

Abstract
This study focused on the activation of cell cycle checkpoint responses in diploid human fibroblasts that were treated with cadmium chloride and the potential roles of ATM and p53 signaling pathways in cadmium-induced responses. The alkaline comet assay indicated that cadmium caused a dose-dependent increase in DNA damage. Cells that were rendered p53-defective by expression of a dominant-negative p53 allele or knockdown of p53 mRNA were more resistant to cadmium-induced inactivation of colony formation than normal and ataxia telangiectasia (AT) cells. Synchronized fibroblasts in S were more sensitive to cadmium toxicity than cells in G1 suggesting that cadmium may target some element of DNA replication. Cadmium produced a dose- and time-dependent inhibition of DNA synthesis. An immediate inhibition was associated with severe delay in progression through S phase and a delayed inhibition seen 24 h after treatment was associated with accumulation of cells in G2. AT and normal cells displayed similar patterns of inhibition of DNA synthesis and G2 delay after treatment with cadmium, while p53-defective cells displayed significantly less of the delayed inhibition of DNA synthesis and accumulation in G2 post-treatment. Total p53 protein and ser15-phosphorylated p53 were induced by cadmium in normal and AT cells. The p53 transactivation target Gadd45α was induced in both p53-effective and p53-defective cells after 4 h cadmium treatment, and this was associated with an acute inhibition of mitosis. Cadmium produced a very unusual pattern of toxicity in human fibroblasts, inhibiting DNA replication and inducing p53-dependent growth arrest but without induction of p21Cip1/Waf1 or activation of Chk1.
Keywords: Cadmium, checkpoint, fibroblasts, DNA synthesis, p53, ataxia telangiectasia, Gadd45α
Introduction
Cadmium is a systemic poison affecting many cellular functions. Cadmium contamination poses a serious health threat throughout the world, and cadmium has been classified as a human carcinogen by the International Agency for Research on Cancer (1994). Toxic responses to cadmium exposure include kidney damage, respiratory diseases, neurological disorders, and lung, kidney, prostate and testicular cancers (Abdulla and Chmielnicka, 1989; Waalkes, 2003).
Cadmium exposure can induce intracellular damage through several mechanisms. In cultured cells cadmium produces direct and indirect genotoxic effects such as DNA strand breaks, DNA-protein crosslinks, oxidative DNA damage, and chromosomal aberrations (Dally and Hartwig, 1997; Hwua and Yang, 1998; Misra et al., 1998). Several cellular factors that respond to DNA damage to regulate proliferation also respond to cadmium exposure. The tumor suppressor gene p53 and proto-oncogenes c-Jun, c-Fos and c-Myc were transcriptionally activated by cadmium (Jin and Ringertz, 1990; Zheng et al., 1996), and cadmium caused an irreversible G2/M arrest (Yang et al., 2004). It was therefore of interest to determine whether cadmium-induced DNA damage causes cells to arrest cell division through p53-dependent cell cycle checkpoints.
Cell cycle checkpoints are biochemical surveillance pathways that actively delay or arrest cell cycle progression in response to DNA damage and activated oncogenes (Kaufmann and Paules, 1996; Kastan and Bartek, 2004). DNA damage checkpoints minimize the probability of replicating and segregating damaged DNA and, therefore, reduce the frequencies of mutations and chromosomal aberrations that are induced by genotoxic stress. p53 and ataxia telangiectasia-mutated (ATM) are important signaling molecules in checkpoint responses to DNA damage (Sancar et al., 2004). ATM is a protein kinase that is activated by autophosphorylation in response to DNA damage and alterations in chromatin structure (Bakkenist and Kastan, 2003). As guardian of the genome and a substrate for ATM, p53 enforces G1 arrest through transactivation of p21Cip1/Waf1 and G2 delay through transrepression of growth-regulated genes such as cyclin B1 and Cdk1 (Agarwal et al., 1998). Germline mutations in p53 and ATM are seen in the familial cancer syndromes, Li-Fraumeni (Malkin et al., 1990) and ataxia telangiectasia (AT) (Savitsky et al., 1995), respectively. Checkpoint responses to environmental carcinogens such as cadmium may suppress cancer development.
Cadmium has the propensity to replace zinc in biological material and several DNA repair factors including p53 (Meplan et al., 1999), XPA (Asmuss et al., 2000), and hMutS-α (Clark and Kunkel, 2004; Banerjee and Flores-Rozas, 2005) are inhibited by cadmium. Inhibition of DNA repair or other elements of DNA damage response such as apoptosis could sensitize cells to carcinogenesis by endogenous or exogenous stresses that damage DNA (Mukherjee et al., 2004; Sancar et al., 2004). Reduced DNA repair increases mutagenesis and clastogenesis by chemical carcinogens and radiations, and reduced apoptosis increases the yields of cells that survive with mutations and chromosomal aberrations.
In the present study, the effects of cadmium chloride (CdCl2) on DNA and cell division were examined in diploid human fibroblasts. Although fibroblasts are not targets of cadmium toxicity in vivo, they represent an excellent in vitro model for elucidating mechanisms of DNA damage response. Unlike transformed and cancer cell lines, diploid human skin fibroblasts express the full repertoire of repair and cell cycle checkpoint gene products that respond to carcinogen-induced DNA damage. Exposure of human fibroblast lines to cadmium caused DNA damage and induced a concentration- and time-dependent inhibition of DNA synthesis and mitosis. Cadmium induced a very unusual pattern of toxicity in fibroblasts, with p53-dependent inactivation of colony formation in the absence of induction of p21Cip1/Waf1, and inhibition of DNA replication without activation of Chk1.
Materials and Methods
Cell lines and culture conditions
The normal human fibroblast strains, F1, F3 and F10, were derived from neonatal foreskin and established in culture according to established methods (Maher and McCormick, 1976; Boyer et al., 1991). AT fibroblasts were isolated from the skin of an affected individual. The original fibroblast strain (GM02052A) was obtained from the NIGMS Human Genetic Cell Repository. Immortalized cell lines from these strains of human fibroblasts were obtained by ectopic expression of the human telomerase reverse transcriptase (hTERT), as previously described (Deming et al., 2002; Heffernan et al., 2002). The immortalized normal human fibroblasts were denoted as F1-hTERT, F3-hTERT, F10-hTERT and the telomerase-expressing AT cell line as GM02052A.
Fibroblasts were cultured in DMEM (Invitrogen) supplemented with 2 mM L-glutamine (Invitrogen) and 10% or 20% (AT cells) fetal bovine serum (Hyclone). All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2, and were tested and shown to be free of mycoplasma contamination using a commercial kit (Gen-Probe).
Plasmids and viruses
F10-hTERT fibroblasts were engineered to express a dominant-negative form of p53 (a histidine to glutamine substitution at amino acid 179 in p53) by infection with a replication-defective retrovirus carrying the mutant p53 cDNA (a generous gift from Dr. Howard Liber, Colorado State University) and the neomycin-resistance gene under the control of the same viral promoter (pLXIN) (Simpson et al., 2005). The resulting derivative line was denoted as F10-hTERT-p53H179Q. An isogenic cell line carrying the empty vector (F10-hTERT-LXIN) was derived in parallel.
The retroviral short interfering RNA (siRNA) vector to inactivate p53 was purchased from Oligoengine. Vesicular stomatitis virus glycoprotein G-pseudotyped, replication-defective retroviruses were produced as previously described following transient transfection of viral vector and helper plasmids into HEK 293T cells (Olsen et al., 1993; Comstock et al., 1997; Johnson et al., 1998). As F10-hTERT was initially selected in 200μg/μl puromycin, stable expression of the p53 RNAi was done by growth in 400μg/μl puromycin. Batch cultures of puromycin-resistant F10-hTERT-p53RNAi were expanded and shown to display 90% inactivation of p53-dependent DNA damage G1 checkpoint function (results not shown).
Cell treatment with cadmium
A stock solution of cadmium chloride (Alfa Aesar) was prepared at 10 mM in sterile H2O. Cadmium was added directly to culture medium. Cells were exposed to cadmium for 4 hours at concentrations ranging from 40–80 μM. After treatment, medium was removed, cells were rinsed with phosphate-buffered saline (PBS) and fresh medium replaced. A sham-treatment control was incorporated in each assay using the same manipulations but without cadmium. All experiments were performed in triplicate in independent trials to assess reproducibility.
Colony formation ability
Colony formation was measured in logarithmically growing cells, plated at 500 cells per 100 mm diameter dish and incubated for 8 hours before the 4 h cadmium treatment. Cells were cultured for 2 weeks, changing medium twice each week. Colonies were fixed and stained with a solution of 40% methanol and 0.05% crystal violet. Colonies of 50 or more cells were counted. Three individual dishes were assayed per treatment and the mean values were used to estimate cytotoxicity. Cytotoxicity was determined as the relative colony-forming ability (the cloning efficiency of the treated cells divided by the cloning efficiency of the untreated cells, multiplied by 100).
Comet assay
The comet assay to detect DNA damage was performed using the method of Sasaki et al. with some modifications (Sasaki et al., 1997). Briefly, fibroblasts were exposed to 40, 60 and 80 μM cadmium for 4 h. At the end of the incubation with cadmium, cells were removed from the plates with trypsin. Trypsin was inactivated with serum-containing medium and cells were collected by sedimentation and resuspension in PBS. Ten microliters of the cell suspension (~100,000 cells/ml) were diluted in 70 μl low-melting-point agarose (0.75% w/v in PBS). The resulting suspension were embedded in previously prepared normal-melting-point agarose (1% w/v in PBS) on frosted slides followed by the addition of 75 μl of normal-melting-point agarose (1%). The slides were then immersed in lysis buffer (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris-HCl, pH 10) for 1 h at 4°C in the dark. After lysis slides were placed in alkaline electrophoresis buffer (0.3 M NaOH, 1 mM Na2EDTA) for 20 min at 4°C to denature DNA and express alkali-labile sites. Electrophoresis was carried out at 4°C for 20 min at 24 V and approximately 300 mA. The slides were then washed twice in neutralizing buffer (0.4 M Tris-HCl, pH 7.4) for 5 min. The DNA was stained by adding 45 μl of ethidium bromide (20 μg/ml) to each slide. More than fifty nuclei on each slide were examined for the presence of comet tails at 200 X magnification using a fluorescence microscope (Zeiss Axioskop) equipped with a green filter. The percent of nuclei with a DNA tail of ≥ 25 μm was used as the measure of DNA damage.
G1 and G2 checkpoint analyses
G1 and G2 checkpoint functions were analyzed at 0, 6 and 24 h after the 4 h cadmium treatment or sham treatment. G1 checkpoint function was assessed by measuring incorporation of 5′-bromo-2′-deoxyuridine (BrdU) by S phase cells (Kaufmann et al., 1995) and G2 checkpoint function was assessed by measuring mitotic cells with 4N DNA that stained positive for mitosis-specific phospho-histone H3 (Juan et al., 1998; Deming et al., 2002). Summit (Cytomation Inc.) flow cytometry analysis software was used to quantify the numbers of unlabeled cells with 2N (G0/G1) and 4N DNA content (G2), BrdU-labeled cells with 2–4N DNA content (S), and 4N cells with phospho-histone H3 (M).
Cell synchronization
F1-hTERT, F3-hTERT and F10-hTERT fibroblasts were synchronized as previously described (Cordeiro-Stone et al., 1986; Kaufmann and Wilson, 1990). Briefly, cells were plated at a density of 1.3×104/cm2 and allowed to grow for 8 days to confluence arrest (G0 phase). Confluence-arrested cells were trypsinized, reseeded at 500 cells per 100 mm dish and incubated for 8 h to allow them to reenter the cell cycle and reach G1 or for 20 h to allow them to reach S. The cells were then treated with different concentrations of cadmium for 4 h followed by washing with PBS and incubation in fresh medium for two weeks to observe colony formation ability.
Cells were similarly released from confluence-arrest, reseeded at 1 million cells per 100 mm dish and incubated with the DNA polymerase inhibitor, aphidicolin (APC), at a concentration of 2 μg/ml for 24 h to collect cells at the beginning of S phase (Kaufmann and Wilson, 1990). APC was washed out and cells incubated in fresh medium containing cadmium for 6 h to observe the increase in DNA content by flow cytometry.
Western immunoblot analyses
Logarithmically growing cells were seeded at 106 per 150-mm dish and incubated for 40 h. Cultures were treated as described above and incubated in fresh medium for 0, 2, 6 and 24 h at 37°C. Cells were harvested by trypsinization, washed once in phosphate-buffered saline, and resuspended in lysis buffer (10 mM sodium phosphate buffer [pH 7.2], 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, and 1% NP-40, supplemented with 10 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 10 mM β-glycerophosphate, 10 mM sodium orthovanadate, and 10 μg/ml leupeptin and aprotinin). Protein concentrations were determined using the Bio-Rad DC protein assay (Bio-Rad Laboratories). Samples containing equal amounts of protein were mixed with an equal volume of 2× Laemmli sample buffer (125 mM Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate (SDS), 20% glycerol) containing 5% β-mercaptoethanol, boiled, and proteins separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to nitrocellulose and probed with antibodies against p21Cip1/Waf1 (Ab-11, Lab vision), p53 (Ab-6, Lab Vision), phospho-ser15 p53 (Cell Signaling), Chk1 (G4, Santa Cruz Biotechnology), phospho-Ser317 Chk1 (Cell Signaling), Chk2 (Upstate Biotechnology Inc.), phospho-Thr68 Chk2 (Cell Signaling), actin (Santa Cruz Biotechnology), caspase3 (Upstate Biotechnology Inc.), PARP (Upstate Biotechnology Inc.), and GADD45α (Santa Cruz Biotechnology).
DNA synthesis assay
Logarithmically growing cells were plated at a density of 2 × 105 cells per 60 mm dish and grown at 37°C for 24 h. The time course of inhibition of DNA synthesis in all cell lines was measured by adding 40 and 80 μM cadmium to culture medium and at various times later pulse-labeling DNA with 10 μCi/ml [3H]-thymidine for 30 min. During the 4 h treatment time, DNA synthesis was measured every hour. After the 4 h treatment, the cadmium was removed and DNA synthesis was measured at 1, 2, 6, 12 and 24 h later. The DNA synthesis assay was described in detail elsewhere (Kaufmann et al., 2003) with some modifications. Briefly, radioactive medium was removed and plates washed twice with cold PBS before adding 3 ml of cold 4% trichloroacetic acid (TCA) and incubating at 4°C for 30 min. After washing the plates with cold 4% TCA at 4°C, the fixed cells were dissolved in 0.4 M NaOH and transferred to test tubes. The Abs260 was measured to estimate nucleic acid content. Acid-insoluble material was then collected on GF/C microfiber glass filters for measurement of radioactivity by liquid scintillation spectrometry. Net 3H radioactivity was normalized for cell number (the concentration of nucleic acids). Mean 3H/nucleic acid ratios from triplicate cultures were determined as DNA synthesis rates.
Statistical analysis
Statistical evaluation was done using the SPSS 11.5 software (SPSS Inc., Lead Technologies, Chicago, IL, USA). In all cases, a P value < 0.05 was considered to represent a significant difference. All data represent the means ± SEM of three or more replicates. Student’s t-test or ANOVA followed by Dunnett’s multiple comparison test was used, as appropriate, to define differences between experimental groups and controls.
Results
Induction of DNA damage by cadmium
The immediate effects of 4 h cadmium treatment on DNA integrity in fibroblasts are shown in Figure 1. DNA damage was assessed by measuring the percent of nuclei with comet tails ≥25 μm. Treatment with 4.5 Gy of IR as a positive control to produce DNA single-and double-strand breaks substantially increased the fraction of nuclei with tails. Incubation with cadmium resulted in a dose-dependent increase of comets of tail length ≥25 μm in normal, p53-defective and AT cells. Significant differences were observed between the untreated cells and those treated with 60 and 80 μM of cadmium. These results support previous studies showing induction of DNA damage in cadmium-treated human lung fibroblasts and HEPG2 cells (Fatur et al., 2002; Mouron et al., 2004).
Figure 1.
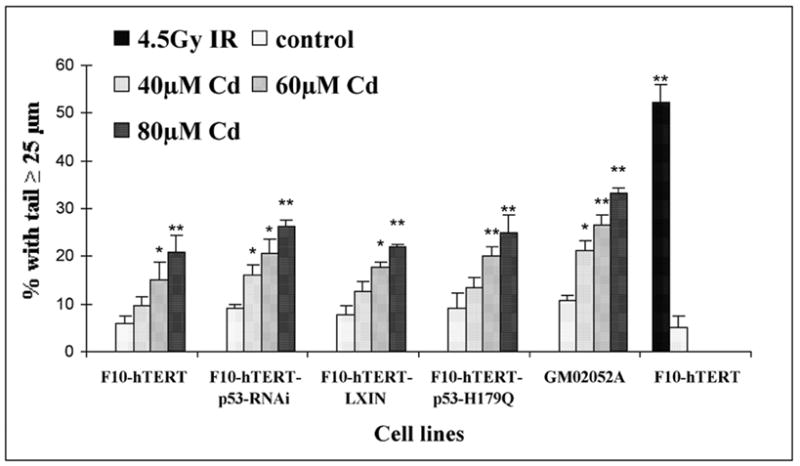
Cadmium-induced DNA damage in human fibroblasts. At the end of the 4 h incubation with 40, 60 and 80 μM cadmium, fibroblasts were removed from the plates with trypsin and collected for comet assay. The percentages of cells with a comet tail ≥ 25 μm were determined as a measure of DNA damage. Fifty cells were analyzed per slide. Mean values of three independent experiments ± S.E. are presented. (*) Denotes values that were significantly different from the untreated control culture (* p<0.05; ** p<0.01). The culture that was treated with 4.5 Gy IR was harvested immediately after irradiation.
Cytotoxicity of cadmium in normal, AT and p53-defective cells
The cytotoxicity of cadmium was determined by colony formation assay. Treatment with cadmium inhibited single cell colony formation by the F1-hTERT, F3-hTERT, F10-hTERT and GM02052A cell lines with similar dose-kinetics (Fig. 2A). For these lines a concentration of 60 μM cadmium inactivated colony formation by about 40%. p53-defective cells were resistant to cadmium in comparison with normal cells, as shown in Figure 2B. The colony formation efficiencies of cells that were treated with 80 μM cadmium were 75% for F10-hTERT-p53-RNAi and 50% for F10-hTERT-p53-H179Q as compared to 21% for F10-hTERT and 20% for F10-hTERT-LX1N.
Figure 2.
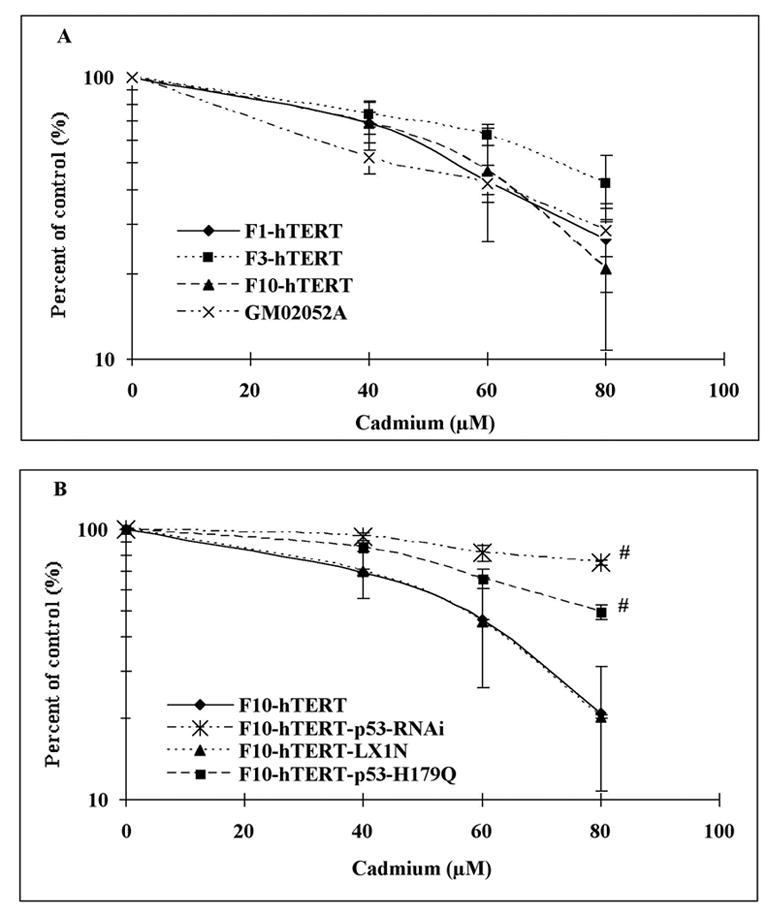
Inactivation of clonogenic survival by cadmium. Colony formation was measured in logarithmically growing cells, plated at 500 cells per 100 mm dish and incubated 8 h before the 4 h cadmium treatment. Fibroblast lines were treated with 0, 40, 60 and 80 μM cadmium, and after a 14- day incubation colonies were stained with crystal violet and counted. Results show the mean colony formation in cadmium-treated cultures relative to the untreated control (Mean±SE, n=3). (A) Colony formation by F1-hTERT, F3-hTERT, F10-hTERT and GM02052A after cadmium treatment. (B) Colony formation in normal and p53-defective cells after cadmium treatment. (#) denotes both F10-hTERT-p53-RNAi and F10-hTERT-p53-H179Q were significantly different than F10-hTERT and F10-hTERT- LX1N (p<0.05).
Sensitivity of G1 and S phase cells to cadmium
In order to compare sensitivity in G1 and S phase cells, F1-hTERT, F3-hTERT and F10-hTERT fibroblasts were released from G0 and the proliferation kinetics of the three cell lines were measured (Fig. 3A). From 8 to 12 h after release from confluence arrest, at least 80% cells were in the pre-replicative G1 phase. Very low fractions of cells were in S or M during this time. After 12 h, cells entered the S phase so that by 20 h about 60% of cells were in S. The fractions of cells in G2 and M remained low at 20 h, but by 24 h increased fractions of G and M cells were seen. According to these results, cells were treated at 8 or 20 h after release when proliferating cells were predominantly in G1 or S. All three cell lines were found to be more sensitive to cadmium when in S than when in G1 (Fig. 3B). In S phase cultures 80 μM cadmium inhibited colony formation by 81–86% while in G1 phase cultures this concentration inactivated colony formation by only 35–41%.
Figure 3.
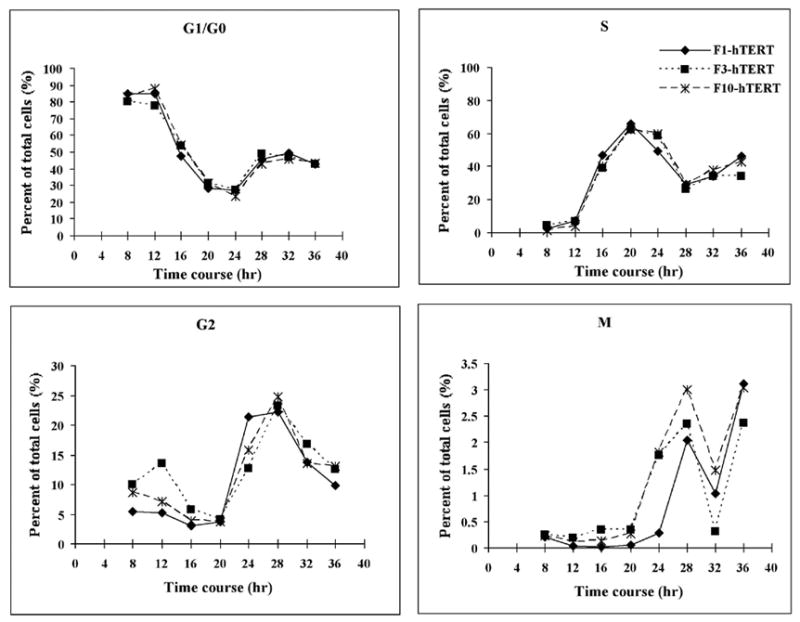
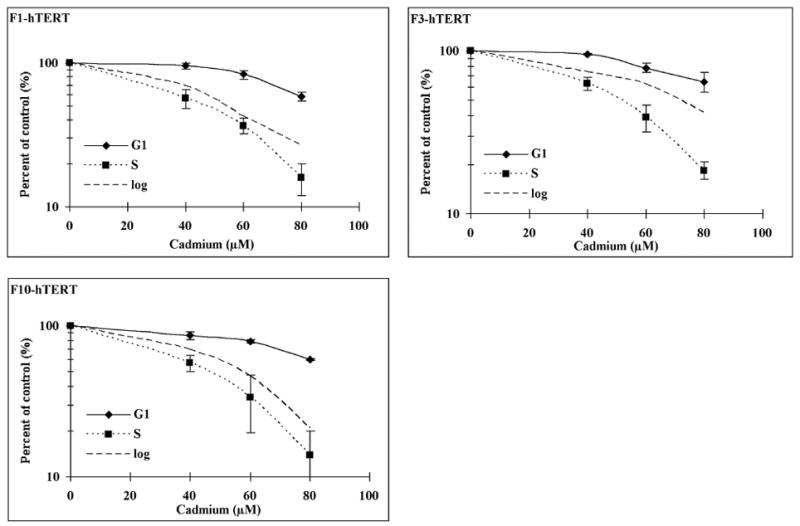
Sensitivity to cadmium of G1 and S phase cells. Cells were allowed to grow for 8 days to confluence arrest (G0 phase). (A) F1-hTERT, F3-hTERT and F10-hTERT fibroblasts were released from G0 and the proliferative activity of the three cell lines was measured by flow cytometry. (B) Confluence-arrested cells were released for 8 h to allow them to reach G1 and 20 h to allow them to reach S phase. The cells were then treated with different concentrations of cadmium for 4 h. The colonies were counted after a 14-day incubation. Results show the mean relative colony formation in cadmium-treated cultures (Mean±SE, n=3).
Cadmium inhibits DNA synthesis
A previous study using Chinese hamster ovary cells showed that cadmium inhibited replicative DNA synthesis (Banfalvi et al., 2000). To examine the inhibition of DNA synthesis in diploid human fibroblasts, incorporation of [3H]-thymidine was monitored during and after incubation with 40 or 80 μM cadmium. Initially the effect of cadmium on DNA synthesis was measured in exponentially growing cells. Normal cells, p53-defective cells and AT cells were all sensitive to inhibition of incorporation of [3H]-thymidine by cadmium (Fig. 4). Cadmium produced a dose- and time-dependent inhibition of DNA synthesis during 4 h incubation. At the end of the 4 h treatment with 80 μM, DNA synthesis in F10-hTERT, F10-hTERT-LX1N, F10-hTERT-p53-RNAi and F10-hTERT-p53-H179Q was 7%, 17%, 12% and 15% of control, respectively. After the 4 h incubation, cadmium was removed and DNA synthesis was measured at various times later. Fig. 4 shows that p53 affected the kinetics of DNA synthesis post-treatment. In normal cells DNA synthesis recovered to 50–80% of control at 6–12 h and then decreased again by 18 and 24 h after treatment with cadmium. In contrast, while p53-defective cells also recovered DNA synthesis they did not display the secondary inhibition. At 24 h after treatment with 80 μM cadmium, DNA synthesis rates in F10-hTERT and F10-hTERT-LX1N were 17% and 21% of control in comparison with 63% and 52% in F10-hTERT-p53-RNAi and F10-hTERT-p53-H179Q, respectively. Thus, relative DNA synthesis rates 24 h after treatment with cadmium were similar to relative colony formation efficiencies.
Figure 4.
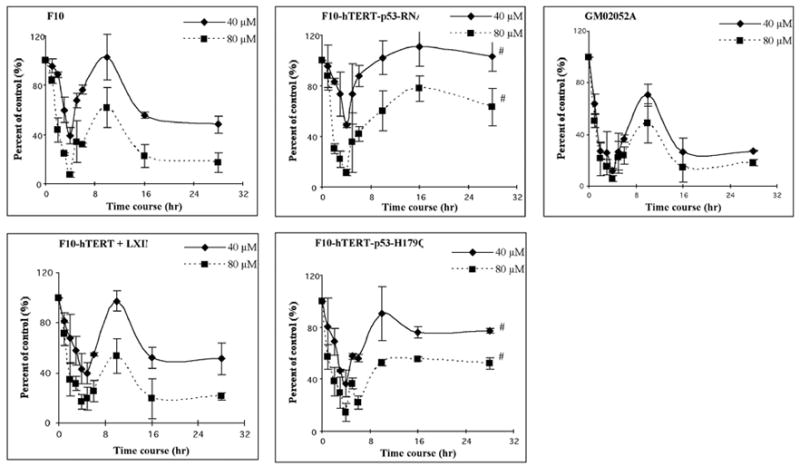
Cadmium inhibits DNA synthesis. The human fibroblast lines, F10-hTERT, F10-hTERT-LX1N, F10-hTERT-53-RNAi, F10-hTERT-p53-H179Q and GM02052A, were treated with cadmium during log-phase growth. During the 4 h treatment, DNA synthesis was measured every hour by incubation with [3H] thymidine for the final 30 min. After the 4 h treatment, the cadmium was removed, fresh medium added and DNA synthesis was measured at 1, 2, 6, 12 and 24 h later. Net [3H] radioactivity was normalized to cell number. Normalized [3H] CPM were graphed as a percent of time-matched, sham-treated controls. (#) denotes both F10-hTERT-p53-RNAi and F10-hTERT-p53-H179Q were significantly different than F10-hTERT and F10-hTERT- LX1N (p<0.05).
It was conceivable that cadmium simply interfered with the uptake of [3H]-thymidine and its conversion to [3H]-TTP. DNA synthesis was measured using a method that did not require uptake and metabolic conversion of a labeled precursor. Aphidicolin was used to collect cells at the beginning of S phase (Kaufmann and Wilson, 1990). After release from aphidicolin the increase in nuclear DNA content served as a measure of DNA synthesis. The flow cytometric profiles of DNA content in untreated and cadmium-treated cells are shown in Fig. 5A. The DNA content of control cells increased from 2N to 3–4N by 6 h after release as an index of DNA synthesis. Incubation of released cells with 40 or 80 μM cadmium for 6 h inhibited DNA synthesis as evidenced by the reduced fraction of nuclei with 3–4N DNA, and this effect was greater with the higher dose. F1-hTERT, F3-hTERT and F10-hTERT behaved similarly in this assay with 80 μM cadmium producing a severe (>90%) inhibition of DNA replication (Fig. 5B).
Figure 5.
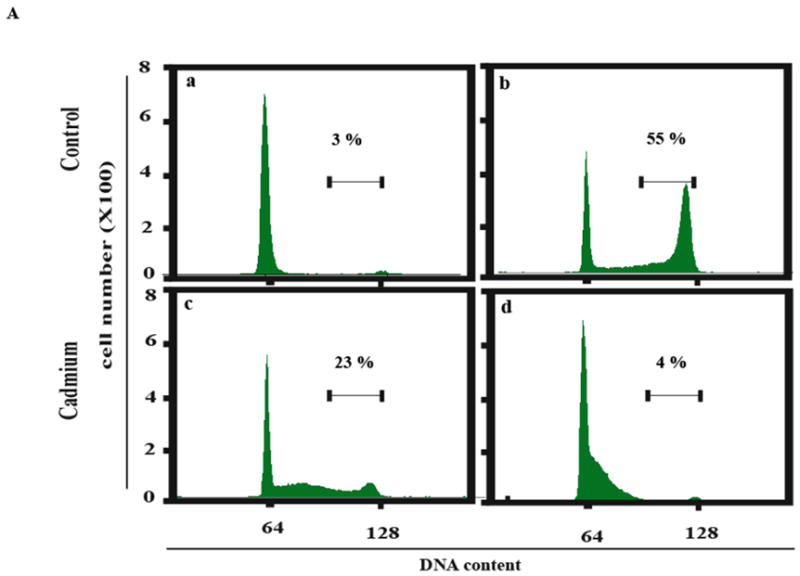
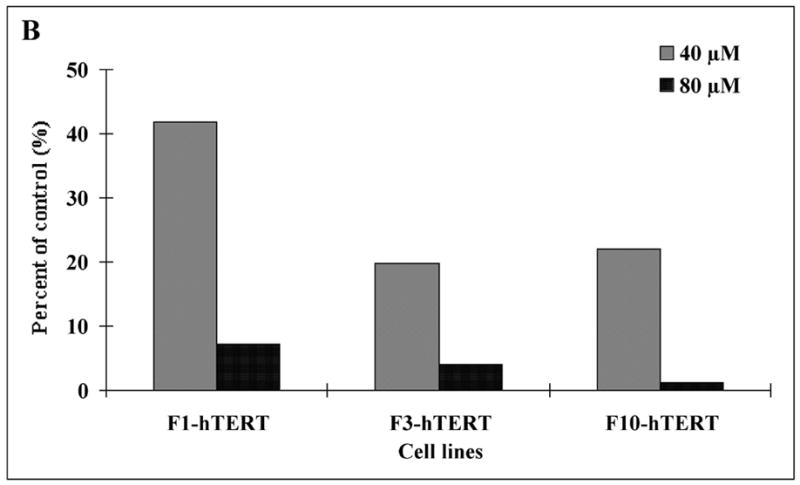
Inhibition of DNA synthesis in S phase cells. Cells were released from confluence-arrest, reseeded at 1 million cells per 100 mm dish and incubated with aphidicolin for 24 h to collect cells at the beginning of S phase. Aphidicolin was then removed and cells incubated in fresh medium with or without cadmium for 6 h to observe the increment of DNA content during S phase progression. Propidium iodide was used to stain DNA. The stained nuclei were analyzed by flow cytometry to determine their DNA content. (A) F1-hTERT cells were released from synchronization and treated with various concentrations of cadmium. Numbers above the bar (3–4 N DNA content) represent the fraction of total cells in this region. (a) Cells synchronized for 24 h with aphidicolin. (b) Cells incubated for 6 h without cadmium after removal of aphidicolin. (c) Cells incubated 6 h with 40 μM cadmium after removal of aphidicolin. (d) Cells incubated 6 h with 80 μM cadmium after removal of aphidicolin. (B) The percentage of nuclei with 3–4 N DNA content after 6 h incubation with 40 or 80 μM cadmium was expressed as a percentage of the sham-treated control.
Rapid and delayed G2 arrest in cadmium-treated fibroblasts
To obtain a more comprehensive view of the effects of cadmium on cell proliferation, flow cytometry was used to quantify cells in G1, S, G2 and M. Analysis of BrdU incorporation demonstrated the expected moderate-to-severe inhibition of DNA replication after 4 h treatment with cadmium (Fig. 6A). Instead of the well-defined arc of positively stained nuclei arrayed between 2N and 4N DNA content as seen in controls, the cadmium-treated cells displayed reduced intensity of labeling with BrdU throughout S phase. This pattern has been observed before in human cells after treatment with cytotoxic fluences of ultraviolet light that severely inhibit DNA replication (Cistulli and Kaufmann, 1998; Cordeiro-Stone et al., 2002). The analysis of cell cycle progression after cadmium exposure also demonstrated both rapid and delayed G2 arrest in cadmium-treated fibroblasts (Fig. 6A). At the end of the 4 h treatment with cadmium, the fraction of mitotic cells was reduced in a dose-dependent fashion with 80 μM producing nearly 80% reduction in the fraction of mitotic cells. This reduction would appear to be due to a G2 arrest blocking the movement of cadmium-damaged cells into mitosis. AT and p53-defective cells showed a similar reduction of mitotic cells after 4 h cadmium treatment (Fig. 6B). Thus, the rapid G2 arrest induced by cadmium was independent of ATM and p53.
Figure 6.
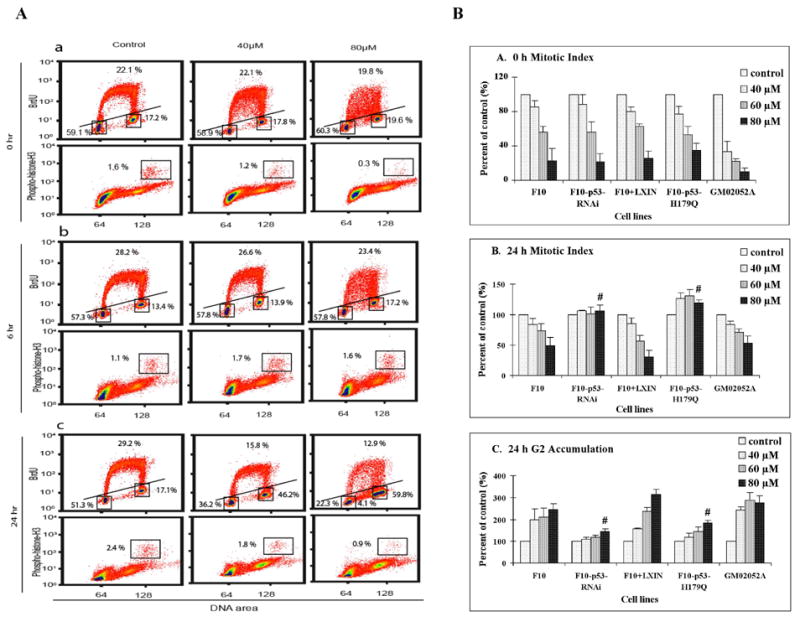
Inhibition of DNA synthesis and mitosis by cadmium. Logarithmically growing cells were treated with cadmium for 4 h. At 0, 6 and 24 h after 4 h cadmium or sham treatment, cells were incubated with 10 μM BrdU for 2 h, then cells were harvested for analysis of BrdU-incorporation by flow cytometry. At 0, 6 and 24 h after cadmium- or sham-treatment, cells were also harvested and processed for determination of mitotic index by flow cytometry. Propidium iodide was used to stain DNA (X-axis). Anti-BrdU-FITC was used to stain BrdU-labeled nuclei and antibody specific for phospho-histone H3 was used to stain mitotic cells. (A) F10-hTERT cells were analyzed 0, 6 and 24 h after cadmium treatment. Boxes and lines separate G1, S, G2 and M cells with the percentages of cells in these phases noted. (B) The cell cycle phase distributions of F10-hTERT, F10-hTERT-p53-RNAi, F10-hTERT-LX1N, F10-hTERT-p53-H179Q and GM02052A cells were analyzed after cadmium treatment. Results show the mean ± SE of three independent experiments. (a) Relative mitotic index immediately after cadmium treatment. (b) Relative mitotic index 24 h after cadmium treatment. (c) Relative percentage of cells in G2 24 h after cadmium treatment. (#) denotes both F10-hTERT-p53-RNAi and F10-hTERT-p53-H179Q were significantly different than F10-hTERT and F10-hTERT-LX1N (p<0.05).
By 6 h after the 4 h cadmium treatment, the mitotic index of all cell lines recovered with mitotic indices equal to or greater than untreated cells (Fig. 6A, B). Incorporation of BrdU by S phase cells also recovered as quantitatively demonstrated in Figure 4. However, by 24 h after the 4 h cadmium treatment, a significantly increased fraction of normal cells accumulated in G2 phase, with a corresponding significant secondary decrease in mitotic cells. Analysis of BrdU incorporation at 24 h revealed that after the severely toxic dose of 80 μM, 4% of cells with 2–4 N DNA content were unable to incorporate BrdU, suggestive of full replicative arrest. The p53-defective cells displayed significantly less accumulation in G2 phase, and no inhibition of mitosis at 24 h after treatment (Fig. 6A, B).
Western immunoblot analysis of cell cycle checkpoint protein expression
To probe the mechanisms of cadmium-induced cell cycle arrest, western immunoblot analyses were performed (Fig. 7). Cadmium increased p53 protein and phospho-ser15-p53 in both normal and AT cells (Fig. 7A) indicating that activation of p53 was independent of ATM. Gadd45α protein levels were increased immediately after cadmium treatment in all cell lines but declined by 6 h after treatment and were undetectable at 24 h (Fig. 7B). In spite of the severe inhibition of DNA replication and G2 arrest induced by cadmium, the levels of Chk1, Chk2, phospho-Ser317 Chk1, phospho-Thr68 Chk2 and p21CIP1/WAF1 did not change in cadmium-treated normal human fibroblasts (Fig. 7C). There was no evidence of apoptosis at 24, 48 and 66h after treatment with 80 μM cadmium as indicated by expression only of intact PARP and caspase 3 (not shown).
Figure 7.
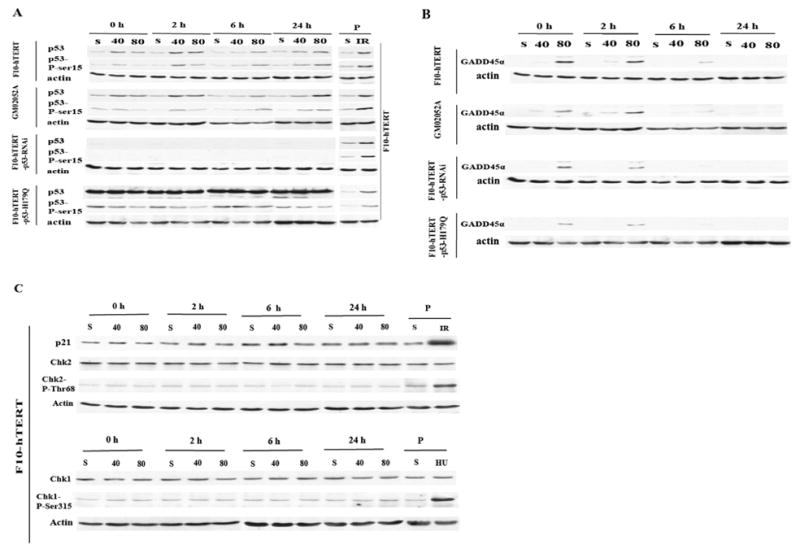
Western immunoblot analysis of DNA damage response in cadmium-treated human fibroblasts. Cells were incubated in fresh medium for 0, 2, 6 and 24 h after 4 h treatment with 40 or 80 μM cadmium or solvent control (S), then harvested and lysed. Protein extracts were prepared and 100 μg of total protein analyzed for expression of selected proteins or phosphorylated species. (A). Expression of p53 and phospho-ser-15-p53. The positive control (P) was F10-hTERT cells harvested 6 h after 1.5 Gy IR. (B). Expression of Gadd45α. (C). Expression of p21Cip1/Waf1, Chk2, phospho-Thr68-Chk2, Chk1 and phospho-ser317-Chk1. The positive control (P) for p21Cip1/Waf1, Chk2 and phospho-Thr68-Chk2 was F10-hTERT cells harvested 6 h after 1.5 Gy IR. The positive control (P) for Chk1 and phospho-ser317-Chk1 was F10-hTERT cells harvested after 24 h incubation with 2 mM hydroxyurea.
Discussion
Cadmium has been found to be carcinogenic to humans and other mammals. However, the mechanisms involved in the carcinogenicity are still unclear (Waalkes, 2003; Waisberg et al., 2003). Indirect genotoxic effects of cadmium have been discussed, e.g. interference with DNA repair and DNA replication processes (Hartwig, 1994; Dally and Hartwig, 1997). We tested the hypothesis that cadmium could cause DNA damage and induce DNA damage checkpoint responses in human fibroblast lines with selected genetic alterations. The survey of the effects of cadmium on DNA metabolism in human fibroblasts confirmed the induction of DNA damage and demonstrated a role for p53 in the cellular responses to cadmium. Cadmium-induced cytotoxicity and the delayed inhibition of DNA replication and mitosis were p53-dependent, and the treatment with cadmium induced p53 protein accumulation and phosphorylation of p53 at ser 15. A high concentration of cadmium also produced a severe inhibition of DNA synthesis in S phase cells and reduced the fraction of mitotic cells seemingly by GADD45α-associated G2 arrest. The results demonstrated profound disturbance in human DNA metabolism and cell division as provoked by acute exposure to toxic concentrations of cadmium.
DNA damage checkpoints are biochemical pathways that delay or arrest cell cycle progression in response to DNA damage (Kaufmann and Paules, 1996; Kastan and Bartek, 2004). It is assumed that checkpoints maintain cell cycle arrest until damaged DNA is repaired when the arresting signal is attenuated or reversed to permit resumption of cell cycle progression (Abraham, 2001). For the rapid G2 arrest, DNA damage likely occurred in G2 cells, as a significant decrease in the mitotic index was observed immediately after 4 h cadmium treatment (Fig. 6A). The rapid induction of Gadd45α could explain the cadmium-induced G2 arrest. Gadd45α is known to inhibit mitosis-promoting-factor (cyclin B1/Cdk1) kinase activity and enforce a G2 checkpoint response to DNA damage (Wang et al., 1999; Zhan et al., 1999; Yang et al., 2000). Upon removal of cadmium, Gadd45α levels diminished and mitosis resumed. Inactivation of p53 did not abrogate the rapid G2 arrest induced by cadmium nor the induction of Gadd45α (Fig. 6B). Cadmium is known to induce phosphorylation of C-Jun apparently through activation of c-Jun NH2-terminal kinase (Lee et al., 2001) and p53-independent induction of Gadd45α after UV-treatment was shown to include mitogen-activated protein kinase interaction with Oct1 and NF-YA transcription factors (Tong et al., 2001). Thus, the ATM- and p53-independent induction of Gadd45α and G2 arrest by treatment with cadmium appears to be mediated by mitogen-activated protein kinase signaling.
Cadmium is also a spindle poison; it depolymerizes microtubules and actins (Li et al., 1993; Wang and Templeton, 1996) and previous studies have shown that G2/M phase cells are more sensitive to challenge with cadmium (Chao and Yang, 2001). Mitotic arrest may thus occur in the presence of cadmium. If the mitotic spindle is a major target for cadmium, a high proportion of cells should be stopped in mitosis after treatment with cadmium. We found that the proportion of mitotic cells decreased in a concentration-dependent manner during cadmium exposure (Fig. 6) implying that cells completed mitosis in the presence of cadmium and the mitotic compartment emptied behind the G2 arrest. This result suggests that microtubules in the mitotic spindle apparatus may not be a major target for cadmium action in diploid human fibroblasts.
p53 protein and phospho-ser15-p53 were induced by cadmium in normal and AT cells. ATM and ATR phosphorylate ser15 of p53 directly and ser20 through activation of Chk2 or Chk1 (Banin et al., 1998; Ryan et al., 2001). Phosphorylation of p53 inhibits its export and degradation, thus increasing its level of expression (Zhang and Xiong, 2001). Cadmium has been shown to induce p53 and phospho-ser15 phosphorylation in MCF-7 cells (Matsuoka and Igisu, 2001). Cadmium is also known to replace zinc in the p53 zinc-finger domain, altering the structure of p53 and inhibiting DNA binding (Meplan et al., 1999). Accordingly, cadmium inhibited the induction of p53 and p21Cip1/Waf1 in human cells treated with the carcinogen, benzo[a]pyrene diolexpoxide I (Mukherjee et al., 2004). The cadmium-induced inhibition of p53 was dose-dependent and at 10–30 μM concentrations, p53 transactivation activity in mouse cells was inhibited by 65–85% (Meplan et al., 1999). We anticipate that metallothionein expression should reduce the concentration of free cadmium inside cells so that p53 function was only attenuated, not fully ablated in normal human fibroblasts. Moreover, the effect of cadmium on p53 was reversible such that by 12 h after removal from culture medium, p53 binding to DNA was restored by 50% (Meplan et al., 1999). Thus, although induction of p21Cip1/Waf1 was not evident in cadmium-treated cells, some function of p53 remained to inactivate clonal expansion and produce the delayed inhibition of DNA synthesis and G2 arrest.
By 24 h after treatment, cadmium induced a p53-dependent but ATM-independent G2 arrest. Gadd45α had returned to control levels and p21Cip1Waf1 was unchanged in cadmium-treated cells (Fig. 7), so Gadd45α and p21Cip1/Waf1 do not appear to contribute to the delayed G2 arrest. p53 activates the transcription of many genes that mediate its downstream functions (Zhou et al., 2006) and can also repress the transcription of different genes through several mechanisms (Taylor and Stark, 2001). Leach et al. reported that the activation of wild-type p53 resulted in a down-regulation of Wee1 expression and dephosphorylation of Cdk1 under conditions of p53-induced G2/M growth arrest and p53-mediated apoptosis (Leach et al., 1998). However, Wee1 expression did not change in cadmium-treated cells (data not shown). p53 may suppress the G2/M transition by negatively regulating the expression of cyclin B1, Cdk1, and topoII-alpha (Passalaris et al., 1999; Taylor and Stark, 2001). Cyclin B1 and Cdk1 are subunits of mitosis-promoting-factor that is required for entry to mitosis, and topoII-alpha is required for timely chromatid decatenation to allow bypass of a decatenation checkpoint that acts in G2 cells (Deming et al., 2001; Deming et al., 2002). Further experiments are needed to determine whether p53 trans-repression of these G2-regulated target genes accounts for the cadmium-induced delayed G2 arrest.
The mechanisms of inhibition of DNA replication by cadmium remain to be determined. Clark and Kunkel reported that cadmium did not inhibit in vitro plasmid DNA replication by human cell extracts under conditions that mismatch repair was inhibited (Clark and Kunkel, 2004). This result implies that cadmium is not directly toxic to the basal DNA replication machinery including DNA primase, DNA polymerases, DNA ligase and required accessory proteins such as RPA. The result also indicates that mismatch repair and DNA replication can be uncoupled in vitro and that DNA replication does not require mismatch repair. One mechanism for inhibition of DNA replication without direct inhibition of the replication machinery is inhibition of dNTP precursor production. Hydroxyurea inhibits DNA replication by this mechanism. The observation that treatment with HU induced phosphorylation of Chk1 while cadmium did not (Fig. 7C) argues against an inhibitory effect of cadmium on DNA precursor production. Further study will be required to determine whether cadmium inhibits any of the enzymes that are required for initiation of DNA replication at replicon origins. As the known mechanisms of inhibition of replicon initiation by DNA damage require activation of Chk1 and Chk2 (Falck et al., 2001; Heffernan et al., 2002), the failure of cadmium to activate Chk1 and Chk2 suggests that the effect of cadmium is not through a recognized S checkpoint.
It is of interest to compare and contrast the mechanisms of growth arrest by cadmium and another carcinogenic metal, chromium. Both metals appear to induce oxidative stress (Valko et al., 2006), activate the mitogen-activated protein kinases p38, JNK and ERK (Chuang and Yang, 2001), and induce inhibition of DNA replication and mitosis (Wakeman et al., 2005). However, while the effects of cadmium on DNA synthesis and mitosis were independent of ATM signaling, chromium activated ATM apparently by inducing DNA dsb in S phase cells (Ha et al., 2004). In keeping with the induction of DNA dsb and activation of ATM, chromium also activated Chk2 (Ha et al., 2003); cadmium did not activate Chk2 even when DNA replication was severely inhibited. Chromium caused ATM-dependent apoptosis in human fibroblasts and AT fibroblasts were more sensitive to inactivation of colony formation by chromium (Ha et al., 2003); cadmium did not induce apoptosis in foreskin fibroblasts and AT fibroblasts were not hypersensitive to cadmium. The enhanced growth arrest and inactivation of colony formation in chromium-treated AT cells may reflect signaling from ATR to p53 (Wakeman and Xu, 2006). Chromium induced GADD45 mRNA (Ceryak et al., 2004) and cadmium induced GADD45α protein consistent with both compounds causing oxidative stress and activating the stress-responsive mitogen-activated protein kinases. This comparison reveals similarities and dissimilarities in the mechanisms of action of cadmium and chromium suggesting that unique properties of the metal salts may contribute to their toxicities. The facility with which cadmium replaces zinc in important proteins such as p53, XPA and hMSH2 may contribute to its biological effects.
In summary the results presented here suggest a model in which cadmium-induced DNA damage or oxidative stress causes p53-independent induction of GADD45α to produce a rapid G2 arrest, and p53-dependent trans-repression of downstream target genes to produce a delayed G2 arrest.
Acknowledgments
We are grateful to Dr. Jacquelin Bower for reading this manuscript and helpful comments. Supported in part by the Chinese national 973 program and by PHS grants ES10126, CA16086 and ES11391. The funding sponsors of this work had no involvement in its design, execution or reporting.
Abbreviations
- APC
aphidicolin
- AT
ataxia telangiectasia
- ATM
ataxia telangiectasia-mutated
- BrdU
5′-bromo-2′-deoxyuridine
- dsb
double-strand breaks
- hTERT
human telomerase reverse transcriptase
- SDS
sodium dodecyl sulfate
- TCA
trichloroacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- IARC monographs on the evaluation of carcinogenic risks to humans: cadmium, mercury, beryllium and exposures in the glass industry. International Agency for Research on Cancer; Lyon: 1994. [PMC free article] [PubMed] [Google Scholar]
- Abdulla M, Chmielnicka J. New aspects on the distribution and metabolism of essential trace elements after dietary exposure to toxic metals. Biol Trace Elem Res. 1989;23:25–53. doi: 10.1007/BF02917176. [DOI] [PubMed] [Google Scholar]
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. The p53 network. J Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
- Asmuss M, Mullenders LH, Eker A, Hartwig A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis. 2000;21:2097–2104. doi: 10.1093/carcin/21.11.2097. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Flores-Rozas H. Cadmium inhibits mismatch repair by blocking the ATPase activity of the MSH2-MSH6 complex. Nucleic Acids Res. 2005;33:1410–1419. doi: 10.1093/nar/gki291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfalvi G, Littlefield N, Hass B, Mikhailova M, Csuka I, Szepessy E, Chou MW. Effect of cadmium on the relationship between replicative and repair DNA synthesis in synchronized CHO cells. Eur J Biochem. 2000;267:6580–6585. doi: 10.1046/j.1432-1327.2000.01751.x. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Boyer JC, Kaufmann WK, Cordeiro-Stone M. Role of postreplication repair in transformation of human fibroblasts to anchorage independence. Cancer Res. 1991;51:2960–2964. [PubMed] [Google Scholar]
- Ceryak S, Zingariello C, O’Brien T, Patierno SR. Induction of pro-apoptotic and cell cycle-inhibiting genes in chromium (VI)-treated human lung fibroblasts: lack of effect of ERK. Mol Cell Biochem. 2004;255:139–149. doi: 10.1023/b:mcbi.0000007270.82431.3e. [DOI] [PubMed] [Google Scholar]
- Chao JI, Yang JL. Opposite roles of ERK and p38 mitogen-activated protein kinases in cadmium-induced genotoxicity and mitotic arrest. Chem Res Toxicol. 2001;14:1193–1202. doi: 10.1021/tx010041o. [DOI] [PubMed] [Google Scholar]
- Chuang SM, Yang JL. Comparison of roles of three mitogen-activated protein kinases induced by chromium(VI) and cadmium in non-small-cell lung carcinoma cells. Mol Cell Biochem. 2001;222:85–95. [PubMed] [Google Scholar]
- Cistulli CA, Kaufmann WK. p53-dependent signaling sustains DNA replication and enhances clonogenic survival in 254 nm ultraviolet-irradiated human fibroblasts. Cancer Research. 1998;58:1993–2002. [PubMed] [Google Scholar]
- Clark AB, Kunkel TA. Cadmium inhibits the functions of eukaryotic MutS complexes. J Biol Chem. 2004;279:53903–53906. doi: 10.1074/jbc.C400495200. [DOI] [PubMed] [Google Scholar]
- Comstock KE, Watson NF, Olsen JC. Design of retroviral expression vectors. Methods Mol Biol. 1997;62:207–222. doi: 10.1385/0-89603-480-1:207. [DOI] [PubMed] [Google Scholar]
- Cordeiro-Stone M, Boyer JC, Smith BA, Kaufmann WK. Effect of benzo[a]pyrene-diol-epoxide-I on growth of nascent DNA in synchronized human fibroblasts. Carcinogenesis. 1986;7:1775–1781. doi: 10.1093/carcin/7.10.1775. [DOI] [PubMed] [Google Scholar]
- Cordeiro-Stone M, Frank A, Bryant M, Oguejiofor I, Hatch SB, McDaniel LD, Kaufmann WK. DNA damage responses protect xeroderma pigmentosum variant from UVC-induced clastogenesis. Carcinogenesis. 2002;23:959–965. doi: 10.1093/carcin/23.6.959. [DOI] [PubMed] [Google Scholar]
- Dally H, Hartwig A. Induction and repair inhibition of oxidative DNA damage by nickel(II) and cadmium(II) in mammalian cells. Carcinogenesis. 1997;18:1021–1026. doi: 10.1093/carcin/18.5.1021. [DOI] [PubMed] [Google Scholar]
- Deming PB, Cistulli CA, Zhao H, Graves PR, Piwnica-Worms H, Paules RS, Downes CS, Kaufmann WK. The human decatenation checkpoint. Proc Natl Acad Sci U S A. 2001;98:12044–12049. doi: 10.1073/pnas.221430898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming PB, Flores KG, Downes CS, Paules RS, Kaufmann WK. ATR enforces the topoisomerase II-dependent G2 checkpoint through inhibition of Plk1 kinase. J Biol Chem. 2002;277:36832–36838. doi: 10.1074/jbc.M206109200. [DOI] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- Fatur T, Tusek M, Falnoga I, Scancar J, Lah TT, Filipic M. DNA damage and metallothionein synthesis in human hepatoma cells (HepG2) exposed to cadmium. Food Chem Toxicol. 2002;40:1069–1076. doi: 10.1016/s0278-6915(02)00058-3. [DOI] [PubMed] [Google Scholar]
- Ha L, Ceryak S, Patierno SR. Chromium (VI) activates ataxia telangiectasia mutated (ATM) protein. Requirement of ATM for both apoptosis and recovery from terminal growth arrest. J Biol Chem. 2003;278:17885–17894. doi: 10.1074/jbc.M210560200. [DOI] [PubMed] [Google Scholar]
- Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- Hartwig A. Role of DNA repair inhibition in lead- and cadmium-induced genotoxicity: a review. Environ Health Perspect. 1994;102(Suppl 3):45–50. doi: 10.1289/ehp.94102s345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol. 2002;22:8552–8561. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwua YS, Yang JL. Effect of 3-aminotriazole on anchorage independence and mutagenicity in cadmium- and lead-treated diploid human fibroblasts. Carcinogenesis. 1998;19:881–888. doi: 10.1093/carcin/19.5.881. [DOI] [PubMed] [Google Scholar]
- Jin P, Ringertz NR. Cadmium induces transcription of proto-oncogenes c-jun and c-myc in rat L6 myoblasts. J Biol Chem. 1990;265:14061–14064. [PubMed] [Google Scholar]
- Johnson LG, Mewshaw JP, Ni H, Friedmann T, Boucher RC, Olsen JC. Effect of host modification and age on airway epithelial gene transfer mediated by a murine leukemia virus-derived vector. J Virol. 1998;72:8861–8872. doi: 10.1128/jvi.72.11.8861-8872.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan G, Traganos F, James WM, Ray JM, Roberge M, Sauve DM, Anderson H, Darzynkiewicz Z. Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry. 1998;32:71–77. doi: 10.1002/(sici)1097-0320(19980601)32:2<71::aid-cyto1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, Frank AR, Zhou Y, Bryant MF, Zhou T, Luche DD, Nikolaishvili-Feinberg N, Simpson DA, Cordeiro-Stone M. Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res. 2003;532:85–102. doi: 10.1016/j.mrfmmm.2003.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WK, Levedakou EN, Grady HL, Paules RS, Stein GH. Attenuation of G2 checkpoint function precedes human cell immortalization. Cancer Res. 1995;55:7–11. [PubMed] [Google Scholar]
- Kaufmann WK, Paules RS. DNA damage and cell cycle checkpoints. FASEB Journal. 1996;10:238–247. doi: 10.1096/fasebj.10.2.8641557. [DOI] [PubMed] [Google Scholar]
- Kaufmann WK, Wilson SJ. DNA repair endonuclease activity during synchronous growth of diploid human fibroblasts. Mutat Res. 1990;236:107–117. doi: 10.1016/0921-8777(90)90038-7. [DOI] [PubMed] [Google Scholar]
- Leach SD, Scatena CD, Keefer CJ, Goodman HA, Song SY, Yang L, Pietenpol JA. Negative regulation of Wee1 expression and Cdc2 phosphorylation during p53-mediated growth arrest and apoptosis. Cancer Res. 1998;58:3231–3236. [PubMed] [Google Scholar]
- Lee SA, Dritschilo A, Jung M. Role of ATM in oxidative stress-mediated c-Jun phosphorylation in response to ionizing radiation and CdCl2. J Biol Chem. 2001;276:11783–11790. doi: 10.1074/jbc.M004517200. [DOI] [PubMed] [Google Scholar]
- Li W, Zhao Y, Chou IN. Alterations in cytoskeletal protein sulfhydryls and cellular glutathione in cultured cells exposed to cadmium and nickel ions. Toxicology. 1993;77:65–79. doi: 10.1016/0300-483x(93)90138-i. [DOI] [PubMed] [Google Scholar]
- Maher VM, McCormick JJ. Effect of DNA repair on the cytotoxicity and mutagenicity of UV irradiation and of chemical carcinogens in normal and xeroderma pigmentosum cells. In: Yuhas JM, et al., editors. Biology of radiation carcinogensis. New York: Raven Press; 1976. pp. 129–45. [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Igisu H. Cadmium induces phosphorylation of p53 at serine 15 in MCF-7 cells. Biochem Biophys Res Commun. 2001;282:1120–1125. doi: 10.1006/bbrc.2001.4700. [DOI] [PubMed] [Google Scholar]
- Meplan C, Mann K, Hainaut P. Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem. 1999;274:31663–31670. doi: 10.1074/jbc.274.44.31663. [DOI] [PubMed] [Google Scholar]
- Misra RR, Smith GT, Waalkes MP. Evaluation of the direct genotoxic potential of cadmium in four different rodent cell lines. Toxicology. 1998;126:103–114. doi: 10.1016/s0300-483x(98)00003-1. [DOI] [PubMed] [Google Scholar]
- Mouron SA, Grillo CA, Dulout FN, Golijow CD. A comparative investigation of DNA strand breaks, sister chromatid exchanges and K-ras gene mutations induced by cadmium salts in cultured human cells. Mutat Res. 2004;568:221–231. doi: 10.1016/j.mrfmmm.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Mukherjee JJ, Gupta SK, Kumar S, Sikka HC. Effects of cadmium(II) on (+/−)-anti-benzo[a]pyrene-7,8-diol-9,10-epoxide-induced DNA damage response in human fibroblasts and DNA repair: a possible mechanism of cadmium’s cogenotoxicity. Chem Res Toxicol. 2004;17:287–293. doi: 10.1021/tx034229e. [DOI] [PubMed] [Google Scholar]
- Olsen JC, Johnson LG, Wong-Sun ML, Moore KL, Swanstrom R, Boucher RC. Retrovirus-mediated gene transfer to cystic fibrosis airway epithelial cells: effect of selectable marker sequences on long-term expression. Nucleic Acids Res. 1993;21:663–669. doi: 10.1093/nar/21.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passalaris TM, Benanti JA, Gewin L, Kiyono T, Galloway DA. The G(2) checkpoint is maintained by redundant pathways. Mol Cell Biol. 1999;19:5872–5881. doi: 10.1128/mcb.19.9.5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332–337. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kaccmaz K, Linn S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Sasaki YF, Izumiyama F, Nishidate E, Ohta T, Ono T, Matsusaka N, Tsuda S. Simple detection of in vivo genotoxicity of pyrimethamine in rodents by the modified alkaline single-cell gel electrophoresis assay. Mutat Res. 1997;392:251–259. doi: 10.1016/s1383-5718(97)00079-x. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase [see comments] Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Simpson DA, Livanos E, Heffernan TP, Kaufmann WK. Telomerase Expression is Sufficient for Chromosomal Integrity in Cells Lacking p53 Dependent G1 Checkpoint Function. J Carcinog. 2005;4:18. doi: 10.1186/1477-3163-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- Tong T, Fan W, Zhao H, Jin S, Fan F, Blanck P, Alomo I, Rajasekaran B, Liu Y, Holbrook NJ, Zhan Q. Involvement of the MAP kinase pathways in induction of GADD45 following UV radiation. Exp Cell Res. 2001;269:64–72. doi: 10.1006/excr.2001.5312. [DOI] [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- Wakeman TP, Wyczechowska D, Xu B. Involvement of the p38 MAP kinase in Cr(VI)-induced growth arrest and apoptosis. Mol Cell Biochem. 2005;279:69–73. doi: 10.1007/s11010-005-8216-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeman TP, Xu B. ATR regulates hexavalent chromium-induced S-phase checkpoint through phosphorylation of SMC1. Mutat Res. 2006;610:14–20. doi: 10.1016/j.mrgentox.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O’Connor PM, Fornace AJ, Jr, Harris CC. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A. 1999;96:3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Templeton DM. Cellular factors mediate cadmium-dependent actin depolymerization. Toxicol Appl Pharmacol. 1996;139:115–121. doi: 10.1006/taap.1996.0149. [DOI] [PubMed] [Google Scholar]
- Yang PM, Chiu SJ, Lin KA, Lin LY. Effect of cadmium on cell cycle progression in Chinese hamster ovary cells. Chem Biol Interact. 2004;149:125–136. doi: 10.1016/j.cbi.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Yang Q, Manicone A, Coursen JD, Linke SP, Nagashima M, Forgues M, Wang XW. Identification of a Functional Domain in a GADD45-mediated G2/M Checkpoint. J Biol Chem. 2000;275:36892–36898. doi: 10.1074/jbc.M005319200. [DOI] [PubMed] [Google Scholar]
- Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJJ. Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999;18:2892–2900. doi: 10.1038/sj.onc.1202667. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 2001;292:1910–1915. doi: 10.1126/science.1058637. [DOI] [PubMed] [Google Scholar]
- Zheng H, Liu J, Choo KH, Michalska AE, Klaassen CD. Metallothionein-I and -II knock-out mice are sensitive to cadmium-induced liver mRNA expression of c-jun and p53. Toxicol Appl Pharmacol. 1996;136:229–235. doi: 10.1006/taap.1996.0029. [DOI] [PubMed] [Google Scholar]
- Zhou T, Chou JW, Simpson DA, Zhou Y, Mullen TE, Medeiros M, Bushel PR, Paules RS, Yang X, Hurban P, Lobenhofer EK, Kaufmann WK. Profiles of global gene expression in ionizing-radiation-damaged human diploid fibroblasts reveal synchronization behind the G1 checkpoint in a G0-like state of quiescence. Environ Health Perspect. 2006;114:553–559. doi: 10.1289/ehp.8026. [DOI] [PMC free article] [PubMed] [Google Scholar]