and vesicle recycling kinetics, play a role in synaptic heterogeneity (25–28). These properties suggest that the assembly and disassembly of the SNARE complex are precisely and tightly regulated, necessitating the existence of cellular signals capable of regulating the release machinery and its recycling after fusion.
SNAP-29 has recently been reported as a candidate molecule regulating the disassembly of the SNARE complex (29). SNAP-29 was initially isolated by yeast two-hybrid selection using syntaxin-3 as the bait and was localized predominantly to the intracellular membrane structures in transiently transfected normal rat kidney cells (30). Due to its intracellular membrane association in non-neuronal cells and its interactions with most members of the syntaxin family, SNAP-29 has been proposed to be a ubiquitous cytoplasmic SNARE protein involved in general membrane trafficking (31). Our previous studies demonstrated that SNAP-29 is present at synapses and regulates recycling of the SNARE complexes by competing with α-SNAP for binding to the SNAREs and consequently inhibiting disassembly of the SNARE complex in vitro. Introduction of SNAP-29 into presynaptic superior cervical ganglion neurons (SCGNs) in culture significantly reduces neurotransmitter release in an activity-dependent manner. Furthermore, the activity-dependent inhibition by SNAP-29 could be reversed by co-injection of α-SNAP, consistent with our in vitro observation that SNAP-29 and α-SNAP compete for the binding to the SNARE complex (29). However, the function of SNAP-29 in synaptic transmission and short-term plasticity in the central nervous system has not yet been examined. In the present study, we overexpressed SNAP-29-EGFP or knocked-down endogenous SNAP-29 expression in cultured rat hippocampal neurons, and we examined the functional role for SNAP-29 in synaptic vesicle turnover, using an approach that combines dual patch clamp whole-cell recording, FM dye imaging, and immunocytochemical assays. Our findings suggest a novel inhibitory role for SNAP-29 in synaptic vesicle recycling.
MATERIALS AND METHODS
Construction of SNAP-29-EGFP Construct
Human SNAP-29 cDNA was amplified by PCR using the forward primer 5′-GATTCTGCAGT-CATGTCAGCTTACCCTAAAAGC-3′ (PstI) and the reverse primer 5′-CGGTGGATCCGAGTTGTCGAACTTTTCTTTC-3′ (BamHI). The construct was then subcloned into the vector pEGFP-N3 (Clontech), and the sequence was confirmed.
Hippocampal Cell Culture and Electrophysiology
Primary hippocampal neuron mixed cultures were prepared from embryonic Sprague-Dawley rat (age, embryonic day 17–18), according to the procedures described previously (32). Cultures were transfected at DIV 6–8 with SNAP-29-EGFP or EGFP control vector using Lipofectamine2000 (Invitrogen) according to the modified version of published protocols (33). All electrophysiological recordings were performed 12–36 h after transfection. Dual patch clamp recordings were performed with whole-cell configuration at room temperature. Postsynaptic currents and potentials were acquired and measured using an EPC9 patch clamp amplifier driven by PatchMaster software, version 1.05 (HEKA Elektronik, Lambrecht, Germany). Data were analyzed by Igor Pro version 4.04 and Origin Pro version 7.0. Extracellular recording solution contained 145 mM NaCl, 3 mM KCl, 3 mM CaCl2, 10 mM HEPES, 2 mM MgCl2, and 8 mM D-glucose (pH 7.3) (310 mosM). Patch pipettes with resistance of 4–8 megaohms were used and backfilled with 105 mM K-gluconate, 30 mM KCl, 9.0 mM NaCl, 4.0 mM ATP-Mg, 10 mM HEPES, and 0.2 mM EGTA (pH 7.3) (310 mosM). Transfected neurons (detected under a fluorescence microscope) forming synapses with untransfected neurons were selected for experiments. To control the timing for PSC measurements, synaptic contact between neurons was confirmed by 5–10 test stimuli, each separated by 30-s intervals. Following the test stimuli, a 3-min rest period was applied before data collection began to ensure that all fusion and recycling machinery had reset to resting levels. Membrane capacitance and series resistance were compensated electronically. Signals were filtered at 10 kHz. Only recordings with access resistance below 20 megaohms and leak current below 100 pA were included in the analysis. Brief depolarization of the transfected neurons elicited action potentials in the axon of presynaptic neurons and, consequently, neurotransmitter release. PSCs were recorded after a short synaptic delay on the untransfected postsynaptic neuron. A hyperpolarizing current (−75 mV, 40 ms) was injected at the end of each trial of stimulation to monitor the steady state of input resistance.
FM Dye Imaging
FM dye imaging was performed on hippocampal cultures 24–48 h after transfection. Presynaptic terminals were loaded with the fluorescent styryl dye FM4-64 (15 μM) by incubating the culture plate for 1 min in high K+solution containing 58 mM NaCl, 90 mM KCl, 10 mM HEPES, 3 mM CaCl2·2H2O, 8 mM glucose, and 2 mM MgCl2·6H2O (pH 7.3). The culture plate was then washed in calcium-free solution for 10 min to reduce nonspecific staining. The nerve terminals of the transfected neuron were identified under a confocal microscope (Olympus IX71 inverted microscope) using an oil immersion UPLAPO ×40 objective lens (Zeiss) by tracing the optically distinguishable axon expressing EGFP or SNAP-29-EGFP. Those EGFP-labeled terminals capable of dye uptake were considered functional release sites. Fluorescence of EGFP and FM dye was excited at 448 and 543 nm, respectively. Only the fluorescent puncta of FM4-64 with an area of ~1.5 × 1.5 μm2 were included in the analysis. During the time-lapse imaging of fluorescence change, consecutive frames were acquired in 5-s intervals, with constant PMT (800 v), HeNe G (12.1%), Gain (1.0), and Offset (0). After 10–20 s of steady-state recording, high K+solution (containing 10 μM 6,7-dinitroquinoxaline-2,3-dione and 50 μM D(−)-2-amino-5-phosphonopetanoic acid) was locally perfused to destain FM4-64 dye. All data were acquired and analyzed by Fluoview 500 software.
Immunocytochemistry
24–48 h after transfection, the hippocampal cultures were fixed with 4% paraformaldehyde (diluted in 0.1 mM PBS) for 10 min at room temperature, permeabilized with 0.2% Triton X-100 for 10 min, and then washed three times with 0.01 mM PBS. The neurons were then pre-incubated with 10% bovine serum albumin (diluted in 0.1 mM PBS) for 1 h at room temperature to block nonspecific binding with the primary antibody. Monoclonal antibody for synaptophysin (Chemicon) was diluted (in 0.1 mM PBS) to 1:1000 and applied to the culture plate. After incubation at 4 °C overnight, the neurons were washed again with PBS and incubated in the dark for 1 h with secondary antibody, CY3-labeled goat anti-mouse IgG (1:600; Jackson ImmunoResearch). Neurons were washed with PBS, mounted with anti-fade mounting medium, and imaged.
Small Interfering RNA Preparation and Transfection
To design a target-specific siRNA duplex, five sequences unique to SNAP-29 were selected from the open reading frame of the rat SNAP-29 sequence and subcloned into pRNAT-H1.1/neo (GenScript), an siRNA expression vector containing a cGFP gene under a separate promoter for tracking transfected neurons. Transfection of siRNA was carried out using Li-pofectamine 2000. Following transfection, cells were cultured for an additional 96 h before immunocytochemistry and electrophysiological recordings. For studies in COS-7 cells, the SNAP-29-EGFP construct was co-transfected into COS-7 cells with either the siRNA expression constructs or vector backbone alone. The effect of siRNA on the expression of heterologous SNAP-29-EGFP in COS-7 cells was assessed by immunoblot with lysates (10 μg of total proteins) from the cells harvested 4 days after transfection. Sequential blots with anti-GFP and beta;-tubulin antibodies from the same membrane tracked the transfection efficiency and assured equal protein loading in each gel lane.
RESULTS
SNAP-29 Inhibits the Efficacy of Synaptic Transmission under Repetitive Stimulation at 0.1 Hz
To evaluate the effect of SNAP-29 on synaptic transmission, we first examined the distribution of SNAP-29-EGFP in cultured rat hippocampal neurons. Whereas SNAP-29-EGFP demonstrated a diffuse pattern of green fluorescence in both soma and dendrites, it expressed as a punctate pattern along the axonal processes and co-localized with the synaptic vesicle marker synaptophysin at synapses (Fig. 1). The distribution profile of SNAP-29-EGFP is similar to that of endogenous SNAP-29 in cultured hippocampal neurons (29), indicating that exogenously expressed SNAP-29-EGFP, like the endogenous protein, is targeted to and localized at synapses.
FIG. 1. SNAP-29-EGFP is present in the synapses of cultured hippocampal neurons.
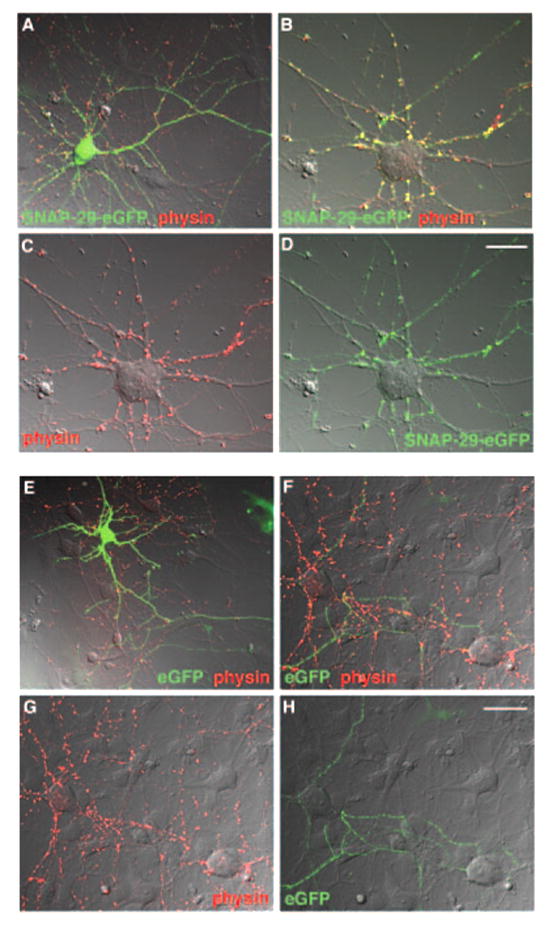
Cultured hippocampal neurons at DIV 7 were transfected with SNAP-29-EGFP (green, A–D) or EGFP (green, E–H) and immunostained with antibody against synaptophysin (physin, red), as a marker of synapses, after 24–48 h of transfection. Images are shown as merged differential interference contrast. A and E, transfected neurons. B–D and F–H, untransfected neurons that form synapses with transfected neurons. Note that presynaptically expressed SNAP-29-EGFP was targeted to the synapses with the untransfected neuron (B) and that EGFP is not significantly co-localized with synaptophysin at synapses (F). Scale bar, 20 μm.
We performed whole-cell dual patch clamp studies only on selected pairs of neurons formed between SNAP-29-EGFP-transfected neurons, which show EGFP signal within the cell body and long axonal processes, and untransfected neurons, which lack EGFP signal (Fig. 2A). Synaptic connections between the selected two neurons were further confirmed by electrophysiological tests. Presynaptic neurons transfected with SNAP-29-EGFP were repetitively stimulated for 15 min at 0.1 Hz. For all synapses tested (n = 9), amplitudes of the PSCs remained steady during the first 80 s (normalized mean PSC, 87 ± 3.7%, mean ±S.E.) and then decreased suddenly to about 60% of the initial amplitude. This low level was maintained throughout the period of recording (15 min) (Fig. 2, B and C). As a control, synapses formed by untransfected or EGFP vector-transfected neurons showed no significant reduction in the efficacy of transmission during the 15-min recording under stimulation at 0.1 Hz (Fig. 2, B and C), suggesting that any effect on synaptic transmission by the transfection procedure is unlikely. The reduced normalized mean PSC measured from the synapses of the SNAP-29-EGFP group (62 ± 4.3%) was significantly smaller than that of the EGFP group (94 ± 4.1%) and untransfected group (108 ± 10.0%) (p < 0.01, Wilcoxon rank-sum test, SAS version 8.0) during the stimulation period from 120 to 240 s and remained significant until the end of the 15-min recording (Fig. 2D). To assess the possibility that SNAP-29 inhibited basal synaptic transmission, we averaged postsynaptic currents recorded from 16 pairs of neurons trans-fected with SNAP-29-EGFP and 12 pairs of neurons transfected with EGFP vector at the same developmental age (Fig. 2E). There was no significant difference in PSC amplitudes (p > 0.05, Wilcoxon rank-sum test) (Fig. 2F), suggesting that overexpression of SNAP-29-EGFP in neurons does not affect the function of the basic release machinery.
FIG. 2. SNAP-29-EGFP inhibits repetitive (0.1 Hz) synaptic transmission but has no effect on basal synaptic transmission in cultured hippocampal neurons.
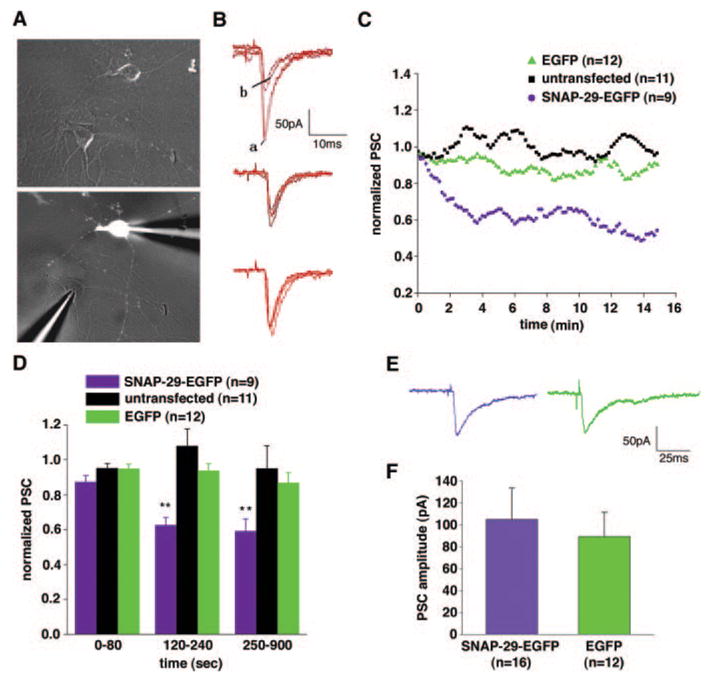
A, a representative pair of neurons demonstrates a synaptic connection between the presynaptic neuron transfected with SNAP-29-EGFP (indicated by the white image in the bottom panel) and the untransfected postsynaptic neuron during patch clamp recording. B, individual traces were recorded from different pairs of synapses. An overlay of four sweeps of PSCs includes the first two (marked as a) and the last two responses (marked as b) during 15 min of repetitive stimulation (0.1 Hz) from the neurons expressing SNAP-29-EGFP (top panel) or EGFP (middle panel) and untransfected control neurons (bottom panel). C, normalized amplitude of PSC plotted against time (in minutes). The tendency curves of three groups of the synapses were smoothed by the adjacent five data points (Origin Pro version 7.0 software). D, histograms of the normalized PSC among groups of synapses during different time periods (in seconds) of recording. Each column represents mean ±S.E. (**, p < 0.01, Wilcoxon rank-sum test). E, representative PSCs from neurons presynaptically expressing SNAP-29-EGFP (left) or EGFP (right). Steady synaptic responses within the first minute of recordings were selected for assessment of basal synaptic transmission. All recordings were performed 12–24 h post-transfection on hippocampal cultures at DIV 8–10. F, histograms of basal synaptic transmission representing mean PSC amplitudes in neurons presynaptically expressing SNAP-29-EGFP (105 ± 29 pA) or EGFP control (89 ± 22 pA) (p > 0.05).
We next determined the density of release sites along axonal processes by means of FM dye staining. Presynaptic terminals were visualized with confocal microscope in axons, which can be identified easily in low density cultures after low efficiency transfection with EGFP constructs (Fig. 1). All functional pre-synaptic terminals were loaded with the membrane-bound styryl dye FM4-64 under stimulation with 90 mM KCl. The FM4-64-loaded release sites were counted only if the FM4-64 puncta could be unloaded by another application of high KCl solution (Fig. 3A) and co-localized with EGFP fluorescence along the axons of transfected neurons (Fig. 3B). The density of the FM dye-loaded synapses was expressed as the number of release sites per micrometer of axonal process. A total of 7175-μm length of axons was counted from 17 neurons expressing SNAP-29-EGFP and a total of 9821-μm length of axons was counted from 17 neurons transfected with EGFP. Analysis by Wilcoxon rank-sum test showed that the density of FM dye-loaded release sites in neurons expressing SNAP-29-EGFP (0.12 ± 0.018/μm) is not significantly different from that in neurons transfected with EGFP (0.11 ± 0.019/μm; p > 0.05, Fig. 3C). To exclude any possibility that SNAP-29 affects the structural maintenance of synapses, we further determined the density of synapses by immunocytochemical staining of the synaptic vesicle marker synaptophysin after transfection (Fig. 3D). A total of 10 neurons transfected with SNAP-29-EGFP and 13 neurons transfected with EGFP were counted (Fig. 3E). Our statistical analysis indicates that expression of SNAP-29-EGFP has no significant effect on the density of either FM dye-loaded or synaptophysin-positive synapses along axons.
FIG. 3. SNAP-29 has no effect on the density of synapses in cultured hippocampal neurons.
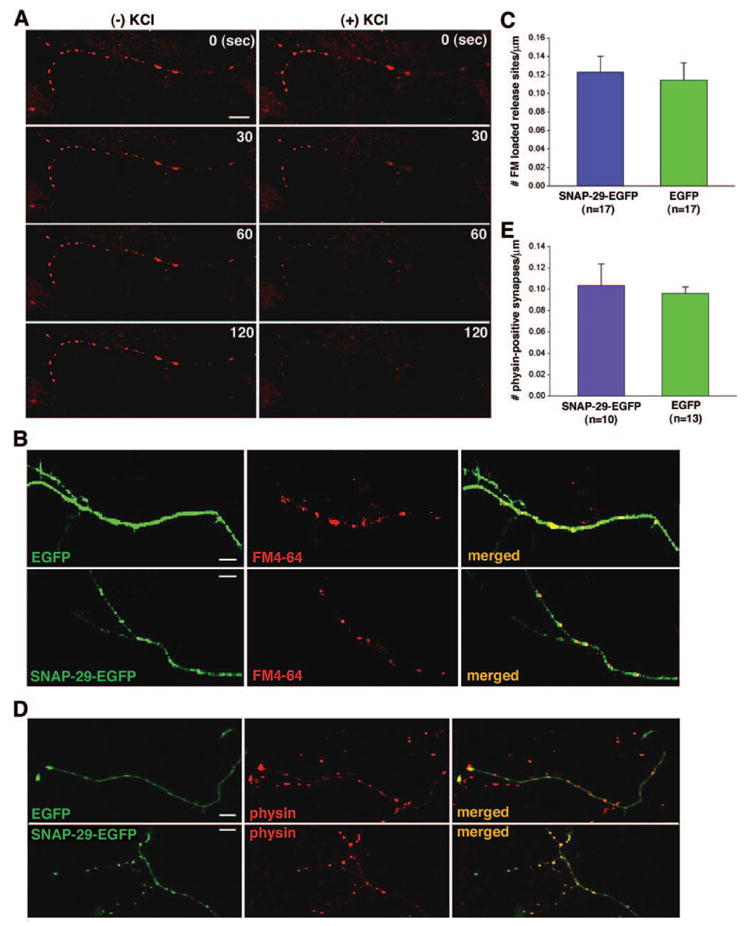
A, high K+-dependent FM4-64 destaining along axonal processes. Axon terminals loaded with FM4-64 were destained in the absence (left column) or presence (right column) of 90 mM KCl. The fluorescence images of FM4-64 were taken at different time points (0, 30, 60, and 120 s). B, axon terminals of hippocampal neurons at DIV 9–10 were loaded with FM4-64 (red) 24–48 h after transfection with SNAP-29-EGFP or EGFP (green). C, quantification of the relative density of FM dye-loaded synapses in neurons transfected with SNAP-29-EGFP (0.12 ± 0.018/μm) or EGFP control (0.11 ± 0.019/μm, mean ±S.E.; p > 0.05). D, immunostaining of the synaptic vesicle marker synaptophysin (physin, red) in neurons expressing SNAP-29-EGFP or EGFP (green) was used to determine the number of synapses. Cultured neurons at DIV 9–10 were obtained 24–48 h after transfection. E, quantification of the relative density of synaptophysin staining puncta in neurons transfected with SNAP-29-EGFP (0.10 ± 0.020/μm) or EGFP (0.096 ± 0.0062/μm; p > 0.05).
Overexpression of SNAP-29 in Neurons Impairs Synaptic Vesicle Turnover
One possible explanation for the effects of SNAP-29 on synaptic transmission observed (Fig. 2D) is interference with the recycling of synaptic vesicles by SNAP-29 (34). We tested this possibility by delivering a set of stimulations to the transfected presynaptic neuron. A 20-Hz train of 80-mV, 2-ms stimuli was given for 500 ms, followed by a test stimuli (80 mV, 2 ms) after various intervals, to allow for vesicle retrieval (Fig. 4A). Postsynaptic currents were rapidly depressed under the train of high frequency stimulation. Recovery ability is expressed as the “recovery ratio” or the ratio of the PSC induced by the test stimulus to the PSC induced by the first stimulus of the 20-Hz train (Fig. 4, B and C). This provides an index for the efficiency of vesicle pool turnover. In the case of 5-s interval, both untransfected and EGFP-transfected synapses showed a complete recovery (99 ± 2.2% (n = 8) and 104 ± 3.4% (n = 7), respectively), whereas the recovery ratio for the SNAP-29-EGFP group was significantly lower (89 ± 1.3% (n = 9); p < 0.01). For all time intervals (0.5, 1, and 3 s, p < 0.05; 5 s, p < 0.01) tested for recovery, neurons transfected with SNAP-29-EGFP exhibited significantly impaired ability, as compared with the EGFP group, to retrieve synaptic vesicles after depletion (Fig. 4C). However, given an extended time interval to 8 s, an almost complete recovery was observed in neurons transfected with SNAP-29-EGFP (98 ± 7.7%, n = 12), suggesting that the inhibitory role of overexpressed SNAP-29 in synaptic transmission could be fully rescued when recycling synaptic vesicles become available for future rounds of priming and fusion.
FIG. 4. SNAP-29 slows synaptic vesicle turnover.
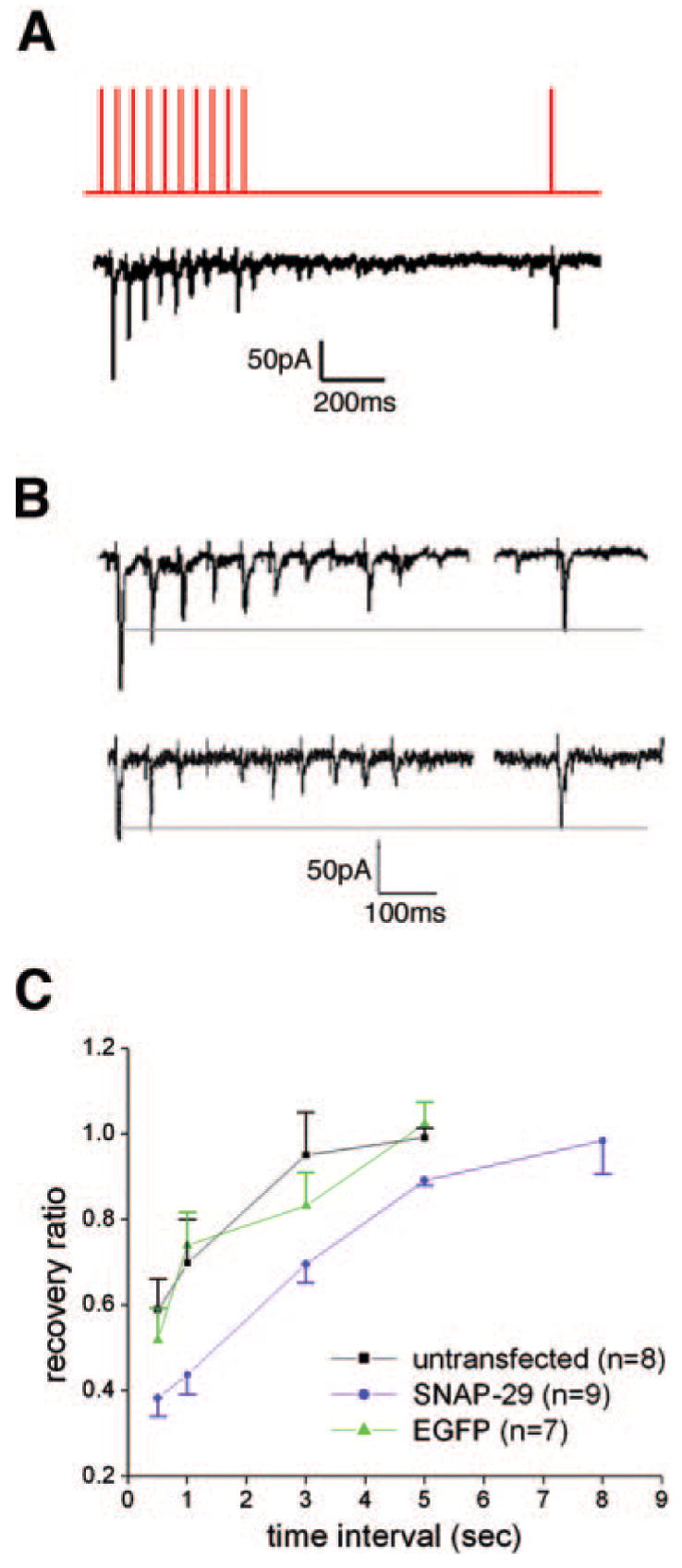
A, the top trace illustrates the stimulation pattern for examining synaptic vesicle turnover. A test stimulus was given following a 20-Hz train on the presynaptic neuron to deplete the vesicle recycling pool. The bottom trace shows PSCs recorded simultaneously from the postsynaptic neuron. B, representative PSCs recorded from neurons expressing SNAP-29-EGFP (top panel) or EGFP (bottom panel) under the same stimulation protocol illustrated in A. C, recovery ratio among three groups of synapses at different time intervals (0.5, 1, 3, 5, and 8 s). The ratio of recovery is expressed as the amplitude of the test response divided by the amplitude of the first response in the train.
The Effect of Overexpressed SNAP-29 Varies with Different Frequencies of Synaptic Activity
Previous studies on synapses formed between cultured SCGNs indicated that microinjection of SNAP-29 inhibited synaptic transmission in an activity-dependent manner, whereas the inhibition caused by SNAP-25 peptides was activity-independent (29). These results suggest that SNAP-29 has a role in modulating the recycling of synaptic vesicles or SNARE release machinery, rather than as a competitor with SNAP-25 for SNARE complex assembly. To address whether the effect of SNAP-29 on synaptic transmission in cultured hippocampal neurons is similarly activity-dependent, we applied various stimulation paradigms using different frequencies. We found a gradual reduction in PSC amplitudes under repetitive stimulations at 1 Hz for 30 s in neurons transfected with SNAP-29-EGFP (n = 10) (Fig. 5A), which is similar to that observed under 0.1-Hz stimulation. However, PSCs of the EGFP-transfected group remained constant under the 1-Hz train for 30 s (n = 10). In contrast, the inhibitory effect of SNAP-29 was absent under 0.05-Hz repetitive stimulations (Fig. 5B). When the last two (at 1 Hz) or five (at 0.05 Hz) PSCs were normalized with the initial two or five PSCs induced by the corresponding repetitive stimuli, a significant difference was observed between the 1- and 0.05-Hz stimulations (p < 0.01) in the synapses presynaptically transfected with SNAP-29-EGFP, but not in the synapses presynaptically transfected with EGFP (p > 0.05) (Fig. 5, C and D).
FIG. 5. An activity-dependent effect of SNAP-29 on synaptic transmission in cultured hippocampal neurons.
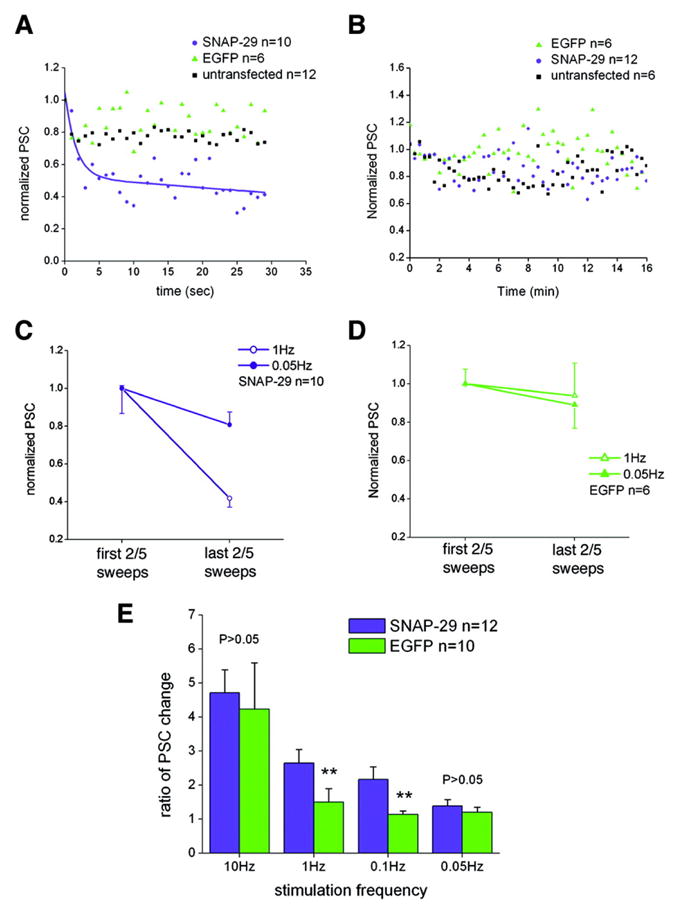
A and B, normalized amplitude of PSC plotted against recoding time under 1-Hz (A) and 0.05-Hz (B) stimulation. The tendency curve in A was fitted by exponential decay 2 (Origin Pro version 7.0 software). C and D, comparing the initial PSCs (averaged first two PSCs for 1-Hz or five PSCs for 0.05-Hz repetitive stimulations) and late PSCs (averaged last two or five PSCs) that were normalized to the mean initial PSC in the SNAP-29-transfected group (C) and EGFP-transfected group (D). Grouped Student’s t test reveals a significant difference between different stimulation paradigms in the later period of recording for the SNAP-29-EGFP-transfected group (p < 0.01), whereas no difference is observed for the EGFP-transfected group (p > 0.05). E, the ratio of PSC change was expressed as “mean amplitude of first two PSCs/mean amplitude of last two PSCs” (10 and 1 Hz) or “mean amplitude of first five PSCs/mean amplitude of last five PSCs” (0.1 and 0.05 Hz). Significant difference of changes was observed between the SNAP-29-EGFP-transfected group (n = 12) and EGFP control group (n = 10) under 0.1- and 1-Hz stimulation (**, p < 0.01, grouped Student’s t test), but not under 0.05- and 10-Hz stimulation (p > 0.05). Each column represents mean ± S.E.
To determine the possible role of SNAP-29 in synaptic transmission under high frequency stimulation, we examined the effect of SNAP-29 on the response to a 10-Hz, 2-s stimulation train. The ratio of PSC changes (mean initial period PSCs/mean late period PSCs) in both groups under 10-Hz stimulation was calculated and plotted contemporarily with those under 1-, 0.1-, and 0.05-Hz stimulations (Fig. 5E). Whereas the ratio of PSC changes was significantly different between groups overexpressing SNAP-29 and EGFP under 1- and 0.1-Hz stimulation (p < 0.05), the ratio was similar under higher (10-Hz) or lower (0.05-Hz) repetitive firing. Taken together, our results on small central synapses suggest that the effect of SNAP-29 on synaptic transmission occurs in an activity-dependent manner with a preferred frequency window for its inhibitory role. This inhibitory role likely occurs in a pattern more complicated than that we previously observed in synapses formed between SCGNs.
Overexpression of SNAP-29 Does Not Change Vesicle Release Probability
It has been hypothesized that the proteins regulating the formation of the SNARE core complex and release machinery in presynaptic terminals modify short-term plasticity (25). Vesicles with different release probabilities may undergo fusion and recycling through different mechanisms (35). Our previous biochemical studies demonstrated that SNAP-29 is involved in the disassembly of the SNARE complex. Thus, examining any potential effect of SNAP-29 on vesicle release probability is of great interest. In contrast to low and moderate frequency stimulation (Fig. 5, A and B), PSCs from synapses transfected with either SNAP-29-EGFP or EGFP showed a similarly marked depression during 10-Hz, 2-s repetitive stimulation (Fig. 6, A and B). No significant difference was observed in the inverted half-decay time between the two groups (p > 0.05, Fig. 6C), suggesting that the kinetics of vesicle release under intense synaptic activity were not affected by overexpression of SNAP-29. Furthermore, paired pulse test (36) at different frequencies (5, 10, and 50 Hz) also showed similar depression tendency curves in synapses formed by neurons transfected with SNAP-29-EGFP or EGFP control (Fig. 6D). To further confirm our electrophysiological results, we next monitored synaptic vesicle fusion and measured the unloading kinetics of the styryl dye FM4-64 (37) using confocal microscopy. All functional presynaptic terminals were loaded with FM4-64 under stimulation with 90 mM KCl followed by unloading with the same high KCl solution (Fig. 3A). The decay curves of FM4-64 intensity were averaged from 101 presynaptic terminals of 10 SNAP-29-EGFP-transfected neurons and 63 presynaptic terminals of 6 EGFP-transfected neurons (Fig. 6E). The similarity of the two decay curves suggests that neither vesicle fusion kinetics nor release probability is markedly affected by overexpression of SNAP-29.
FIG. 6. Overexpression of SNAP-29 does not change vesicle release probability.
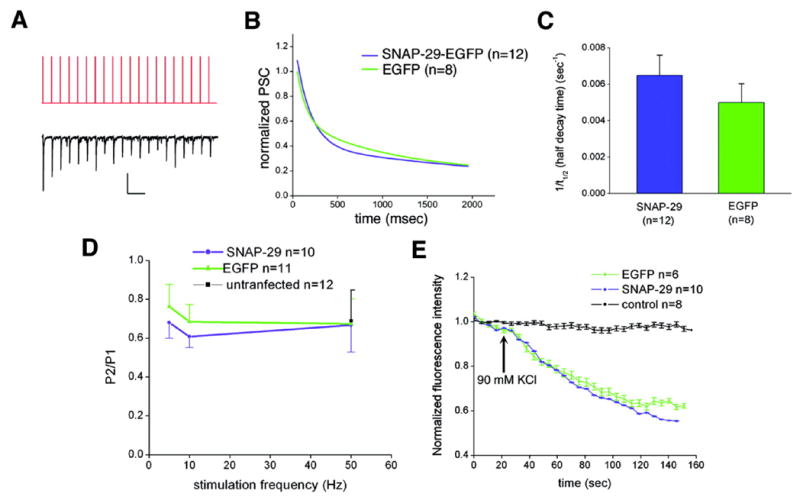
A, the stimulus pattern of a representative 10-Hz train (top trace) and the corresponding postsynaptic current (bottom trace). Scale bar, 200 pA, 200 ms. B, normalized PSCs recorded from synapses of neurons presynaptically transfected with SNAP-29-EGFP (blue) and EGFP (green) plotted against time. Decay curves of normalized PSCs were fitted by exponential decay 2 (Origin Pro version 7.0 software). C, histogram (mean ± S.E.) of the inverted half-decay time (1/t½) of the two decay curves in B. No significant difference (p > 0.05) was detected by grouped Student’s t test. D, paired pulse ratio calculated as the amplitude of second response (P2) divided by that of the first response (P1) in the SNAP-29-EGFP group (blue) and the EGFP group (green) at different frequencies (5, 10, and 50 Hz). Grouped Student’s t test reveals no significant difference between the two groups under all stimulation paradigms. E, the destaining kinetic curves of FM4-64 taken up by presynaptic terminals expressing SNAP-29-EGFP (blue) and EGFP (green) in the presence of 90 mM KCl. Normalized fluorescence intensities were plotted against time (in seconds). Real-time imaging by confocal microscopy did not reveal significant differences in destaining kinetics between the two groups. As a control, the styryl dye did not bleach under repetitive laser scanning without high K+stimulation (black).
Knockdown of SNAP-29 Expression by Small Interfering RNA Increases the Efficiency of Synaptic Transmission during Repetitive Firing
To further test whether SNAP-29 is involved in modulation of neurotransmitter release in cultured hippocampal neurons, we reduced endogenous SNAP-29 expression at the cellular level with SNAP-29-targeted siRNAs (SNAP-29-siRNAs). Five 21-nt siRNA duplexes specific to the SNAP-29 sequence were constructed into the siRNA expression vector with a GFP marker and tested extensively and rigorously under multiple controls. SNAP-29-siRNA-3, which corresponds specifically to amino acid residues 240–246 of rat SNAP-29, was selected for the current study because it was able to knockdown efficiently and specifically the expression of exogenously expressed SNAP-29-EGFP in COS-7 cells (Fig. 7A). To test whether the SNAP-29-siRNA-3 could effectively knockdown endogenous SNAP-29 expression, cultured hippocampal neurons were transfected at DIV 5 with the siRNA expression construct. Cells were immunostained with antibody against SNAP-29 4 days post-transfection. As shown in Fig. 7B, transfection of SNAP-29-siRNA-3 resulted in a significant reduction of endogenous SNAP-29 staining within cGFP-positive neurons compared with that of untransfected neurons in the same image. In contrast, the control siRNA had no effect on the expression of SNAP-29 in transfected cells (data not shown). The specificity of the siRNA was further confirmed by staining syntaxin-1, SNAP-25, and synaptophysin. The SNAP-29-targeted siRNA-3 has no inhibitory effect on the expression of these proteins in neurons (data not shown). The results from hippocampal and COS-7 cells indicate that the SNAP-29-siRNA-3 is able to efficiently and specifically knockdown the expression of both endogenously and exogenously expressed SNAP-29 in mammalian cells.
FIG. 7. Knockdown of SNAP-29 by siRNA resulted in a change of short-term synaptic plasticity.
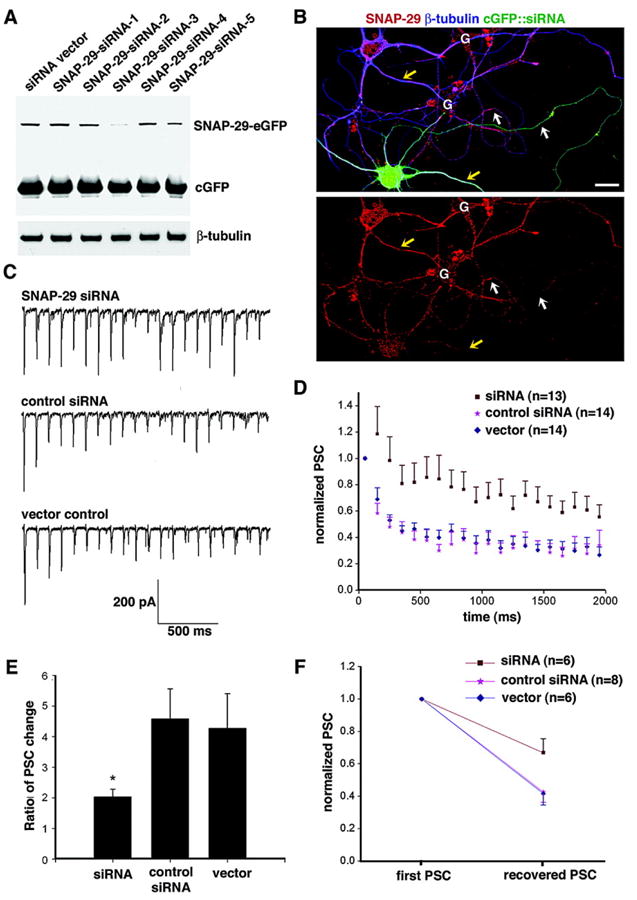
A, siRNA knocks down exogenously expressed SNAP-29 in COS cells. SNAP-29-EGFP was co-transfected into COS cells with SNAP-29-targeted siRNA constructs containing a cGFP marker or with cGFP vector alone. The level of expressed SNAP-29-EGFP was determined by immunoblotting cell lysates prepared 4 days after transfection. To monitor transfection efficacy and equal protein loading (10 μg), SNAP-29-EGFP, cGFP, and β-tubulin were sequentially immunoblotted after stripping the blot between each antibody application. Whereas SNAP-29-siRNA-1 (nt 565–585), SNAP-29-siRNA-2 (nt 432–452), SNAP-29-siRNA-4 (nt 195–215), and SNAP-29-siRNA-5 (nt 364–384) showed no significant effect on the expression of SNAP-29-EGFP in COS cells, SNAP-29-siRNA-3 (nt 719–739) exhibited a remarkable reduction in exogenously expressed SNAP-29-EGFP. B, siRNA knocks down endogenous SNAP-29 in neurons. Hippocampal neurons at DIV 5 were transfected with SNAP-29-siRNA-3 expression construct containing a cGFP marker. The neurons were fixed and stained with an antibody against SNAP-29 (red) and neuron-specific class III β-tubulin (blue) 4 days after transfection. Note that in neurons transfected with SNAP-29 siRNA, fluorescent density of SNAP-29 staining is significantly lower in soma, dendrites (yellow arrows), and axon (white arrows) compared with that of untransfected neuron in the same image (G, glia cells; scale bar, 10 μm). C, a 10-Hz, 2-s train of stimuli was given, as described in Fig. 5A, to individual pairs of connected neurons. Representative PSCs recorded from neurons receiving synaptic input from neurons transfected with SNAP-29-siRNA-3 (top trace), control siRNA (middle trace), and vector control (bottom trace). D, normalized amplitudes of PSCs recorded from different groups transfected with SNAP-29-targeted siRNA (brown), control siRNA (pink), and siRNA vector (blue) plotted against time. Each data point represents mean ± S.E. E, ratio of PSC change was calculated as the mean amplitudes of the first two traces/mean amplitudes of the last two traces. Wilcoxon rank-sum test reveals a significant reduction in the ratio of PSC change in the SNAP-29-siRNA group as compared with the control siRNA group and vector group (both p < 0.05). Each column represents mean ± S.E. F, stimuli testing vesicle turnover rate are given as illustrated in Fig. 4, A and B. Amplitude of the PSC in response to the test stimulus was normalized to that of its initial PSC recorded in the train for all three groups. Grouped Student’s t test reveals significant differences (p < 0.05) between the SNAP-29 knockdown group and its two control groups.
Our previous and current studies by injection or overexpression of SNAP-29 in presynaptic neurons suggest that SNAP-29 acts as a negative modulator of neurotransmitter release. Thus, reduced expression of endogenous SNAP-29 by siRNA would be expected to relieve its negative effect on vesicle release and consequently increase the efficacy of synaptic transmission during repetitive firing. Because we observed that PSCs from both untransfected and EGFP-transfected synapses remained constant under 0.1 Hz for 15-min stimulations and under 1 Hz for 30-s stimulations (Fig. 5, A and B), we selected 10 Hz as the stimulation paradigm under which PSCs from control synapses showed a marked depression during 2-s repetitive stimulation (Fig. 6A). Cultured neurons (DIV 5–7) were transfected with siRNA constructs, and whole-cell dual patch clamp recordings were performed 96 h after transfection from selected synapses in which presynaptic neurons expressed cGFP marker of the siRNA vector. Representative traces recorded under 10-Hz train were shown for each transfection group (Fig. 7C). Whereas PSCs of both control siRNA and vector groups showed immediate decay under repetitive stimulation, that of the SNAP-29 siRNA group appeared much more steady. Normalized amplitudes of PSCs were plotted against time in different transfection groups (Fig. 7D). The decay curves of control siRNA and vector-transfected groups exhibited similar depression kinetics. However, the depression of PSCs was significantly slower in the SNAP-29 knockdown group, and facilitation was observed during the first two pulses in the train. The ratio of averaged amplitude of the first two to that of the last two PSCs was calculated as an indicator of the percentage of recycling vesicles being depleted. As shown in Fig. 7E, the ratio of PSC change for the knockdown group (siRNA, 2.0 ± 0.25) is significantly smaller than that of other groups (control siRNA, 4.6 ± 0.98, p < 0.05; vector, 4.3 ± 1.1, p < 0.05), implicating an up-regulated vesicle turnover mechanism in neurons deficient in SNAP-29. To further confirm this observation, the 500-ms interval recovery stimulation as described in Fig. 4 was given to SNAP-29 knockdown group and control group. The recovered PSC as normalized to the initial PSC in SNAP-29 knockdown group (67 3 8.7%) was significantly higher than that in the control siRNA group (43 3 6.4%, p < 0.05) and the vector control (42 ± 7.0%, p < 0.05) (Fig. 7F), indicating a more efficient nature of vesicle turnover in SNAP-29 knockdown neurons. Our results from the siRNA knockdown approach are consistent with observations taken from previous and current studies using injection and overexpression of SNAP-29 and further suggest that SNAP-29 is a negative modulator of synaptic transmission, probably by slowing recycling of the SNARE-based fusion machinery and synaptic vesicle turnover.
DISCUSSION
Many proteins in the presynaptic terminal, including the synapsins (38) and RIM1α (39), have been reported to play regulatory roles in neurotransmitter release and thereby contribute to various forms of presynaptic plasticity. In the present study, we investigated the role of a recently identified protein, SNAP-29, in modulation of synaptic transmission and vesicle recycling in cultured rat hippocampal neurons. Previous biochemical studies suggest that SNAP-29 competes with α-SNAP for binding to syntaxin-1A, thereby inhibiting disassembly of the SNARE complex. The activity-dependent inhibition of synaptic transmission in cultured SCGNs further implicates a potential regulatory role in synaptic vesicle recycling. Here, we took advantage of small central synapses, which have a limited vesicle supply and rely heavily on vesicle recycling to sustain repetitive synaptic activity (40). Our study demonstrates that overexpression of SNAP-29 inhibits sustained synaptic transmission within a specific frequency window (0.1–1 Hz) and that the impairment in transmission was not caused by an intrinsic defect in basal synaptic function, but rather a defect in synaptic vesicle turnover. Furthermore, reduced expression of endogenous SNAP-29 using siRNA significantly enhances the efficacy of synaptic transmission under repetitive stimulation.
The restricted inhibitory role of overexpressed SNAP-29 that we observed in cultured hippocampal neurons implies that vesicle recycling may be regulated differently under different stimulation frequencies. It has been suggested that only vesicles in the recycling pool in the vicinity of the active zone are involved in transmitter release under low frequency repetitive stimulation, whereas vesicles in the reserve pool are mobilized only after strong stimulations such as high K+ or high frequency trains. Our findings that overexpression of SNAP-29 inhibits synaptic activity at 0.1 and 1 Hz, but not at 10 Hz, suggest that SNAP-29 acts as a modulator for rapid recruitment of vesicles from the recycling pool. After both 0.1- and 1-Hz stimulations, overexpressed SNAP-29-induced inhibition occurred in an especially rapid manner, implying that there are only few release-ready vesicles docked at these active zones in cultured hippocampal neurons. This is in accordance with previous optical studies, which demonstrated that ~2–6 vesicles are docked at small boutons and that no more than 20 vesicles are docked at large ones (3–4). Thus, vesicles available for recycling are limited. The hippocampal neurons used in this study were at age DIV 8–11, at which presynaptic boutons are unlikely to be fully mature. Thus, the release-ready vesicles are quickly depleted under relatively low frequencies of stimulation. An immediate replenishment of release-ready vesicles from the recycling pool is critical for efficient synaptic transmission during repetitive synaptic activity. It is presently believed that SNARE complex dissociation and vesicle budding could be directly coupled (41). The cis-SNARE complexes are likely dissociated immediately after fusion to permit the direct recycling and specific uptake of the vesicle-associated SNARE into budding vesicle. The priming step of docked vesicles corresponds to the assembly of the trans-SNARE complex by active (dissociated) cognate SNAREs. Our previous biochemical studies suggested an inhibitory role for SNAP-29 on synaptic transmission by slowing recycling of the SNARE core complex. SNAP-29 competes with α-SNAP for binding to the SNARE protein syntaxin and consequently inhibits the disassembly of the SNARE complex by N-ethylmaleimide-sensitive factor after fusion (29). Given the presence of enriched target membrane SNAREs (syntaxin-1A and SNAP-25) at the presynaptic targeting membrane, the observed impairment of exocytosis was most likely the result of a deficiency of free vesicle-associated SNAREs (VAMP) on recycling-competent synaptic vesicles. However, vesicle recycling differs under intensive stimulation, such as a 10-Hz train and high K+. Under these conditions, vesicles in the reserve pool quickly mobilize to replenish the recycling pool in order to meet the needs of sustained transmitter release. We hypothesize that more free or active vesicle-associated SNAREs carried by recycling vesicles from the reserve pool are recruited to replenish VAMP/synaptobrevin at the active zone and facilitate the priming of the docked vesicles. This assumption is consistent with our observation that pre-synaptic neurotransmitter release was unaffected by overexpression of SNAP-29 during intensively repetitive stimulation. The results that low frequency activity (0.05 Hz) was not affected by SNAP-29 (Fig. 5) and that synaptic depression can be fully rescued under 8-s interval recovery test suggest that the inhibition was, to some extent, reversible.
Release probability is critically dependent on the size of the readily releasable pool (42–44) and is closely related to the number of recycling vesicles present during firing at 10-Hz stimulation in hippocampal neurons (45). Thus, the efficiency of vesicle turnover in the presynaptic terminal and recycling of the SNARE proteins after fusion would make a significant contribution to the efficacy of synaptic transmission under repetitive stimulation. Although overexpression of SNAP-29 in hippocampal neurons revealed no statistically significant difference in depression kinetics under 10-Hz stimulation, knockdown of endogenous SNAP-29 by siRNA slowed depression of synaptic responses, suggesting that this protein plays an inhibitory role in vesicle turnover under physiological conditions. These observations are in perfect accordance with our previous biochemical findings that SNAP-29 inhibits SNARE complex disassembly (29). A deficiency of SNAP-29 in neurons would allow the efficient dissociation of the SNARE core complex following fusion to occur, consequently speeding up the recycling of the SNARE proteins and turnover of synaptic vesicles, resulting in an increased pool size of release-ready vesicles. Thus, our studies with SNAP-29-targeted siRNA suggest that certain amounts of endogenous SNAP-29 localized in synapses are sufficient in regulating synaptic vesicle turnover and short-term plasticity in mammalian central nervous system.
Our observation that overexpressed SNAP-29 inhibits synaptic transmission at frequencies of 0.1 and 1 Hz could be due to impaired turnover of synaptic vesicles and/or the fusion machinery; however, one cannot exclude the possibility that impairment could be caused by a SNAP-29-induced reduction in presynaptic N- or P/Q-calcium channel-mediated calcium transients at the nerve terminals. It has been well documented in the literature that syntaxin-1A interacts with N- and P/Q-type calcium channels through a channel motif termed the “synprint” site (46, 47) and influences the synchronous transmitter release by changing the inactivation kinetics of these calcium channels (48, 49). However, our studies clearly show that the inhibitory effect of overexpressed SNAP-29 does not coincide with the calcium influx required for excitation-secretion coupling but rather is confined to a specific frequency window of synaptic activity (Fig. 5E). As shown in Fig. 2, E and F, overexpression of SNAP-29 in neurons does not affect basal synaptic transmission. In addition, neither vesicle fusion kinetics nor release probability is affected by overexpression of SNAP-29. Taken together, our results well argue against the possibility that the observed effect of overexpressed SNAP-29 on synaptic transmission could have resulted from modulation of calcium channel inactivation.
Due to their specific structural characteristics, different synapses may utilize different mechanisms to regulate synaptic efficacy (50). For example, at the neuromuscular junction, the same stimulus protocol yields quite different results compared with those observed in the present study, possibly due to profuse vesicles docked at neuromuscular junction terminals, making depletion less likely. The present study using hippocampal synapses complements our previous results with rat SCGNs and helps us to better understand the role of SNAP-29 in different brain regions. In addition, our studies may have implications for a molecular mechanism to correlate recycling processes of SNARE-based fusion machinery with turnover of synaptic vesicles under repetitive synaptic activity. However, several key questions remain to be addressed. Does the recycling of the SNARE proteins remain unchanged while vesicle recycling is impaired and vice versa? Does the SNARE complex undergo dissociation in fusion models such as “kiss-and-run” and “kiss-and-stay” (35)? Are synaptic vesicle recycling and activation of the SNAREs or dissociation of cis-SNARE complex coupled and, if so, are they regulated differently under different stimulation conditions? Addressing these questions will provide insights into the regulation of synaptic vesicle turnover and shed new light on our understanding of presynaptic mechanisms for modulating synaptic efficacy.
Acknowledgments
We thank S. Mochida for insightful advice on data analysis; J. Rettig, L.-G. Wu, M. Leenders, J.-H. Xu, B. McNeil, and C. Gerwin for critical reading of the manuscript; and X.-Y. Zhu and C.-H. Wu for providing the hippocampal neuron culture.
Abbreviations used
- VAMP
vesicle-associated membrane protein
- PSC
postsynaptic current
- siRNA
small interfering RNA
- GFP
green fluorescent protein
- EGFP
enhanced green fluorescent protein
- SCGN
superior cervical ganglion neuron
- nt
nucleotide(s)
- PBS
phosphate-buffered saline
- DIV
days in vitro
Footnotes
This work was supported in part by the intramural research program of NINDS National Institutes of Health (Z.-H. S.) and Major State Basic Research Program of China Grant G200077800 and National Natural Science Foundation of China Grant 30321002 (to S. D.).
References
- 1.Zucker RS, Regehr WG. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 2.Richards DA, Guatimosim C, Betz WJ. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 3.Harris KM, Sultan P. Neuropharmacology. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-s. [DOI] [PubMed] [Google Scholar]
- 4.Schikorski T, Stevens CF. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothman JE. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 6.Bajjalieh SM, Scheller RH. J Biol Chem. 1995;270:1971–1974. doi: 10.1074/jbc.270.5.1971. [DOI] [PubMed] [Google Scholar]
- 7.Sü dhof TC. Nature. 1995;375:645–653. [Google Scholar]
- 8.Trimble WS, Cowan DM, Scheller RH. Proc Natl Acad Sci U S A. 1988;85:4538–4542. doi: 10.1073/pnas.85.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC. J Cell Biol. 1989;109:3039–3052. doi: 10.1083/jcb.109.6.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett MK, Calakos N, Scheller RH. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- 11.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 12.Calakos N, Bennett MK, Peterson K, Scheller RH. Science. 1994;263:1146–1149. doi: 10.1126/science.8108733. [DOI] [PubMed] [Google Scholar]
- 13.Fasshauer D, Eliason WK, Brü nger AT, Jahn R. Biochemistry. 1998;37:10354–10362. doi: 10.1021/bi980542h. [DOI] [PubMed] [Google Scholar]
- 14.Augustine GJ. Curr Opin Neurobiol. 2001;11:320–326. doi: 10.1016/s0959-4388(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 15.Chapman ER. Nat Rev Mol Cell Biol. 2002;3:498–508. doi: 10.1038/nrm855. [DOI] [PubMed] [Google Scholar]
- 16.Mayer A, Wickner W, Haas A. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 17.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi T, Yamasaki S, Nauenburg S, Binz T, Niemann H. EMBO J. 1995;14:2317–2325. doi: 10.1002/j.1460-2075.1995.tb07226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 20.Barnard RJ, Morgan A, Burgoyne RD. J Cell Biol. 1997;139:875–883. doi: 10.1083/jcb.139.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Littleton JT, Barnard RJ, Titus SA, Slind J, Chapman ER, Ganetzky B. Proc Natl Acad Sci U S A. 2001;98:12233–12238. doi: 10.1073/pnas.221450198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- 23.Sanyal S, Tolar LA, Pallanck L, Krishnan KS. Neurosci Lett. 2001;311:21–24. doi: 10.1016/s0304-3940(01)02125-5. [DOI] [PubMed] [Google Scholar]
- 24.Kawasaki F, Mattiuz AM, Ordway RW. J Neurosci. 1998;18:10241–10249. doi: 10.1523/JNEUROSCI.18-24-10241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rettig J, Neher E. Science. 2002;298:781–785. doi: 10.1126/science.1075375. [DOI] [PubMed] [Google Scholar]
- 26.Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- 27.Sippy T, Cruz-Martin A, Jeromin A, Schweizer FE. Nat Neurosci. 2003;6:1031–1038. doi: 10.1038/nn1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakur P, Stevens DR, Sheng ZH, Rettig J. J Neurosci. 2004;24:6476–6481. doi: 10.1523/JNEUROSCI.0590-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su Q, Mochida S, Tian JH, Mehta R, Sheng ZH. Proc Natl Acad Sci U S A. 2001;98:14038–14043. doi: 10.1073/pnas.251532398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steegmaier M, Yang B, Yoo JS, Huang B, Shen M, Yu S, Luo Y, Scheller RH. J Biol Chem. 1998;273:34171–34179. doi: 10.1074/jbc.273.51.34171. [DOI] [PubMed] [Google Scholar]
- 31.Hohenstein AC, Roche PA. Biochem Biophys Res Commun. 2001;285:167–171. doi: 10.1006/bbrc.2001.5141. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Wang H, Ye C, Ge W, Chen Y, Wu CP, Poo M, Duan S. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- 33.Lao G, Scheuss V, Gerwin CM, Su Q, Mochida S, Rettig J, Sheng ZH. Neuron. 2000;25:191–201. doi: 10.1016/s0896-6273(00)80882-x. [DOI] [PubMed] [Google Scholar]
- 34.Galli T, Haucke V. Sci STKE. 2001;88:RE1. doi: 10.1126/stke.2001.88.re1. [DOI] [PubMed] [Google Scholar]
- 35.Gandhi SP, Stevens CF. Nature. 2003;423:607–613. doi: 10.1038/nature01677. [DOI] [PubMed] [Google Scholar]
- 36.Stevens CF, Wang Y. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- 37.Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 38.Chi P, Greengard P, Ryan TA. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- 39.Calakos N, Schoch S, Sudhof TC, Malenka RC. Neuron. 2004;42:889–896. doi: 10.1016/j.neuron.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Harata N, Pyle JL, Aravanis AM, Mozhayeva M, Kavalali ET, Tsien RW. Trends Neurosci. 2001;24:637–643. doi: 10.1016/s0166-2236(00)02030-0. [DOI] [PubMed] [Google Scholar]
- 41.Sollner TH. Mol Membr Biol. 2003;20:209–220. doi: 10.1080/0968768031000104953. [DOI] [PubMed] [Google Scholar]
- 42.Dobrunz LE, Stevens CF. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 43.Murthy VN, Sejnowski TJ, Stevens CF. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 44.Schikorski T, Stevens CF. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- 45.Waters J, Smith SJ. J Physiol. 2002;541:811–823. doi: 10.1113/jphysiol.2001.013485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheng ZH, Rettig J, Takahashi M, Catterall WA. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 47.Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Proc Natl Acad Sci U S A. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bezprozvanny I, Scheller RH, Tsien RW. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 49.Degtiar VE, Scheller RH, Tsien RW. J Neurosci. 2000;20:4355–4367. doi: 10.1523/JNEUROSCI.20-12-04355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atwood HL, Karunanithi S. Nat Rev Neurosci. 2002;3:497–516. doi: 10.1038/nrn876. [DOI] [PubMed] [Google Scholar]