

Abstract
In the present study, we examined the role of S. aureus protein A (SpA) in inducing inflammatory response in HCECs. Exposure of HCECs to SpA induces rapid NF-κB activation and secretion of proinflammatory cytokine/chemokines (TNF-α and IL-8) in both concentration and time-dependent manner. Challenge of HCECs with live SpA−/− mutant S. aureus strains resulted in significantly reduced production of the cytokines when compared to the wild-type S. aureus strain. SpA also elicited the activation of MAP Kinases P38, ERK, but not JNK, in HCECs. SpA-induced production of proinflammatory cytokine were completely blocked by the NF-κB and p38 inhibitors and partially inhibited by the Jnk inhibitor. Pretreatment with anti-TLR2 neutralizing antibody had no effect on SpA induced inflammatory response in HCECs, suggesting that this response is independent of TLR2 signaling. Moreover, unlike TLR2 ligands, SpA failed to induce the expression of antimicrobial peptides (hBD2 and LL-37) in HCECs. These studies indicate that SpA is a S. aureus virulence factor that stimulates HCEC inflammatory response through a pathway distinct from TLR2 in HCECs.
Introduction
The cornea, a transparent window in front of the eye provides two-thirds of the refractive properties of the eye [1] and constitutes a vital barrier preventing infectious agents, such as bacteria, viruses or fungi from entering the eye. The integrity of the corneal ocular surface barrier is crucial for the protection of the eye and hence preservation of sight. Studies from our laboratory and others have shown that corneal epithelium, like other mucosal linings in the body, constitutes the first line of defense and plays a critical role in corneal innate immunity[2]. The ability corneal epithelial cells to respond to pathogens is now known to be dependent largely on Toll-like receptors (TLRs), the recently discovered pattern recognizing type 1 membrane receptors. In the eye, several tissues/cells including corneal epithelial cells express several functional TLRs in vitro and in vivo and their individual role(s) in corneal innate immunity are summarized in a recent review [2].
Staphylococcus aureus causes a variety of local and systemic infections in humans and is one of the most important community- and nosocomially acquired pathogens. In the eye, it remains one of the leading causes of microbial keratitis. The ability of S. aureus to establish infections in a wide range of body sites probably depends on synthesis of large number of extracellular and cell-bound virulence factors. These include cell wall components (peptidoglycan, lipoproteins, lipoteichoic acid), and proteins such as coagulase, SpA, alpha-, beta-, gamma-, and delta-toxin, and leukocidin. Among these factors, protein A, a 42-kDa molecule existing in a secreted and membrane-associated form, interacts with a variety of human and animal immunoglobulins [3]. Due to this unique binding property, SpA is widely used for various laboratory techniques such as affinity purification of polyclonal and monoclonal antibodies, immunoprecipitation, ELISA, and in western Immunoblotting. The role of SpA as a virulence factor of S. aureus in various staphylococcal diseases including that in keratitis has been documented [4, 5].
In the present study, we investigated the role of staphylococcal protein A (SpA) in inducing inflammatory response and compared the responses of the cells to SpA with that to the TLR2 ligand Pam3Cys in corneal epithelial cells. Our results indicate that SpA induces an inflammatory response in HCECs characterized by secretion of proinflammatory cytokine and chemokine via a mechanism distinct from that of TLRs.
Material and Methods
Bacterial Strains
SpA-null or SpA overexpressor mutants of S. aureus, constructed on strain RN6390, were kindly provided by Professor Ambrose Cheung (Dartmouth Medical School, Hanover, NH).. Bacteria were grown in the presence of chloramphenicol (10 μg/ml) to an optical density of 0.5 (OD 600), and tetracycline (250 ng/ml) was added (to SpA overexpressor mutants) for 5 h to induce SpA expression.
Corneal Epithelial Cell Lines
Human telomerase-immortalized corneal epithelial (HUCL) cells were maintained in a defined keratinocyte-serum-free medium (SFM, Invitrogen Life Technologies, Carlsbad, CA) in a humidified 5% CO2 incubator at 37 °C. Before treatment, cells were cultured in growth factor-free and antibiotic-free keratinocyte basic medium (KBM, BioWhittaker, Walkersville, MD) for 16 h (growth factor starvation). To verify the results obtained from HUCL cells, HCECs were isolated from human donor corneas obtained from the Michigan Eye Bank. The epithelial sheet was separated from the underlying stroma after overnight dispase treatment. The dissected epithelial sheet was trypsinized and the epithelial cells were collected by centrifugation (500 × g, 5 min). HCECs were cultured in keratinocyte growth medium (KBM supplemented with growth factors, BioWhittaker) in T25 flasks coated with FNC and used at passage 3.
Western Blotting
Cells challenged with either SpA or bacteria were lysed with RIPA buffer, and protein concentration was determined with the BCA assay (Micro BCA; Pierce). Proteins were separated by SDS-PAGE in Tris/glycine/SDS buffer (25 mM Tris, 250 mM glycine, 0.1% SDS) and electro-blotted onto nitrocellulose membranes (0.45 μ, Bio-Rad). After blocking for 2 hr in PBST (20mM Tris-HCl, 150 mM NaCl, 0.5% Tween) containing 5% non-fat milk, the blots were probed with first antibodies overnight at 4°C. Membranes were washed with 0.05% (vol/vol) Tween 20 in PBS (pH 7.6) and incubated with a 1:2,000 dilution of horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) for 60 min at room temperature. Protein bands were visualized by Supersignal reagents (Pierce).
NF-κB DNA-binding activity
HUCL cells were grown to 80% confluency in 100-mm culture dishes and treated with or without 100μg/ml protein for various time points. To prepare nuclear extracts, cells were lysed by incubation on ice for 15 min in a hypotonic buffer (10 mM HEPES, 1.5 mM inhibitor mixture) and centrifuged. Further MgCl2, 10 mM KCl, 1 mM DTT, and protease cell disruption was achieved by repetitive filling and flushing of cell suspensions with a syringe having a narrow-gauge needle. The resulting suspensions were centrifuged. The crude nuclear pellets were then resuspended in extraction buffer (20 mM HEPES, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 1 mM DTT, and protease inhibitor mixture) and the nuclei were disrupted using a fresh syringe as above. The nuclear suspensions were incubated on ice for 30 min with occasional tapping, and centrifuged. A total of 30 μg of the nuclear extracts was added to an assay wells and ELISA was performed as per manufacturer’s instruction (BD Mercury TransFactor NF-κB p65 Kit, BD Biosciences). The absorbance was read at 450 nm on a microplate reader.
ELISA measurement of cytokines
Secretion of TNF-α and IL-8 was determined by ELISA. HCECs were plated at 1 × 106 cells/well in six-well plates. After growth factor starvation, cells were pretreated with or without flagellin and then further challenged for various times. At end of culture, the media were harvested for measurement of cytokines. The ELISA was performed according to the manufacturer’s instructions (R & D Systems). The amounts of cytokines in culture media were expressed as picograms of cytokine per ml. All values were expressed as mean ± standard deviation (SD). Statistical analysis was performed using analysis of variance (Anova), and each pair showed a significant difference in secretion of cytokines compared with untreated HUCL or primary cells at various concentration or time-points (P < 0.05).
RNA isolation and semi-quantitative RT-PCR
Total RNA was isolated from HUCL cells using the TRIzol solution according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA) and 2 μg of total RNA was reverse-transcribed with a first-strand synthesis system for RT-PCR (SuperScript; Invitrogen). cDNA was amplified by PCR using primers for human hBD2, LL-37, A20 IL-8, TNF-α and GAPDH. The PCR products and internal control GAPDH were subjected to electrophoresis on 1% agarose gels containing ethidium bromide. Stained gels were captured using a digital camera and band intensity was quantified using 1D Image Analysis Software (EDAS 290 system; Eastman Kodak, Rochester, NY).
Results
SpA induces NF-κB and MAPKs activation in HCECs
To investigate whether SpA induces NF-κB activation, we treated HUCL cells with SpA and determined NF-κB activation by assessing IκB-α phosphorylation and degradation. As shown in Figure 1, HUCL cells were responsive to SpA concentrations as low as 50 μg/ml, as measured by IκB-α phosphorylation and degradation (decrease in immune-reactivity of the protein); the treatment of HUCL cells with 100 μg/ml or higher SpA resulted in maximal phosphorylation of IκB-α (Fig. 1A). Time–course studies of the HCEC response to SpA (100 μg/ml) revealed that IκB-α phosphorylation was detectable as early as 15 min, and reaching a peak at 120 min post-stimulation (Fig. 1B). Accompanying the increase in IκB-α phosphorylation, IκB-degradation was observed 15 min poststimulation and afterwards. We further tested the effect of SpA on primary HCECs and observed that SpA (100 μg/ml) also stimulated NF-κB activation in time-dependent manner similar to that observed in HUCL cells (Fig. 1C). NF-κB activation was also assessed be measuring DNA binding ability of p65 in the nuclear extracts of HUCL cells using ELISA. While no p65 DNA binding activity was observed in the control, untreated cells, HUCL exposed to SpA exhibited increased p65 DNA binding in a time dependent manner (Fig. 1D).
Fig 1. Effect of SpA on NF-κB activation in HUCL and primary HCECs: dose and time-dependent course studies.
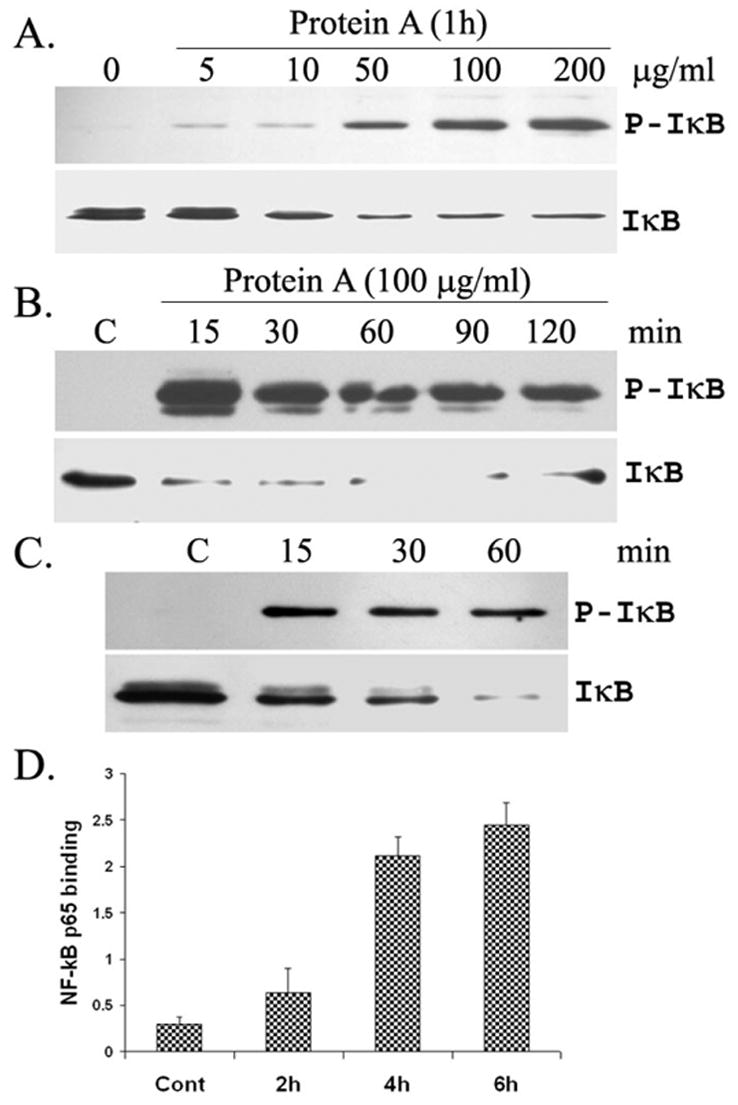
HUCL (A, B & D) and Primary (C) cells were incubated with various concentrations of SpA for 1 h (A), or 100 μg/ml for different time points (B & C) with unstimulated cells as control. A, B, and C, Western blotting of total cell lysate for phospho-IκB-α (IκB-α) and total IκB-α. D. NF-κB p65 DNA binding activity in HUCL nuclear extracts measured by ELISA using a Colorimetric Transfactor p65 kit.. Results shown are representative of three independent experiments.
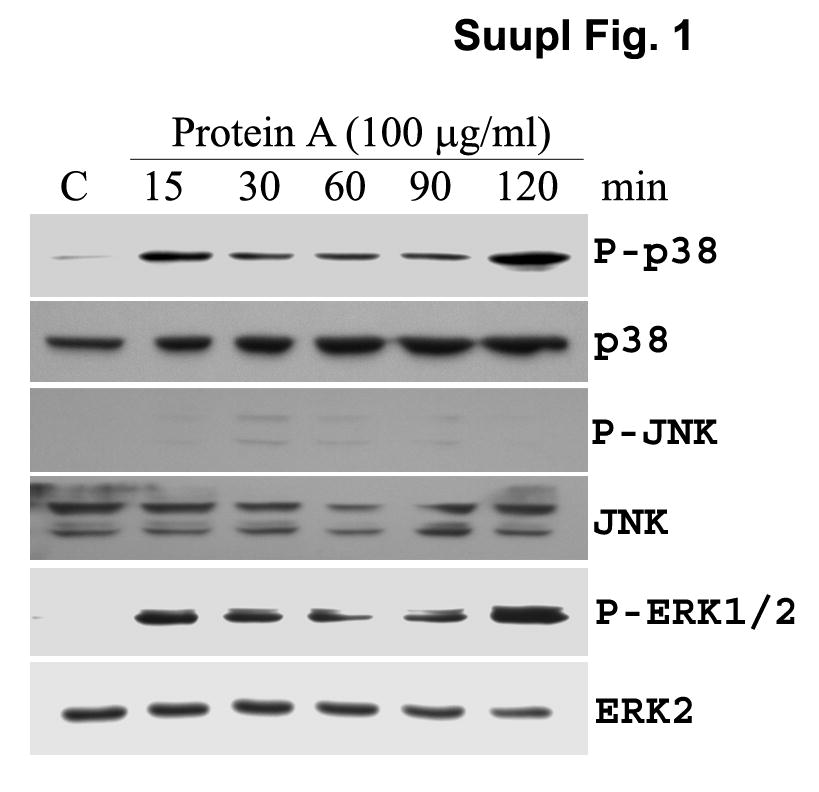
We next examined the effect of SpA on the activation of MAPKs in HCECs. SpA stimulated the activation of p38, and ERK½, but not JNK1 in a time-dependent manner (Supplementary Fig. 1). Take together; these results suggest that SpA induces activation of NF-κB, ERK, and p38, but not JNK, pathways in HCECs
SpA induces TNF-α and IL-8 production in HCECs
To assess the biological relevance of induced signaling pathways, we determined the effects of SpA on the production of proinflammatory cytokines. The effect of SpA on IL-8 and TNF- α secretion in HUCL cells was assessed using ELISA (Fig. 2). SpA at 5 or 10 μg/ml had no apparent effect on cytokine production whereas 50 μg/ml or higher significantly increased amounts of IL-8 and TNF-α in the culture media of HUCL cells (Fig. 2A). SpA induced significant increases in the production of IL-8 and TNF-α as early as 4 h post stimulation (Fig. 3B). Interestingly, after 4 h, the levels of TNF-α started to decline with significant decrease at 24 h whereas the levels of IL-8 remained elevated at 24 h post stimulation. Like HUCL cells, SpA also induced production of IL-8 and TNF-α in a time-dependent manner in primary HCECs (Fig. 2C).
Fig 2. Effect of SpA stimulation on TNF-α and IL-8 production in HCECs.
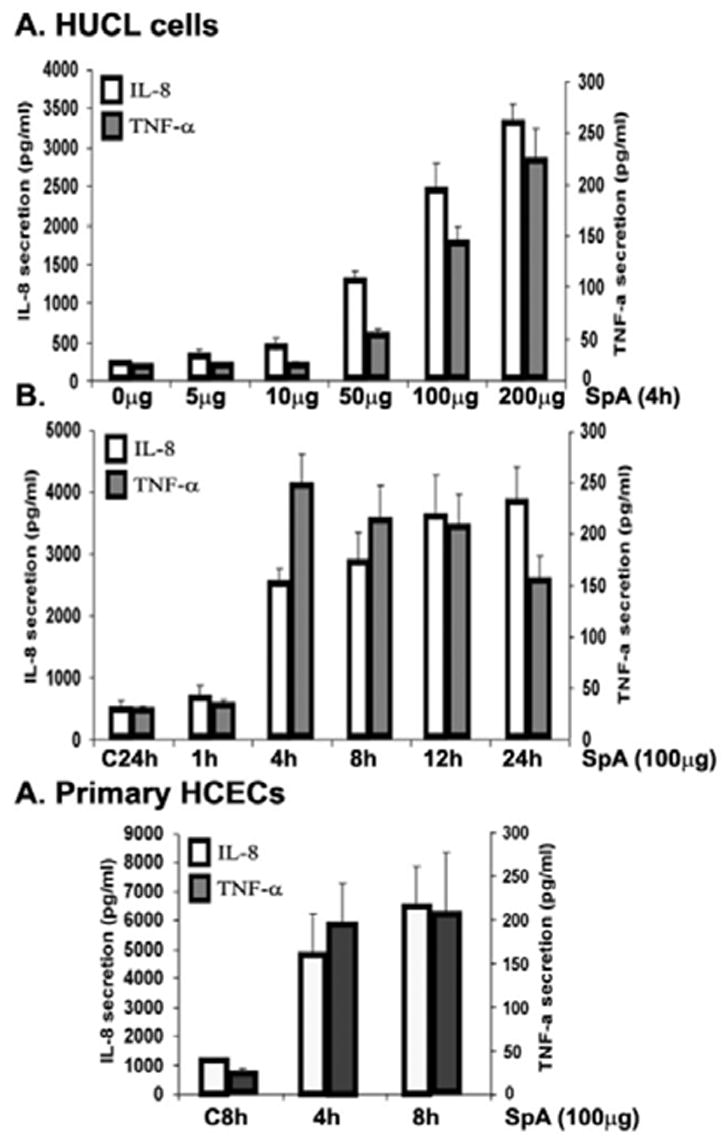
HUCL (A&B) and Primary HCEC (C) were cultured with various concentration of SpA for 4 h (A) or with 100 μg/ml SpA for different time periods up to 24 h (B). Secretion of IL-8 (open bars) or TNF-α (gray bars) into culture media was assayed by ELISA and presented as pg/ml for 1 × 106 cells. Data represent means ± SD of n = 3 independent experiments; *P < 0.05.
Fig 3. Effect of MAPKs inhibitors and TLR2 neutralizing antibody on SpA-induced IL-8 secretion.
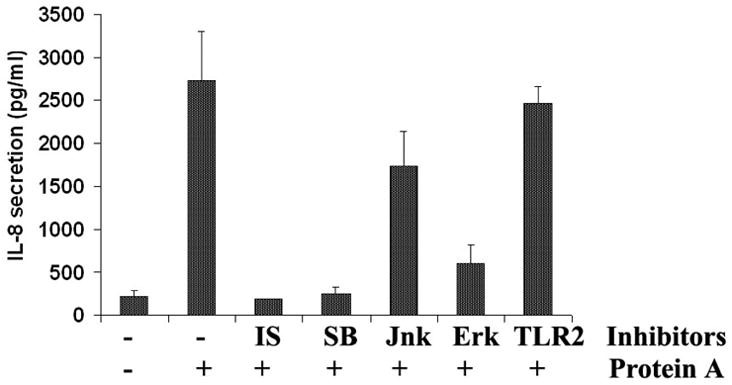
HUCL cell were incubated with inhibitors of MAPKs-mediated signal pathways for 30 min prior to the addition of SpA and the induced secretion of IL-8 after 4 h was assessed by IL-8 cytokine ELISA. The inhibitors used include the NF-κB inhibitor isohelenin (IS, 25 μM) and three MAP kinase pathways, SB (p38 MAP kinase inhibitor, 5 μM); SP (JNK inhibitor, 25 μM) and U0126 (ERK inhibitor, 10 μM). Anti-TLR2 antibody (20 μg/ml) was used to block the TLR2 pathway. The data is representative of three individual experiments.
Having demonstrated that purified SpA induces inflammatory response in HCECs, we next sought to determine the effect of live bacteria expressing different levels of SpA on production of proinflammatory cytokines. Both the wild-type and a strain overexpressing SpA significantly increased the secretion of TNF-α and IL-8 in HUCL cells. On the other hand, cells challenged with SpA null-strain produced significantly reduced amount (~50%) of both TNF- α (Supplementary Fig. 2A) and IL-8 (Supplementary Fig. 2B), compared to that induced by the wild-type. Taken together these results suggest that SpA induces inflammatory response in HCECs.
SpA-induced secretion of TNF-α and IL-8 is NF-κB, p38, and ERK dependent
In order to investigate the role of SpA-stimulated downstream signaling pathways on the secretion of proinflammatory cytokine/chemokines in HCECs, the specific inhibitors for their effectors were used (Fig. 3). Pre-incubation of HCECs with the NF-κB inhibitors isohelenin and the p38 MAP kinase inhibitor SB203580, completely blocked SpA-induced TNF-α secretion, the ERK inhibitor UO126 also had profound effect on IL-8 production (80% inhibition). The JNK inhibitor SP600125, on the other hand, had only partial effect on TNF-α secretion (<40% inhibition). We previously showed that TLR2 neutralizing antibody inhibited Pam3Cys induced inflammatory response in HCECs [6]. To investigate the involvement of TLR2 in SpA induced HCEC activation, HCECs were pretreated with the TLR2 neutralizing antibody and then stimulated with SpA in the presence of the same antibody. Presence of the TLR2 neutralizing antibody had no effect on SpA-induced production of IL-8 in HCECs.
SpA does not interfere with TLR2-signaling in HCECs
We have previously shown that HCECs abundantly express TLR2 at both mRNA and protein levels and stimulation with synthetic TLR2 ligand Pam3Cys results in secretion of proinflammatory cytokine/chemokines. Furthermore, pre-exposure of HCECs to TLR ligands such as flagellin (TLR5 ligand) or Pam3Cys induces a state of tolerance or hyporesponsiveness, characterized by no further activation of NF-κB upon second challenge with the same ligand even at higher concentration. To test the hypothesis whether SpA interferes with TLR2 signaling, we pretreated the HUCL cells with different concentration of Pam3Cys or SpA for 24 h to induce tolerance and then re-challenged with SpA or Pam3Cys. As shown in Supplementary Fig. 3, SpA is able to induce IκB phosphorylation and its degradation in HCECs pretreated with Pam3Cys. Similarly, the SpA exposure of HCECs also had no effect on Pam3Cys mediated NF-κB activation.
TLR2 ligand and SpA induce differential gene expression in HCECs
Activation of TLRs is known to induce the expression of proinflammatory cytokines and genes with antimicrobial properties. The foregoing results suggest that both TLR2 agonist Pam3Cys and SpA induces NF-κB activation and production of proinflammatory cytokines in a non-overlapping manner. We next compared the effects of these stimuli on expression of proinflammatory cytokines and antimicrobial peptides in HCECs. The RT-PCR analysis revealed that transcripts of both IL-8, and TNF-α, were not detectable in non-treated cells and both SpA and Pam3Cys induced the expression of TNF-α and IL-8 in time-dependent manner. Increased levels of TNF-α were detectable as early as 30 minutes and IL-8 expression was detectable at 2 h after stimulation with SpA. Similarly Pam3Cys also upregulated the mRNA levels of both IL-8 and TNF-α with significant increase at 4 h. Interestingly, the expression of genes with antimicrobial properties such as hBD2 and LL-37 was augmented only in cells challenged with Pam3Cys but not with SpA (Fig. 4).
Fig 4. SpA and TLR2 agonist Pam3Cys mediated gene expression in HCECs.
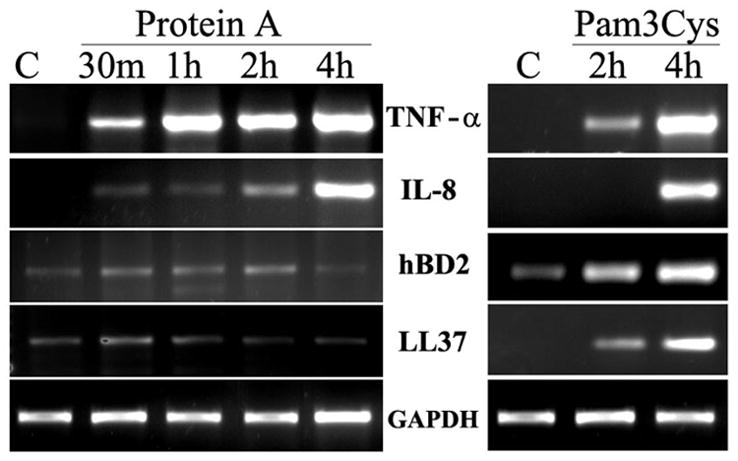
HUCL cells cultured in KBM were challenged with SpA (100 μg/ml) or Pam3Cys (10μg/ml). At the indicated times, cells were processed for semiquantitative RT-PCR to assess mRNA expression of hBD2, LL-37, IL-8 and TNF-α with GAPH as internal control. Results shown are representative of two independent experiments.
Discussion
Previous studies from our and other laboratories suggest that TLR2 is a major receptor in the cornea for recognition of Gram-positive bacteria such as S. aureus [6–8]. Herein, we provide the first evidence to show that S. aureus protein A induces an inflammatory response in HCECs and reported that HCECs possess an ability to recognize the SpA and initiate an innate immune response. This response is characterized by activation of multiple signaling pathways (NF-κB, JNK and ERK) and production of proinflammatory cytokine/chemokines. We also demonstrate and the SpA-induced inflammatory response is independent of TLR2 activation, implying that a distinct cell surface receptor is involved in innate response against S. aureus in HCECs.
Several studies have demonstrated a role for SpA in staphylococcal virulence. SpA-defective (SpA−/−) mutants have reduced virulence in murine models of septic arthritis, septicemia and skin abscesses, most likely due to the antiphagocytic effect of SpA binding IgG Fc [9]. In a murine pneumonia model, SpA mutants showed reduced virulence [10]. In these studies, tumor necrosis factor receptor-1 (TNFR-1) was implicated in the pro-inflammatory properties of SpA. The possible contribution of SpA to S. aureus virulence has also been investigated in a rabbit keratitis model, and this study has shown that SpA does not contributes significantly to the severity of the diseases [4, 11]. Although S. aureus is a common corneal pathogen, efforts to achieve a corneal infection by application of these bacteria to the scarified corneas have been unsuccessful. Whereas the topical application of Pseudomonas aeruginosa to a scarified rabbit cornea results in keratitis, S. aureus does not elicit keratitis in scarified rabbit eyes. Several studies revealed that topical application of a large number of S. aureus to scarified rabbit cornea resulted in inflammation, but not actual infection since bacterial replication, a characteristic of bacterial keratitis, was not produced in these animal models [12]. Therefore, to date, the animal models of S. aureus keratitis have relied on intrastromal injection of log phase bacteria into the cornea to study the role of specific staphylococcal surface components in corneal virulence and in triggering host inflammation and defense. As such, these models represent the characteristics of keratitis as compare to topical models but they do not mimic the sequential events of corneal infection in human that the invading bacteria first contact to the surface epithelial cells [4, 11]. Under normal circumstances, the cornea is highly impermeable to pathogens due to intact epithelium, however when the epithelial barrier is broken such as due to contact lens wearing or trauma [13], the bacteria gain access to the ocular surface, resulting in their intracellular multiplication and severe inflammation, leading to keratitis. Because SpA is a major surface protein present in almost all strains of S. aureus we reasoned that it is likely to possess ability to interact with and trigger epithelial response, contributing corneal inflammation and pathogenesis [14]. Indeed, we showed that SpA induced an inflammatory response in HCECs. This response is characterized by production of proinflammatory cytokine such as TNF-α and chemokine such as IL-8 which are known to recruit the immune cells in the cornea during microbial keratitis [15]. Moreover, while wild type S. aureus and a strain that overproduce SpA induce severe inflammatory response, the production of inflammatory cytokines was significantly attenuated in HCECs challenged with bacterial strains lacking SpA. Thus, SpA contributes to the induced inflammation during S. aureus keratitis.
Since IgG Fc, a well known SpA receptor, is unlikely to be expressed in epithelial cells, the receptor for SpA in non-myeloid cells remains to be elusive. Recently, the role of resident epithelial cells in mediating S. aureus-induced corneal inflammation in a mouse model of staphylococcal keratitis has been suggested [8]. The ability of epithelial cells to recognize and to respond to S. aureus infection in the cornea is related to the action of TLRs and MyD88. TLR2 recognizes a broad spectrum of pathogen associated molecular patterns (PAMPs) including PGN, LTA and lipoproteins derived from Gram-positive bacteria [16]. The interaction of TLRs with its PAMP results in the activation of multiple intracellular signaling events such as NF-κB activation and subsequent production of cytokines such as IL-6, IL-8, and TNF-α [2]. The finding that TLR2 neutralizing antibody failed to attenuate SpA-induced inflammatory response suggests that TLR2 is not involved in SpA-mediated inflammatory response in HCECs. Furthermore, preexposure of cells to TLR2 ligand has been shown to induce cell tolerance, leading to hyporesponsiveness of the cells to secondary challenge of the same or different TLR ligands. The facts that Pam3Cys pretreated cells are still responsive to SpA challenge provides an additional evidence that SpA induces HCEC inflammatory response is through a TLRs independent pathway. Recently, SpA has been shown to interact with TNF-α receptors (TNFR) and to induce the production of inflammatory cytokines in a TNFR1 dependent manner. Since TNFR1 is expressed in the cornea, further study to determine whether this cytokine receptor also functions as cell surface receptor for SpA in the ocular surface is warranted.
In addition to NF-κB activation, TLR ligands also induce the activation of other MAPKs such as p38, JNK and ERK and these signaling molecules are known to contribute towards inflammatory response in epithelial cells [17]. Our results that SpA induces the activation of p38 and ERK and inhibition of these two pathways completely blocked the SpA-induced cytokine secretion in HCECs suggesting that TLR ligands and SpA share some homology in terms of intracellular signaling pathway activation. TLR2 activation in HCECs not only induces the activation of NF-κB and the production of proinflammatory cytokines but also up-regulates the expression of several antimicrobial genes such as hBD2, and LL-37. The expression of these antimicrobial molecules renders epithelial and infiltrated PMN the ability to kill invading bacteria. This enhanced innate defense is believed to play a role in limiting infection when host-pathogen interaction occurs. Interestingly, SpA, although is able to induce NF-κB activation and proinflammatory cytokine production, does not trigger the expression of antimicrobial peptides in HCECs. Thus, targeting SpA triggered inflammatory response may have some advantage over TLR pathways as it may reduce inflammatory response without significantly affecting the innate defense mechanism in S. aureus-infected corneas. Thus, a better understanding of host-pathogen interaction in the cornea might provide molecular bases for the development of novel, specific therapies for prevention or treatment of infective keratitis.
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by NIH grants NIH/NEI EY14080, EY10869 (F.S.Y), Fight for Sight (A.K) and Mid West Eye banks (A.K) and an unrestricted grant from the Research to Prevent Blindness to the Department of Ophthalmology, Wayne State University School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kurpakus-Wheater M, Kernacki KA, Hazlett LD. Maintaining corneal integrity how the “window” stays clear. Prog Histochem Cytochem. 2001;36:185–259. [PubMed] [Google Scholar]
- 2.Kumar A, Yu FS. Toll-like receptors and corneal innate immunity. Curr Mol Med. 2006;6:327–337. doi: 10.2174/156652406776894572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman GJ, Goodyear CS. Confounding B-cell defences: lessons from a staphylococcal superantigen. 2006;6:465–475. doi: 10.1038/nri1853. [DOI] [PubMed] [Google Scholar]
- 4.Callegan MC, Engel LS, Hill JM, O’Callaghan RJ. Corneal virulence of Staphylococcus aureus: roles of alpha-toxin and protein A in pathogenesis. Infect Immun. 1994;62:2478–2482. doi: 10.1128/iai.62.6.2478-2482.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster TJ. Immune evasion by staphylococci. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Zhang J, Yu FSX. Toll-like receptor 2-mediated expression of [beta]-defensin-2 in human corneal epithelial cells. Microbes and Infection. 2006;8:380–389. doi: 10.1016/j.micinf.2005.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Ophthalmol Vis Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y, Hise AG, Kalsow CM, Pearlman E. Staphylococcus aureus-Induced Corneal Inflammation Is Dependent on Toll-Like Receptor 2 and Myeloid Differentiation Factor 88. Infect Immun. 2006;74:5325–5332. doi: 10.1128/IAI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmqvist N, Foster T, Tarkowski A, Josefsson E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microbial Pathogenesis. 2002;33:239–249. doi: 10.1006/mpat.2002.0533. [DOI] [PubMed] [Google Scholar]
- 10.Gomez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, Cheung A, Prince A. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10:842–848. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- 11.Jonsson P, Lindberg M, Haraldsson I, Wadstrom T. Virulence of Staphylococcus aureus in a mouse mastitis model: studies of alpha hemolysin, coagulase, and protein A as possible virulence determinants with protoplast fusion and gene cloning. Infect Immun. 1985;49:765–769. doi: 10.1128/iai.49.3.765-769.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girgis DO, Dajcs JJ, O’Callaghan RJ. Phospholipase A2 Activity in Normal and Staphylococcus aureus-Infected Rabbit Eyes. Invest Ophthalmol Vis Sci. 2003;44:197–202. doi: 10.1167/iovs.02-0548. [DOI] [PubMed] [Google Scholar]
- 13.Schornack MM, Peterson D. Staphylococcus aureus ulcer associated with continuous wear of silicone hydrogel contact lenses. Eye Contact Lens. 2006;32:72–74. doi: 10.1097/01.icl.0000174754.45463.84. [DOI] [PubMed] [Google Scholar]
- 14.Bochud PY, Hawn TR, Aderem A. Cutting edge: a Toll-like receptor 2 polymorphism that is associated with lepromatous leprosy is unable to mediate mycobacterial signaling. J Immunol. 2003;170:3451–3454. doi: 10.4049/jimmunol.170.7.3451. [DOI] [PubMed] [Google Scholar]
- 15.Hazlett LD. Corneal response to Pseudomonas aeruginosa infection. Prog Retin Eye Res. 2004;23:1–30. doi: 10.1016/j.preteyeres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005;18:521–540. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mossman BT, Lounsbury KM, Reddy SP. Oxidants and Signaling by Mitogen-Activated Protein Kinases in Lung Epithelium. Am J Respir Cell Mol Biol. 2006;34:666–669. doi: 10.1165/rcmb.2006-0047SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.