

Abstract
Cannabinoids are known to interact with CB1 and CB2 receptors expressed in the nervous and immune system, respectively and mediate a wide range of effects, including anti-inflammatory properties. However, cannabinoids that bind CB1 are also psychoactive thereby limiting their clinical use. In this study, we investigated the immunosuppressive properties of JWH-015, a synthetic CB2-selective agonist. We found that JWH-015 triggered apoptosis in thymocytes in vitro and inhibited the proliferative response of T and B cells to mitogens through induction of apoptosis. JWH-015 induced cross-talk between extrinsic and intrinsic pathways of apoptosis involving caspase-8, caspase-9, and caspase-3 as well as loss of mitochondrial membrane potential. Finally, administration of JWH-015 in vivo caused thymic atrophy, apoptosis, and decreased peripheral T cell response to mitogens. Together, this study suggests that CB2 selective agonists, devoid of psychotropic effect, may serve as novel anti-inflammatory/immunosuppressive agents.
Keywords: cannabinoids, CB2 receptor, immunosuppression, apoptosis, anti-inflammatory agent, non-psychoactive agent
Introduction
Marijuana has been used for recreational and medicinal purposes for centuries [1]. Its medicinal use can be traced back to ancient Chinese and Egyptian civilizations [1]. Cannabinoids, the active ingredients in cannabis, have many distinct pharmacological properties, including anticonvulsive, analgesic, antianxiety, and antiemetic [2]. Cannabinoids have been suggested as potential therapeutic agents in the treatment of ailments such as nausea, cachexia and pain in AIDS and cancer patients, and intraocular pressure in glaucoma [1,3–6]. More recently, cannabinoids have been shown to induce apoptosis and inhibit tumor cell growth, thereby suggesting the potential use of cannabinoids in the treatment of gliomas, prostate and breast cancers, and malignancies of immune origin [1,3–9]. On the other hand, abuse of marijuana or exposure to cannabinoids has also been shown to increase the susceptibility to infections that has encouraged the study of the effect of cannabinoids on the immune system [10–12].
Δ9-tetrahydrocannabinol (THC), the major psychoactive component of marijuana, and other cannabinoids have been shown to be immunosuppressive both in vitro and in vivo, affecting macrophages, natural killer cells, B cells, T cells, mast cells and dendritic cells (DCs) [1,13,14]. It is generally believed that cannabinoids alter the immune functions through their ability to mediate an effect on cytokine production and to exert anti-proliferative properties [15] (Review). Recent studies from our laboratory suggested that the anti-proliferative activity of THC may result from its ability to trigger apoptosis in immune cells [16]. To this end, we demonstrated that administration of THC into mice caused a reduction in the cellularity of the thymus and spleen, and decreases the ability of T and B cells to divide upon activation, which correlated with induction of apoptosis [16]. Furthermore, a recent study from our laboratory demonstrated that THC or anandamide, an endogenous cannabinoid, can induce apoptosis in dendritic cells (DCs) in vitro and treatment of mice with THC caused depletion of splenic DCs [13]. While such studies on one hand suggest that abuse of marijuana or clinical use of THC in AIDS or cancer patients may cause an undesired effect namely immunosuppression. On the other hand, the ability of cannabinoids to suppress the immune functions can be potentially exploited for the treatment of inflammatory and autoimmune diseases by inhibiting the proliferative response of B and T cells, and by decreasing antigen-presentation by DCs [16] [13]. However, the clinical use of marijuana and cannabinoids such as THC is still very controversial due to the psychotropic side effect associated with the treatment. These effects are due to the fact that THC can signal not only through the cannabinoid receptor (CB) 2, mainly expressed on cells of the immune system and accounting for the immunosuppressive effects, but also through the CB1 receptor found on cells of the central nervous system, causing psychotropic effects [1,17]. Therefore, it is of great interest to test whether synthetic CB2-selective cannabinoid receptor agonists would induce apoptosis in immune cells and mediate immunosuppression in vivo. Such CB2-selective agonists would be ideal candidates for clinical use as immunosuppressive drugs because of their minimal binding to CB1 receptors and therefore devoid of psychotropic effects.
In the current study, we tested the ability of a selective CB2 agonist to induce apoptosis in lymphoid cells and cause immunosuppression. The CB2-selective agonist that we chose to study was the commercially available synthetic cannabinoid (2-Methyl-1-propyl-1H-indol-3-yl)-1-naphtalenylmethanone also known as JWH-015, which has Ki values of 13.8 nM (CB2) and 383 nM (CB1). We demonstrate for the first time that administration of JWH-015, a CB2-selective agonist into mice inhibits the proliferative responsiveness of T and B cells, and induces thymic atrophy via the induction of apoptosis. Looking deeper into the mechanism of JWH-015-induced apoptosis, we find that JWH-015 signals primarily through the death receptor pathway and through crosstalk, with the mitochondrial pathway.
Materials and Methods
Mice
Adult female (5–7 weeks of age) C57BL/6 wild-type mice were purchased from the National Institutes of Health (NIH) and maintained in our animal facility. Adult female C57BL/6lpr/lpr mice were originally purchased from Jackson Laboratories (West Grove, PA) and bred in our facility.
Reagents
JWH-015 was purchased from Tocris Cookson (Ellisville, MO), initially diluted to a concentration of 20 mM using DMSO (Sigma, St Louis, MO) and stored at –20°C. It was then further diluted as needed by the experiments using the appropriate medium. The following CB2 specific antagonists were also used: SR144528 (Sanofi Research, Montpellier, France), and AM630 (Tocris).
In vitro proliferation assay
Spleens were aseptically harvested from C56BL/6 mice, placed into a sterile plastic bag with 10 ml of complete RPMI medium and single cell suspension was prepared using a laboratory homogenizer (Stomacher, Tekmar, Cincinnati, OH). Contaminating erythrocytes were lysed by resuspending the cells in 3 ml lysing buffer (Sigma). After 2 washes in complete RPMI medium, the cellularity was determined using the trypan blue dye exclusion method. The cells were resuspended to a concentration of 5x106cells/ml in complete medium. The cells (5x105 in 100 μl) were cultured in 96 well plates with various concentrations of JWH-015 (0, 5, 10, and 20 μM) and either left unstimulated or stimulated with 2 μg/ml concanavalin A (Con A) (Sigma), 2 μg/ml anti-CD3 mAbs (Pharmingen, San Diego, CA), or 5 μg/ml lipopolysaccharide (LPS) (Sigma) for 48 hrs, as we have found this time point to correspond to the peak of the mitogen-induced proliferative response. Eight hours prior to the end of the assay, the cells were pulsed with 2 μCi of 3H-thymidine. DNA synthesis was determined by β-scintillation counting.
Cell cycle analysis
T cells were purified from C57BL/6 spleens by plastic adherence followed by passage over a nylon wool column. The cells (5x105 in 100 μl) were cultured in 96 well plates with 20 μM JWH-015 or the vehicle and stimulated with 2 μg/ml Con A for 24 hrs. The cells were then harvested and fixed using ice-cold 70% ethanol. The cells were then stained with propidium iodide (PI) in the presence or RNAse A for 30mn at 37oC and analyzed by flow cytometry.
Evaluation of JWH-015-induced apoptosis in thymocytes and splenocytes
Spleens and thymi were aseptically removed from euthanized C57BL/6 mice and reduced to single cell suspensions as described above. After lysis of contaminating erythrocytes and appropriate washes, splenocytes and thymocytes were readjusted to a concentration of 5x106 cells/ml. Splenocytes (5x105cells in 100 μl) were then cultured in 96 well plates with various concentrations of JWH-015 and left unstimulated or stimulated with Con A, anti-CD3 mAbs or LPS for 48 hrs as described above. Thymocytes (5x106cells/well) were cultured in 24-well plates with 20 μM JWH-015, as this concentration happened to be effective in inducing apoptosis in splenocytes. The thymocytes were only kept in culture overnight because extended periods of culture favor spontaneous apoptosis of the cells, which makes it more difficult to assess the effects of JWH-015. After the appropriate incubation time, splenocytes and thymocytes were harvested, washed twice in PBS, and fixed with 100 μl of 4% paraformaldehyde at room temperature for 30 mn. The level of apoptosis was then evaluated using the TUNEL method followed by flow cytometry.
Detection of apoptosis using the TUNEL method
Apoptotic events were quantified at the single cell level using the TUNEL method (Boerhinger-Mannheim, Indianapolis, IN), as described [18]. The cells were washed with PBS twice and permeabilized on ice for 2 mn using 100 μl of 0.1% Triton X-100 in 0.1% sodium citrate. After two additional washes, the cells were incubated with 25 μl of TUNEL reaction composed of FITC-dUTP and TdT at 37°C with 5% CO2. After 1hr, the cells were washed two more times, resuspended in PBS, and fluorescence was determined by flow cytometry.
Assessment of JWH-015-induced activation of caspase-3 and/or –7
Splenocytes and thymocytes were cultured with the vehicle or various concentrations of JWH-015 (5, 10, or 20 μM) in a total volume of 100 μl for 24 hrs (splenocytes) or overnight (thymocytes). The induction of apoptosis was then detected using the Apo-ONE homogeneous caspase-3/7 assay (Promega Corporation, Madison, WI)) according to the manufacturer’s recommendation. Briefly, 100 μl of a 1:100 substrate / buffer solution was added to the plate. The plate was then shaken for 30 sec and incubated at room temperature for 6 hrs before reading the results on a Wallac Victor2 instrument (Perkin Elmer life sciences, Boston, MA).
Assessment of JWH-015-induced loss of mitochondrial membrane potential
Thymocytes and splenocytes (5x106/well) were cultured with various concentrations of JWH-015 (5, 10, and 20 μM) or vehicle for 24 hrs. In some experiments, cells were also cultured with caspase-8 inhibitor (Z-IETD-FMK) (R&D systems, Minneapolis, MN). Fifteen minutes prior to the end of the incubation, 3,3-dihexyloxacarbocyanine iodide (DiOC6) was added to a final concentration of 40 nM. Mitochondrial membrane potential (Δψm) was then assessed by flow cytometric analysis, as described [19].
Blockade of JWH-015-induced apoptosis using a CB2 antagonist
For the caspase-3/7 assay, thymocytes were cultured in 96-well plates with 20 μM JWH-015 in the presence or absence of CB2 specific antagonist for 24 hrs. The induction of apoptosis was then detected using the Apo-ONE homogeneous caspase-3/7 assay (Promega Corporation) as described above. For the TUNEL assay, cells were cultured overnight in 24-well plates with JWH-015 in the presence of absence of CB2 specific antagonist. The cells were then harvested, fixed and analyzed by TUNEL staining followed by flow cytometry as described above.
Western blot analysis
Thymocytes and splenocytes were cultured with various concentrations of JWH-015 (5, 10, or 20 μM) or the vehicle for 18 hrs. Proteins (total or cytosolic, depending on the experiment) were extracted and the concentration was determined using the BCA protein assay kit (Pierce, Rockford, IL). Proteins were then separated by SDS-PAGE and transferred to a membrane. Blots were blocked with 5% (w/v) nonfat milk in TBS-T (0.5 M NaCl, 20 mM Tris-HCl, 0.1% (v/v) Tween 20, pH 7.6), and probed with primary antibodies against caspase-3 (Cell Signaling Technology, Beverly, MA) (total proteins), or cytochrome c (Pharmingen) (cytosolic proteins). Beta-actin (Sigma) was used as a loading control. Finally, blots were washed and incubated with HRP-conjugated secondary antibodies and the proteins of interest visualized with ECL reagent (Amersham Biosciences, Piscataway, NJ).
Caspase inhibition studies
Thymocytes were pre-treated with caspase-8 and caspase-9 inhibitors (R&D systems) at various concentrations (12.5 μM to 100 μM) for one hour. The cells were then either left untreated or cultured with 20 μM JWH-015 or the vehicle overnight. Finally the cells were harvested, fixed and analyzed for apoptosis using the TUNEL method followed by flow cytometry as described above.
Determination of the effect of in vivo exposure to JWH-015 on in vitro splenocyte proliferation
C57BL/6 mice were treated with a daily dose (100 mg/kg body weight) of JWH-015 or vehicle for three days (days 0 to 2). On day 3, the mice were sacrificed, the spleens were harvested, prepared into a single-cell suspension as described above, and adjusted to 5x106 cells/ml in complete medium. The cells (100 μl/well) were cultured in 96-well flat-bottomed plates and either left unstimulated or stimulated with 2 μg/ml Con A for 48 hrs. The cells were cultured with 2 μCi of 3H-thymidine for the final 8 hrs, and DNA synthesis was determined by β-scintillation counting.
Analysis of JWH-015-induced thymic atrophy and apoptosis in vivo
Groups of four C57BL/6 mice were treated with a daily dose (150 mg/kg body weight) of JWH-015 or vehicle for three days (days 0 to 2). On day 3, the mice were sacrificed, the thymi were harvested, prepared into a single-cell suspension as described above, and the total number of viable cells was determined by trypan blue dye exclusion. The cells were then adjusted to 5x106 cells/ml in complete medium, and cultured in vitro in 96-well flat-bottomed plates (1x106 cells/well in 0.2ml of medium) overnight at 37oC. The cells were then harvested, fixed, and analyzed for apoptosis using the TUNEL method followed by flow cytometry.
Determination of the effect of exposure to JWH-015 on T cell activation with staphylococcal enterotoxin A (SEA) in vivo
Mice were injected with 10 μg SEA (Toxin Technology, Sarasota, FL) into the rear footpads on day 0 to activate T cells in the draining popliteal lymph nodes as described [20]. Mice were then injected with a daily dose (150 mg/kg body weight) of JWH-015 or vehicle once a day for three days. Twenty four hours after the last injection, the mice were sacrificed. The draining lymph nodes were harvested, pooled and prepared into a single cell suspension as described above. The cells were stained with 1 μg of PE-conjugated anti-Vβ3 or PE-conjugated anti-Vβ11 mAbs (Pharmingen). The cells were stained for 30 mn on ice, washed twice in PBS and fixed. Some of the cells were then further stained using the TUNEL method. Both single-stained and double-stained samples were then analyzed using a flow cytometer.
Statistical Analysis
Each experiment using flow cytometry was repeated at least twice with consistent results and representative data were plotted. In other experiments, JWH-015-treated group was compared to vehicle controls using Student’s t test and p<0.05 was considered to be statistically significant.
Results
Exposure to JWH-015 reduces the proliferative response of splenocytes to mitogens in vitro
Splenocytes from C57BL/6 mice were cultured with various concentrations of JWH-015 and left unstimulated or stimulated with anti-CD3 mAbs, Con A or LPS for 48 hrs, as described in the Materials and Methods. The resulting proliferation was assessed by 3H-thymidine incorporation. As shown in Fig. 1A, JWH-015 treatment significantly impaired the ability of splenocytes to proliferate in response to mitogenic stimulation, in a dose-related manner. It should be noted that the response to both T cell (Con A and anti-CD3 mAbs) and B cell (LPS) mitogens was reduced, suggesting that JWH-015 might affect both subsets of lymphocytes. Our splenocyte preparation does contain macrophages, which express high level of cannabinoid receptors. However, LPS treatment of macrophages typically triggers the release of pro-inflammatory cytokines rather than proliferation. Therefore, we attribute most of the proliferative response to LPS to B cells. Finally, cell cycle analysis of purified T cells stimulated with Con A in the presence of JWH-015 revealed an increase in the sub G0/G1 peak, which is characteristic of apoptotic cells, when compared to cells stimulated with Con A and treated with the vehicle (Fig. 1B). Together these data suggest that JWH-015 may inhibit proliferation through the induction of apoptosis.
Figure 1. The effect of JWH-015 on the proliferative response of splenocytes to stimulation with Con A, anti-CD3 mAbs, and LPS in vitro.
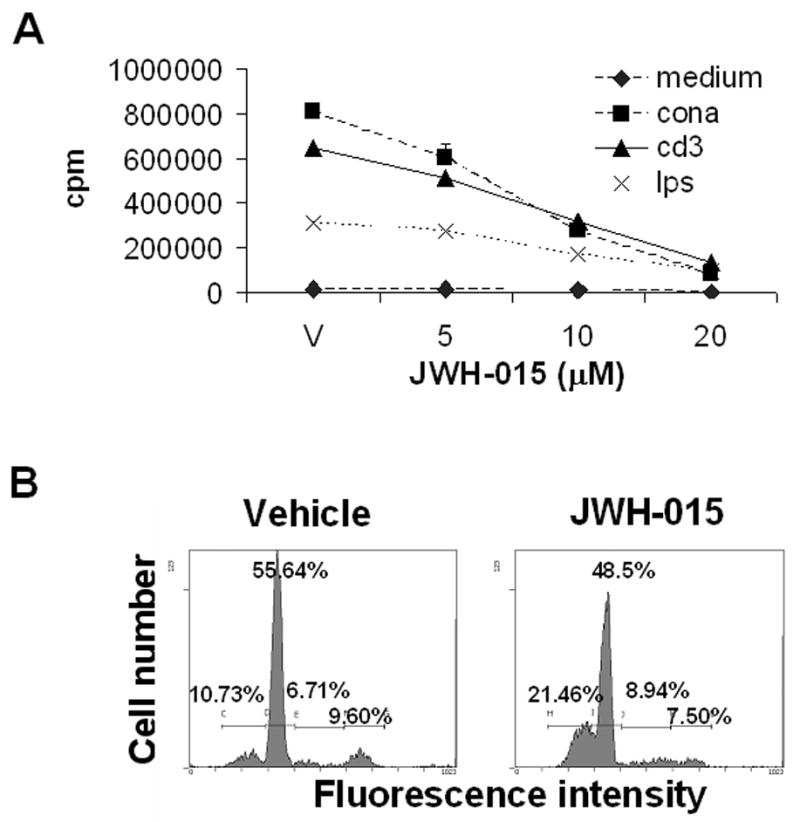
A) Splenocytes (5x105) from C57BL/6 mice were incubated with various concentrations of JWH-015 (0, 5, 10, and 20 μM) in the absence or presence of 2 μg/ml Con A, 5 μg/ml anti-CD3 mAbs, or 5 μg/ml LPS for 48hrs. The proliferative response was measured by pulsing with 2 μCi of [3H]-thymidine during the last 8 hrs of incubation. Thymidine incorporation was determined by β-scintillation counting. The data points represent the mean ± SEM of triplicate cultures. The error bars are not visible because they fall within the symbols. JWH-015 at all concentrations tested caused a significant decrease in cell proliferation (p<0.05) when compared to vehicle control except for the LPS response at 5 μM JWH-015. B) T cells were purified from C57BL/6 spleens by plastic adherence followed by passage over a nylon wool column. The cells (5x105 / well) were stimulated with 2 μg/ml Con A, in the presence of JWH-015 (20 μM) or the vehicle for 24 hrs. The cells were then harvested, fixed and stained with PI. Cell cycle analysis was performed by flow cytometry. The percentage of cells in each stage of the cell cycle has been depicted in each histogram.
Exposure to JWH-015 leads to induction of apoptosis in both naïve and activated splenocytes in vitro
In order to confirm that the impaired proliferative response to mitogens resulting from JWH-015 treatment was due to the induction of apoptosis, splenocytes were cultured with JWH-015 in the presence or absence of mitogens, as described in the Materials and Methods. Analysis of the samples after TUNEL staining revealed an increase in the number of apoptotic events upon JWH-015 treatment, in splenocyte cultures stimulated with medium or various mitogens (Fig. 2A). Of note, the levels of apoptosis resulting from mitogenic stimulation were lower than the corresponding levels detected for the same concentrations of JWH-015 when the cells were incubated in medium alone. This is consistent with our previous findings that lymphocytes down-regulate cannabinoid receptor expression upon activation and are therefore slightly less sensitive to apoptosis [16]. To further corroborate that JWH-015 treatment induces apoptosis in splenocytes, we studied caspase activity and found that JWH-015 caused a dose-related increase in caspase-3/7 activation in splenocytes, confirming the induction of apoptosis in these cells (Fig. 2B).
Figure 2. JWH-015 induces apoptosis in naïve and activated splenocytes in vitro.
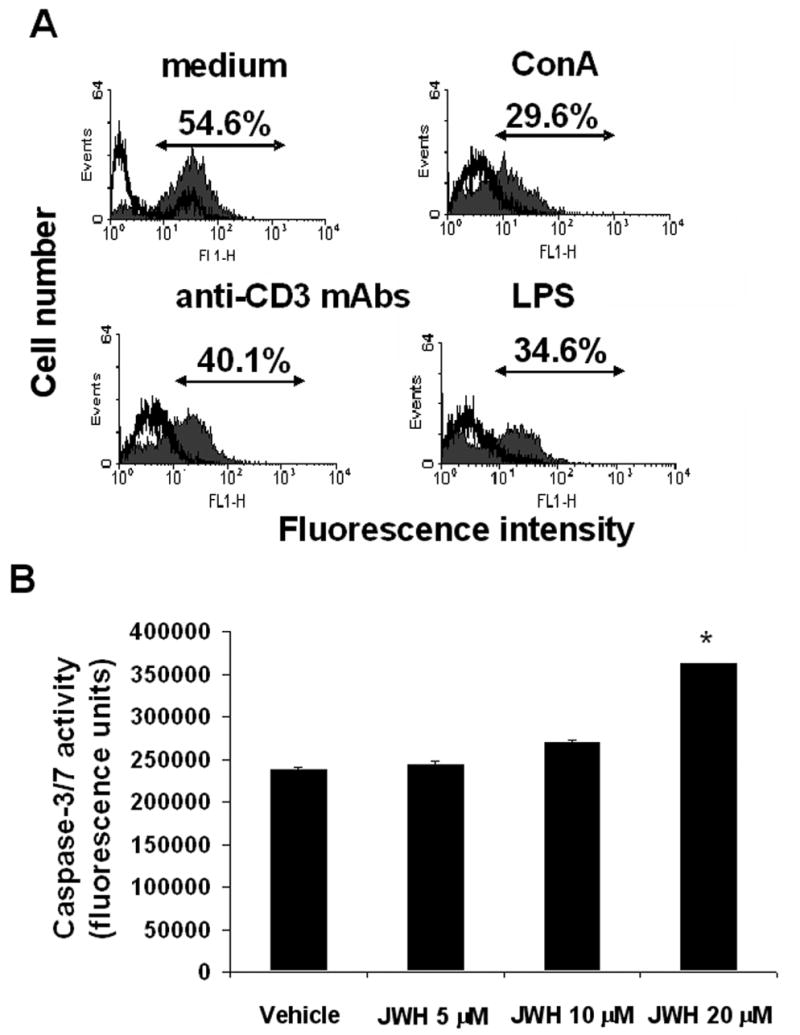
A) Splenocytes from C57BL/6 were cultured in medium alone or in the presence of 2 μg/ml Con A, 5 μg/ml anti-CD3 mAbs, or 5 μg/ml LPS for 48hrs. In addition, these cultures received 20 μM JWH-015 or the vehicle. The cells were harvested and analyzed for apoptosis using the TUNEL assay followed by flow cytometry. The percentage of apoptotic cells after exposure to JWH-015, obtained by subtracting the empty histogram (vehicle control) from the filled histogram (cells treated with JWH-015) has been depicted in each histogram. B) Splenocytes from C57BL/6 mice were incubated with various concentrations of JWH-015 (0, 5, 10, and 20 μM) or the vehicle for 24hrs. The induction of apoptosis was then analyzed using the caspase-3/7 Apo-One fluorimetric assay. The data represent the mean ± SEM of duplicate cultures. Asterisk indicates statistically significant difference when compared to vehicle control.
In vitro treatment of splenocytes with JWH-015 induces mitochondrial injury
In order to determine whether JWH-015 affects the mitochondria, splenocytes from C57BL/6 mice were cultured with JWH-015 for 24hrs. Fifteen minutes prior to the end of the incubation, DiOC6 was added to the cultures as described in the Materials and Methods. The data showed that JWH-015 treatment led to increased proportion of splenocytes exhibiting loss of mitochondrial membrane potential (Δψm) as indicated by a decrease in mean fluorescence intensity, when compared to vehicle controls (Fig. 3A), In addition, Western blot analysis of cytosolic proteins extracted from splenocytes cultured with JWH-015 or the vehicle showed a marked release of cytochrome c from the mitochondria following JWH-015 treatment, confirming damage to the organelle (Fig. 3B).
Figure 3. The effect of JWH-015 on loss of Δψm in splenocytes in vitro.
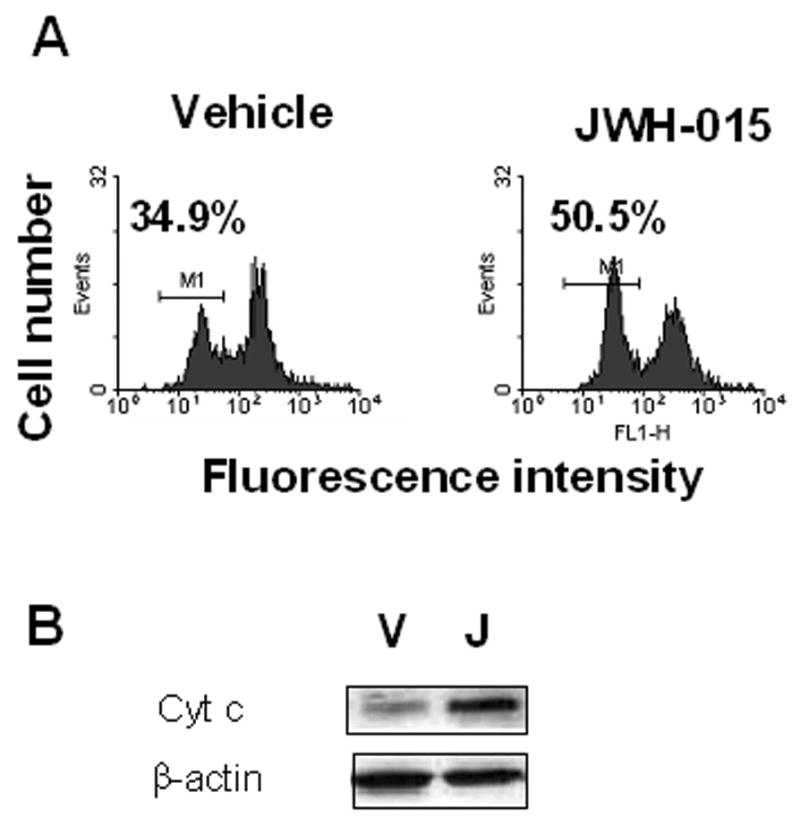
A) Splenocytes from C57BL/6 mice were incubated with 5 μM of JWH-015 or the vehicle for 24hrs. The loss of mitochondrial membrane potential was analyzed using DiOC6 staining followed by flow cytometry. The percentage of cells showing loss of Δψm was depicted in each histogram. B) Splenocytes from C57BL/6 mice were treated with 20 μM JWH-015 for 18hrs. Cytosolic proteins were then harvested and analyzed by Western blotting for cytochrome c expression. Beta-actin was used as a loading control.
JWH-015 induces apoptosis in thymocytes in vitro
Analysis of thymocytes cultured overnight with JWH-015 or vehicle using the TUNEL method showed that JWH-015 caused a significant increase in apoptosis when compared to vehicle controls (Fig. 4A). In addition, we also found a dose-related increase in caspase-3/7 activity upon JWH-015 treatment (Fig. 4B). Finally, Western blot analysis of proteins extracted from thymocytes cultured with various concentrations of JWH-015 for 1hr showed an increase in caspase-3 cleavage following JWH-015 treatment (Fig. 4C), confirming that JWH-015 induces apoptosis in thymocytes.
Figure 4. Thymocytes exposed to JWH-015 in vitro undergo apoptosis.
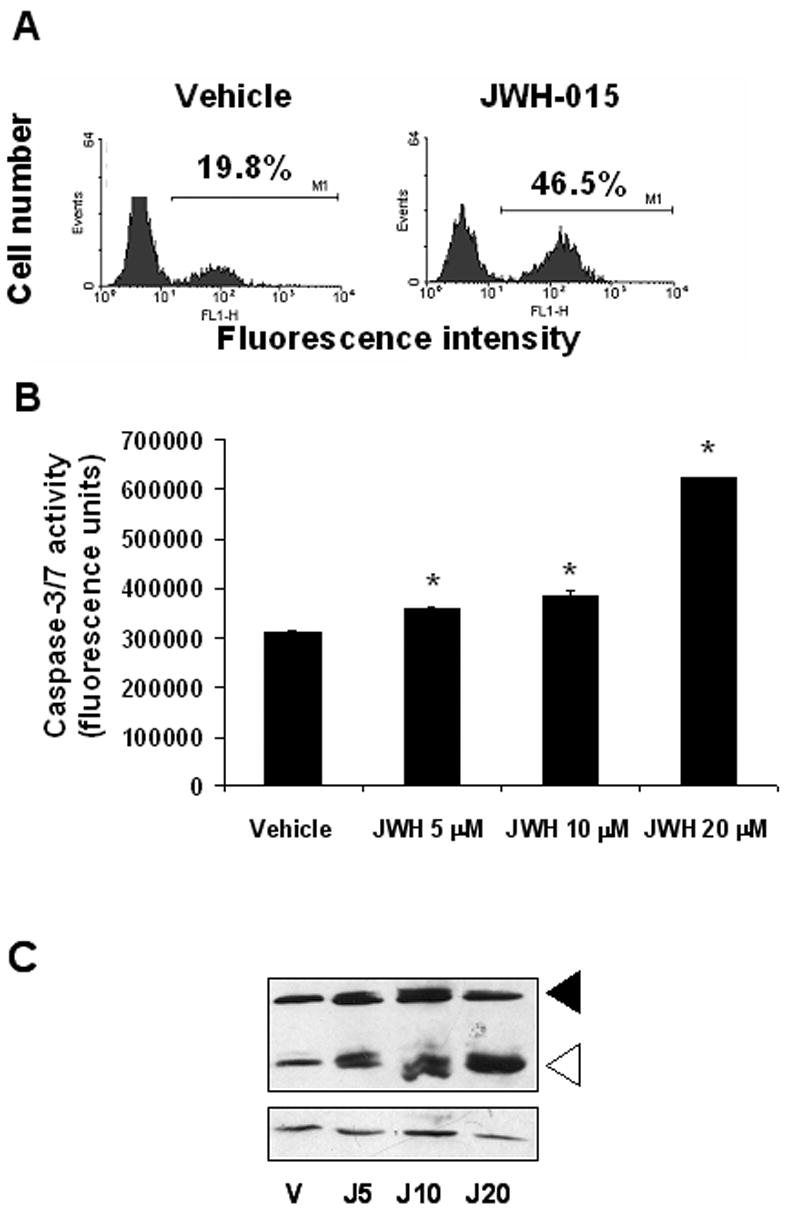
A) Thymocytes (5x106) from C57BL/6 mice were incubated with 20 μM JWH-015 or the vehicle for 24hrs. The cells were harvested and analyzed for apoptosis using the TUNEL assay followed by flow cytometric analysis. The percentage of apoptotic cells was depicted on each histogram. B) Thymocytes from C57BL/6 mice were incubated with various concentrations of JWH-015 (5, 10, and 20 μM) or the vehicle overnight. The induction of apoptosis was then analyzed using the caspase-3/7 Apo-One fluorimetric assay. The data represent the mean ± SEM of duplicate cultures. Asterisk indicates statistically significant difference when compared to vehicle control. C) Thymocytes from C57BL/6 mice were cultured with various concentrations of JWH-015 (5, 10, or 20 μM) or the vehicle for 18hrs, after which, whole proteins were extracted and analyzed for caspase-3 cleavage by Western blotting. Beta-actin was used as a loading control.
In vitro treatment of thymocytes with JWH-015 induces mitochondrial damage
Δψm analysis of thymocytes cultured with various concentrations (0, 5, 10, and 20 μM) of JWH-015 for 24 hrs using DiOC6 staining showed a dose-related loss of Δψm when compared to vehicle-treated cells (Fig. 5), suggesting damage to the organelle.
Figure 5. The effect of JWH-015 on mitochondrial injury in thymocytes in vitro.
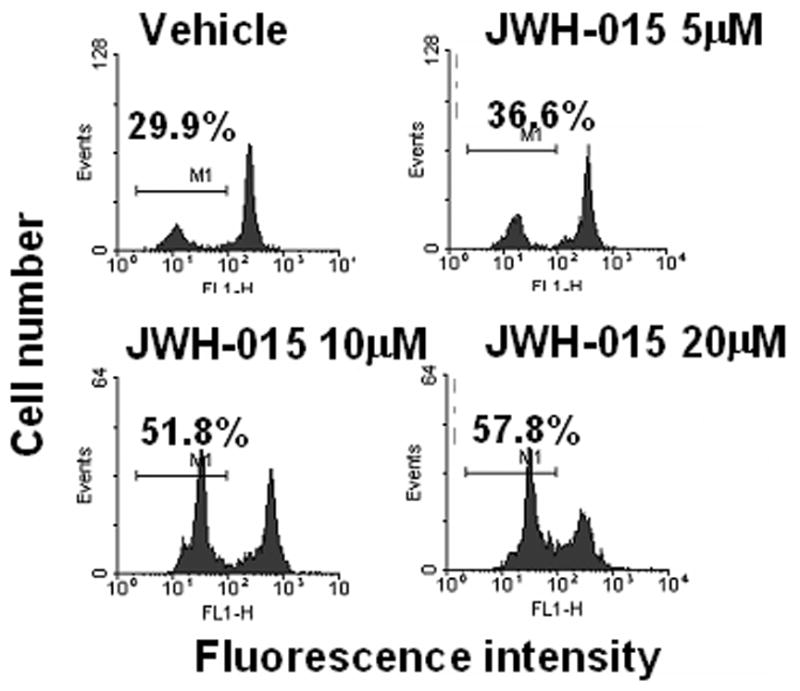
Thymocytes from C57BL/6 mice were incubated with various concentrations of JWH-015 (5, 10, and 20 μM) or the vehicle for 24hrs. The loss of mitochondrial membrane potential was analyzed using DiOC6 staining followed by flow cytometric analysis. The percentage of cells showing loss of Δψm was depicted in each histogram.
CB2 antagonist blocks JWH-015-induced apoptosis
In order to confirm the specificity of JWH-015 for CB2, we undertook to block its effect using a CB2-specific antagonist. Thymocytes were therefore cultured with JWH-015, in the presence or absence of CB2 antagonist. Analysis of the samples using the caspase-3/7 assay showed a decrease in caspase activity in cells cultured with JWH-015 and SR144528, when compared to cells cultured with JWH-015 alone (Fig. 6A). In addition, when analyzing the cells by the TUNEL method, we found that pre-treatment with CB2 antagonist could prevent JWH-015-induced apoptosis (Fig. 6B). Together, these data show that JWH-015’s effect is indeed mediated through CB2.
Figure 6. The effect of the CB2 antagonist on JWH-015-induced apoptosis thymocytes in vitro.
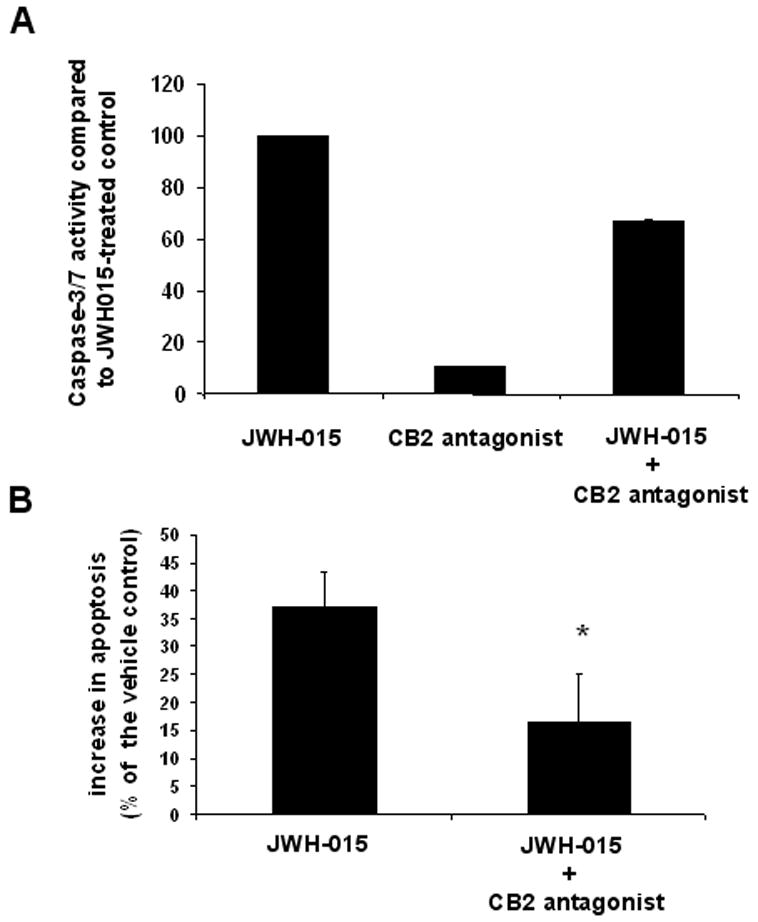
A) Thymocytes from C57BL/6 mice were cultured with the vehicle, 20 μM JWH-015, 5 μM SR144528 or 20 μM JWH-015 + 5 μM SR144528 for 24 hrs. The induction of apoptosis was then analyzed using the caspase-3/7 Apo-One fluorimetric assay. The data represent the mean ± SEM of duplicate cultures and is expressed as a percentage of caspase-3/7 activity in JWH-015-treated groups, considering this response as 100%. B) Thymocytes from C57BL/6 mice were cultured with the vehicle, 20 μM JWH-015, 2.5 μM AM630 or 20 μM JWH-015 + 2.5 μM AM630 for 24 hrs. The cells were then harvested, fixed, and analyzed for apoptosis using the TUNEL assay followed by flow cytometry. Vertical bars represent the average increase in apoptosis following treatment compared to the vehicle control, calculated from four experiments. Asterisk indicates statistically significant difference when compared to JWH-015 control.
Role of caspase-8, caspase-9, and Fas in JWH-015-induced apoptosis
To further directly address the role of intrinsic and extrinsic pathways in JWH-015-induced apoptosis, we pre-treated thymocytes with various concentrations of caspase-8 or caspase-9 inhibitors (12.5 μM to 100 μM) for one hour. The cells were then incubated with JWH-015 or the vehicle overnight. Cells cultured with caspase-8 or caspase-9 inhibitor alone did not show an increase in TUNEL positive cells when compared to the DMSO-treated control (data not shown). Treatment of thymocytes with JWH-015 induced 66.82% apoptosis, which was inhibited in a dose-dependent manner both by caspase-8 and by caspase-9 inhibitors, suggesting the involvement of both pathways of apoptosis (Fig. 7A). The fact that higher concentrations of caspase-8 inhibitor could almost completely block apoptosis suggested that JWH-015-induced apoptosis may involve cross-talk between the death receptor and the mitochondrial pathways. To further corroborate this, we studied the effect of caspase-8 inhibitor on JWH-015-induced loss of Δψm. The data shown in Fig 7B suggested that addition of caspase 8 inhibitor significantly decreased the loss of Δψm caused by JWH-015 thereby, suggesting that activation of caspase-8 may play a critical role in the induction of mitochondrial damage. The data however does not completely prove the involvement of the death receptor pathway as loss of mitochondrial membrane potential could result from activation of caspase-3 or -9, which could be non-specifically blocked by the caspase-8 inhibitor. Next, we investigated whether JWH-015 could activate the death receptor pathway through Fas signaling. To this end, we used C57BL/6lpr/lpr thymocytes, which lack Fas, and noted that Fas-deficient thymocytes were sensitive to JWH-015-induced apoptosis, thereby suggesting that Fas was not involved (Fig. 7C).
Figure 7. The effect of caspase inhibitors on JWH-015-induced apoptosis and loss of Δψm in thymocytes in vitro.
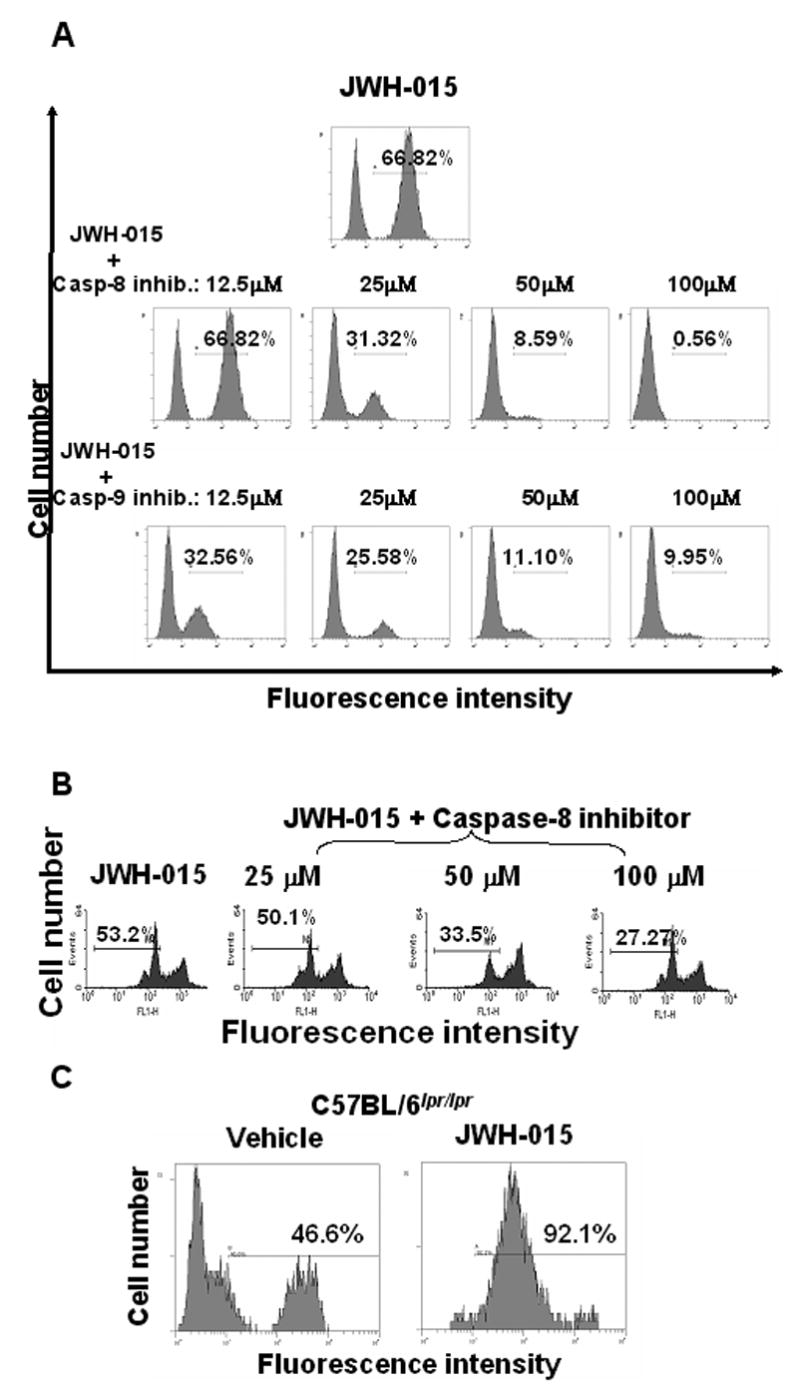
A) Thymocytes from C57BL/6 mice were cultured with 20 μM JWH-015, 20 μM JWH-015 + various concentrations of caspase-8 inhibitor, or 20 μM JWH-015 + various doses of caspase-9 inhibitor for 24hrs. The cells were harvested and analyzed for apoptosis using the TUNEL assay followed by flow cytometry. The percentage of apoptotic cells was depicted in each histogram. B) Thymocytes from C57BL/6 mice were cultured with 20 μM JWH-015, or 20 μM JWH-015 + various concentrations of caspase-8 inhibitor for 24hrs. The loss of Δψm was analyzed using DiOC6 staining followed by flow cytometric analysis. The percentage of cells showing loss of Δψm was depicted in each histogram. C) Thymocytes from C57BL/6lpr/lpr mice were cultured with 20 μM JWH-015 or the vehicle for 24hrs. The cells were harvested and analyzed for apoptosis using the TUNEL assay followed by flow cytometric analysis. The percentage of apoptotic cells was depicted on each histogram.
In vivo exposure to JWH-015 leads to inhibition of the in vitro proliferative response of splenocytes to Con A, as well as thymic atrophy due to the induction of apoptosis
To test if JWH-015 would cause immunosuppression in vivo, we injected 150 mg/kg body weight of JWH-015 or vehicle into C57BL/6 mice for 3 consecutive days and studied various immune parameters. JWH-015-treated mice showed a decrease in splenic (Fig. 8A) and thymic cellularity (Fig. 8B) as well as increased levels of apoptosis in the thymus (Fig. 8C). In addition, treatment with JWH-015 significantly decreased the ability of splenocytes to proliferate in vitro in response to Con A (Fig. 8D). Together these data suggested that JWH-015 was immunosuppressive in vivo.
Figure 8. The effect of JWH-015 in vivo on lymphoid organs and T cell proliferation.
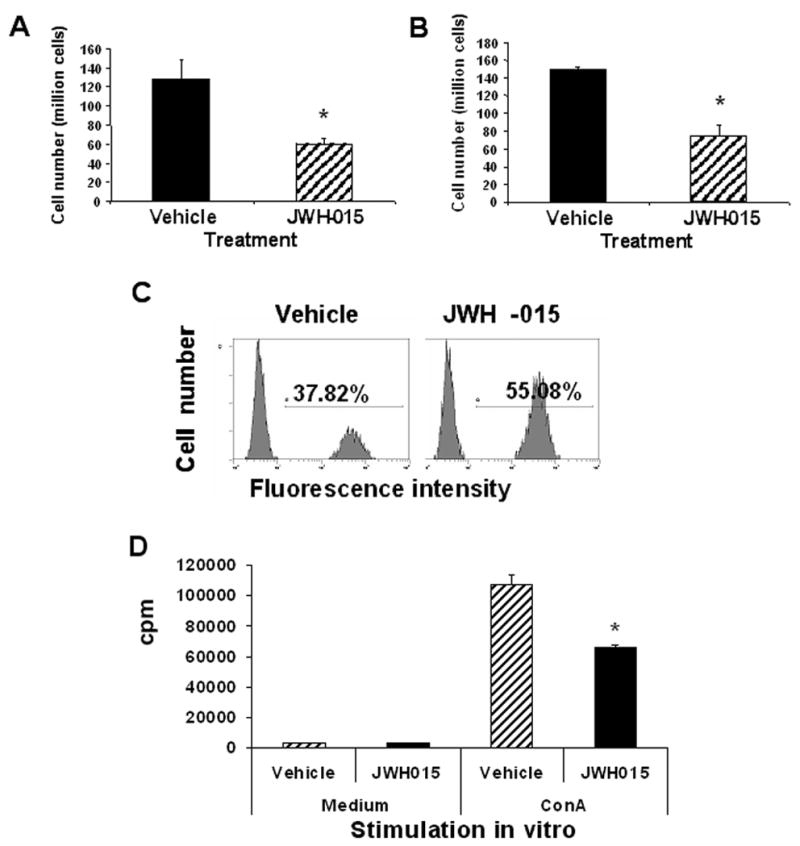
Adult female C57BL/6 mice were injected i.p. with 150 mg/kg body weight of JWH-015 or the vehicle once a day for 3 days. The spleen and thymus were harvested on day 4 and made into a single-cell suspension. Splenic (A) and thymic (B) cellularity was determined by trypan blue dye exclusion. Vertical bars represent mean cellularity ± SEM from 4 individual mice. Asterisk indicates statistically significant difference when compared to vehicle control. C) The thymocytes were cultured overnight, harvested, and analyzed for apoptosis using the TUNEL method followed by flow cytometric analysis. The percentage of apoptotic cells has been depicted in each histogram. D) The splenocytes (5x105) were cultured in medium in the presence or absence of 2 μg/ml Con A for 48 hrs. The proliferative response was measured by pulsing with 2 μCi of [3H]-thymidine during the last 8 hrs of incubation. Thymidine incorporation was determined by β-scintillation counting. The data points represent the mean ± S.D. of triplicate cultures.
In vivo exposure to JWH-015 leads to decreased proliferative response of Vβ3+ and Vβ 11+ T cells to SEA
To further investigate the effect of JWH-015 treatment in vivo on antigen-specific T cell response, mice were injected with 10 μg SEA in each rear footpad. The mice were also treated with 150 mg/kg body weight of JWH-015 for 3 consecutive days, after which the draining popliteal lymph node cells were analyzed for the expression of Vβ3 and Vβ11, as T cells bearing these markers are known to be specifically activated by SEA [20]. Injection of SEA into vehicle-treated mice caused a significant increase in the percentage and absolute numbers of Vβ3+ and Vβ11+ T cells in the draining lymph nodes (Fig. 9A and B). Moreover, treatment with JWH-015 caused a significant decrease in the percentage and the absolute number of Vβ3+ and Vβ11+ T cells in the draining lymph nodes of SEA-treated mice (Fig. 9A and B). In addition, analysis of the SEA-treated samples using the TUNEL method revealed an increase in apoptosis in both Vβ3+ and Vβ11+ T cells upon in vitro treatment with JWH-015, as shown by the increase in the percentage of TUNEL positive cells as well as in MFI depicted in Fig. 9C. It has to be noted that the high level of apoptosis detected in the vehicle control is likely due to the induction of AICD as the cells have been stimulated with SEA in vivo for 3 days. Together these data confirmed that JWH-015 could suppress the T cell response to a bacterial superantigen in vivo, via the induction of apoptosis.
Figure 9. In vivo exposure to JWH-015 leads to a decreased T cell response to SEA treatment.
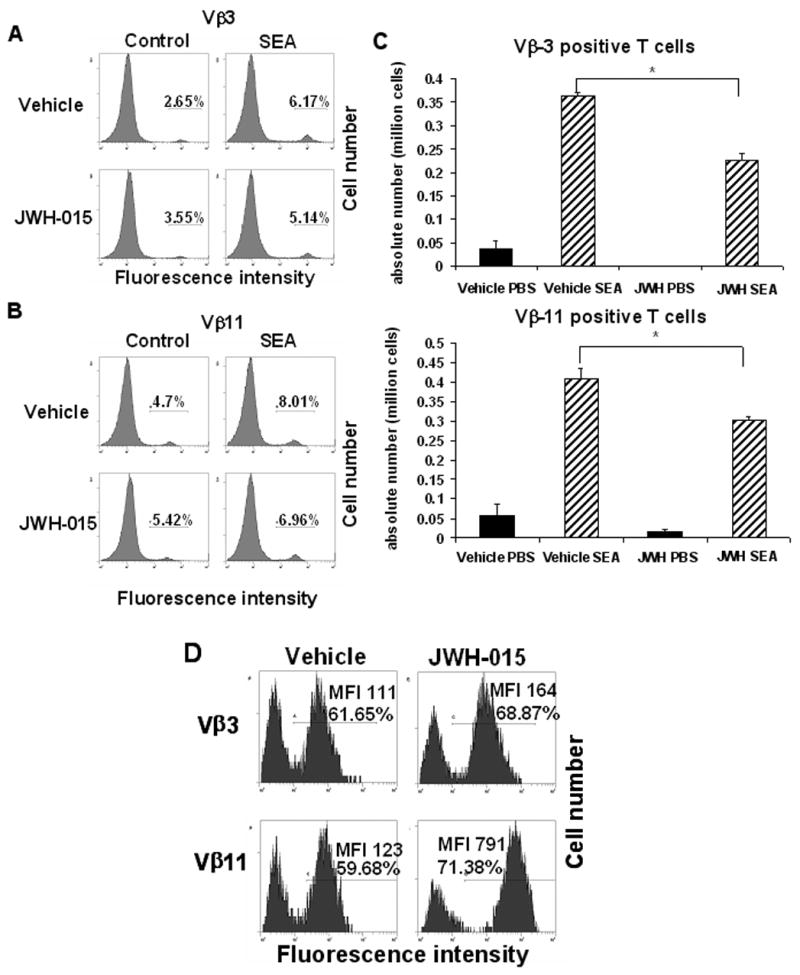
C57BL/6 mice were treated with SEA (10 μg / footpad) on day 0 and received a daily injection with 150 mg/kg body weight of JWH-015 or the vehicle once a day for 3 days. On day 3, the draining popliteal lymph nodes were removed and the cells were stained for Vβ3 (A) and Vβ11 (B) expression. The percentage of marker positive cells has been depicted in each histogram. C) Absolute numbers of Vβ3 and Vβ11 positive cells recovered from the draining popliteal lymph nodes. Vertical bars represent mean number of SEA-reactive T cells per lymph node calculated from groups of 4 mice. Asterisk indicates statistically significant difference when compared to vehicle control. D) Cells were stained for Vβ3 and Vβ11 and analyzed for apoptosis using the TUNEL method followed by flow cytometry. The histograms represent the result of the TUNEL staining, gated on the Vβ3+ or Vβ11+ population. The percentage of TUNEL positive cells and the mean fluorescence intensity (MFI) have been depicted on each histogram
Discussion
The cannabinoid receptor, CB2, is almost exclusively expressed on immune cells and the role of this receptor in immunoregulation remains unclear. In the current study, we demonstrated that JWH-015, a CB2 selective agonist, inhibited proliferation of splenocytes stimulated with either B cell or T cell mitogens in vitro. We also found that the decrease in lymphocyte proliferation correlated with an increase in the level of apoptosis. JWH-015 induced apoptosis in thymocytes in vitro as well. Treatment of immune cells with JWH-015 appeared to trigger both the extrinsic and intrinsic pathways of apoptosis. Moreover, administration of JWH-015 into mice caused thymic atrophy which correlated with increased levels of apoptosis in thymocytes and decreased ability of peripheral T cells to proliferate when stimulated with ConA. In addition, JWH-015 prevented the expansion of Vβ3+ and Vβ11+ T cells following SEA stimulation in vivo. Together, the current study demonstrated that ligation of CB2 receptors can induce apoptosis in immune cells and cause immunosuppression and that synthetic CB2 selective agonists may serve as ideal candidates for clinical immunosuppression.
Apoptosis is mediated primarily through two pathways each involving specific caspases [21–23]. The extrinsic (death receptor) pathway is characterized by the activation of a death receptor leading to the recruitment of an adaptor molecule and procaspase-8 to form a Death-Inducing Signaling Complex (DISC) [23–25]. Formation of the DISC results in the autocatalytic cleavage and subsequent activation of caspase-8, which in turn activates caspase-3 or caspase-7 leading to apoptotic death of the cell [23–25]. In the intrinsic (mitochondrial) pathway, damage to the mitochondria results in the release of cytochrome c, which in turn forms the apoptosome with apoptosis protease activator factor-1 (Apaf-1), dATP, and procaspase-9 [26–29]. Formation of the apoptosome is followed by activation of caspase-9, activation of caspase-3 or caspase-7, and apoptosis [26–29]. In some cases where the amount of caspase-8 activated is insufficient to activate caspase-3 or caspase-7, caspase-8 cleaves the pro-apoptotic Bcl2 family member Bid, which then translocates to the mitochondria to cause release of cytochrome c and the subsequent events characteristic of the intrinsic pathway [30–32]. In this study, we showed that JWH-015 induced a loss of Δψm, suggesting damage to the mitochondria. Furthermore, the involvement of the extrinsic pathway of apoptosis was suggested by the ability of caspase-8 inhibitor to block JWH-015-induced apoptosis as well as JWH-015-induced loss of mitochondrial membrane potential. However, we used a wide range of caspase-8 inhibitor concentrations (from 12.5 to 100 μM), and it is possible that at the highest concentrations the inhibitor may act non-specifically and inhibit other caspases such as caspase-9 or caspase-3.
Interestingly, there appeared to be a possible cross-talk between the two pathways of apoptosis induced by JWH-015 that was evident from caspase inhibition studies. We observed that caspase-8 inhibitor could almost completely block JWH-015-induced apoptosis, thereby suggesting that the extrinsic pathway of apoptosis played a crucial role in JWH-015-induced apoptosis. Moreover, because caspase-9 inhibitor could also block the apoptosis completely, this suggested that the extrinsic pathway, by itself, was not able to trigger apoptosis and that it depended on the intrinsic pathway to complete the apoptotic process. The importance of the cross-talk between the two pathways was further substantiated by the direct observation that addition of caspase-8 inhibitor was able to block JWH-015-induced loss of Δψm in thymocytes, demonstrating that caspase-8 activation occurred upstream of the mitochondria and was essential, at least in part, for triggering mitochondrial injury by JWH-015. While we are not certain as to which of the precise death receptors/ligands are involved in JWH-015-induced apoptosis, we demonstrated that Fas was not involved inasmuch as, thymocytes from C57BL/6lpr/lpr mice were sensitive to JWH-015-induced apoptosis.
The use of cannabinoids as therapeutic agents has been proposed in the treatment of many disorders ranging from pain, nausea, and cachexia to multiple sclerosis and epilepsy [1,3–6]. An oral synthetic form of THC has actually been approved in the treatment of cancer and AIDS patients [33]. In addition, studies from our laboratory have suggested the potential use of cannabinoids in the treatment of immunological disorders [16]. However, the use of THC is still controversial due to the fact that THC signals through both CB1 and CB2 receptors, and activation through CB1 is accompanied by psychotropic side effects. Some investigators have tried to bypass this problem by using enantiomers of psychoactive cannabinoids. For example, dexanabinol (HU-211), the enantiomer of the very potent cannabinoid HU-210 has been shown to be devoid of THC-like effects and proposed in the treatment of Experimental Allergic Encephalomyelitis (EAE) [34,35]. However, studies from our laboratory have shown that the immunosuppressive effect of THC was mediated at least in part by signaling through CB2 [16]. Because these non-psychoactive enantiomers have a different configuration, they may not be able to bind the cannabinoid receptors as well as the psychoactive compounds. For example, HU-211 (dexanabinol), a synthetic analog of tetrahydrocannabinol, does not bind CB1 or CB2 [36]. Consequently, such enantiomers may not be able to induce apoptosis in lymphocytes and prevent their proliferation. Therefore, we hypothesized that it would be beneficial to find a CB2-specific agonist with immunosuppressive properties as it would facilitate limiting psychotropic side-effects. In this study, we show that JWH-015 is capable of inducing apoptosis in lymphocytes, and preventing their proliferative response to mitogens in vitro, but we also show that JWH-015 can be immunosuppressive in vivo, implying the potential use of JWH-015 or other CB2 selective agonists as an alternative to THC, with limited psychoactive side-effects, in the treatment of inflammatory disorders.
It is interesting to note that JWH-015 was recently found to improve the neurologic defect induced by Theiler’s virus infection in which decreased numbers of CD4+ infiltrating T cells were noted in the spinal cord [37]. Although the precise mechanism of action of JWH-015 was unknown, the authors suggested that JWH-015 and other cannabinoids may decrease the number of CD4+ T cells thereby enhancing the myelin repair. Bacterial superantigens are considered to be the most powerful T cell activators [38]. Thus, exposure to such superantigens results in a dramatic induction of cytokines and consequent fever, shock and death. Our study suggests that targeting CB2 may constitute a novel approach to deplete superantigen-reactive T cells, thereby reducing the pathogenesis. Currently, to treat inflammatory diseases, non-steroidal anti-inflammatory drugs (NSAIDs) have been widely used. However, they cause significant gastro-intestinal toxicity because of which Cox2 inhibitors were developed [39]. Recent studies suggested that Cox2 inhibitors increase the risk of developing cardiovascular disease [39]. Thus, there is growing need for development of novel approaches to treat inflammatory diseases. The current study suggests that targeting CB2 receptors may constitute a unique treatment modality against inflammatory diseases.
Footnotes
This work was supported in part by National Institutes of Health grants: R01-AI058300, R01-HL058641, R01-AI53703, R01-ES09098, R21-DA014885, and R01-DA016545.
Publisher's Disclaimer: This is a PDF le of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its nal citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berdyshev EV. Cannabinoid receptors and the regulation of immune response. Chem Phys Lipids. 2000;108:169–190. doi: 10.1016/s0009-3084(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 2.Baker D, Pryce G, Giovannoni G, Thompson AJ. The therapeutic potential of cannabis. Lancet Neurol. 2003;2:291–298. doi: 10.1016/s1474-4422(03)00381-8. [DOI] [PubMed] [Google Scholar]
- 3.Beal JE, Olson R, Laubenstein L, Morales JO, Bellman P, Yangco B, Lefkowitz L, Plasse TF, Shepard KV. Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS. J Pain Symptom Manage. 1995;10:89–97. doi: 10.1016/0885-3924(94)00117-4. [DOI] [PubMed] [Google Scholar]
- 4.Grinspoon L, Bakalar JB, Doblin R. Marijuana, the AIDS wasting syndrome, and the U.S. government. N Engl J Med. 1995;333:670–671. doi: 10.1056/nejm199509073331020. [DOI] [PubMed] [Google Scholar]
- 5.Voth EA, Schwartz RH. Medicinal applications of delta-9-tetrahydrocannabinol and marijuana. Ann Intern Med. 1997;126:791–798. doi: 10.7326/0003-4819-126-10-199705150-00008. [DOI] [PubMed] [Google Scholar]
- 6.Watson SJ, Benson JA, Jr, Joy JE. Marijuana and medicine: assessing the science base: a summary of the 1999 Institute of Medicine report. Arch Gen Psychiatry. 2000;57:547–552. doi: 10.1001/archpsyc.57.6.547. [DOI] [PubMed] [Google Scholar]
- 7.McKallip RJ, Lombard C, Fisher M, Martin BR, Ryu S, Grant S, Nagarkatti PS, Nagarkatti M. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood. 2002;100:627–634. doi: 10.1182/blood-2002-01-0098. [DOI] [PubMed] [Google Scholar]
- 8.Ruiz L, Miguel A, Diaz-Laviada I. Delta9-tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. 1999;458:400–404. doi: 10.1016/s0014-5793(99)01073-x. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez C, Galve-Roperh I, Canova C, Brachet P, Guzman M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998;436:6–10. doi: 10.1016/s0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- 10.Morahan PS, Klykken PC, Smith SH, Harris LS, Munson AE. Effects of cannabinoids on host resistance to Listeria monocytogenes and herpes simplex virus. Infect Immun. 1979;23:670–674. doi: 10.1128/iai.23.3.670-674.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Specter S, Lancz G, Westrich G, Friedman H. Delta-9-tetrahydrocannabinol augments murine retroviral induced immunosuppression and infection. Int J Immunopharmacol. 1991;13:411–417. doi: 10.1016/0192-0561(91)90011-u. [DOI] [PubMed] [Google Scholar]
- 12.Cabral GA, Mishkin EM, Marciano-Cabral F, Coleman P, Harris L, Munson AE. Effect of delta 9-tetrahydrocannabinol on herpes simplex virus type 2 vaginal infection in the guinea pig. Proc Soc Exp Biol Med. 1986;182:181–186. doi: 10.3181/00379727-182-42325. [DOI] [PubMed] [Google Scholar]
- 13.Do Y, McKallip RJ, Nagarkatti M, Nagarkatti PS. Activation through Cannabinoid Receptors 1 and 2 on Dendritic Cells Triggers NF-{kappa}B-Dependent Apoptosis: Novel Role for Endogenous and Exogenous Cannabinoids in Immunoregulation. J Immunol. 2004;173:2373–2382. doi: 10.4049/jimmunol.173.4.2373. [DOI] [PubMed] [Google Scholar]
- 14.Samson MT, Small-Howard A, Shimoda LM, Koblan-Huberson M, Stokes AJ, Turner H. Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. J Immunol. 2003;170:4953–4962. doi: 10.4049/jimmunol.170.10.4953. [DOI] [PubMed] [Google Scholar]
- 15.Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, Friedman H. The cannabinoid system and immune modulation. J Leukoc Biol. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- 16.McKallip RJ, Lombard C, Martin BR, Nagarkatti M, Nagarkatti PS. Delta(9)-tetrahydrocannabinol-induced apoptosis in the thymus and spleen as a mechanism of immunosuppression in vitro and in vivo. J Pharmacol Exp Ther. 2002;302:451–465. doi: 10.1124/jpet.102.033506. [DOI] [PubMed] [Google Scholar]
- 17.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 18.Kamath AB, Xu H, Nagarkatti PS, Nagarkatti M. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol Appl Pharmacol. 1997;142:367–377. doi: 10.1006/taap.1996.8049. [DOI] [PubMed] [Google Scholar]
- 19.Gastman BR, Yin XM, Johnson DE, Wieckowski E, Wang GQ, Watkins SC, Rabinowich H. Tumor-induced apoptosis of T cells: amplification by a mitochondrial cascade. Cancer Res. 2000;60:6811–6817. [PubMed] [Google Scholar]
- 20.Camacho IA, Nagarkatti M, Nagarkatti PS. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces Fas-dependent activation-induced cell death in superantigen-primed T cells. Arch Toxicol. 2002;76:570–580. doi: 10.1007/s00204-002-0390-2. [DOI] [PubMed] [Google Scholar]
- 21.Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002;21:2283–2294. doi: 10.1038/sj.onc.1205258. [DOI] [PubMed] [Google Scholar]
- 22.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 23.Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998;10:545–551. doi: 10.1016/s0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- 24.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) Embo J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 26.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 27.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 28.Cain K, Brown DG, Langlais C, Cohen GM. Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J Biol Chem. 1999;274:22686–22692. doi: 10.1074/jbc.274.32.22686. [DOI] [PubMed] [Google Scholar]
- 29.Cain K, Bratton SB, Langlais C, Walker G, Brown DG, Sun XM, Cohen GM. Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J Biol Chem. 2000;275:6067–6070. doi: 10.1074/jbc.275.9.6067. [DOI] [PubMed] [Google Scholar]
- 30.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. Embo J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 32.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998:94, 481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 33.Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 34.Mechoulam R, Feigenbaum JJ, Lander N, Segal M, Jarbe TU, Hiltunen AJ, Consroe P. Enantiomeric cannabinoids: stereospecificity of psychotropic activity. Experientia. 1988;44:762–764. doi: 10.1007/BF01959156. [DOI] [PubMed] [Google Scholar]
- 35.Little PJ, Compton DR, Mechoulam R, Martin BR. Stereochemical effects of 11-OH-delta 8-THC-dimethylheptyl in mice and dogs. Pharmacol Biochem Behav. 1989;32:661–666. doi: 10.1016/0091-3057(89)90014-2. [DOI] [PubMed] [Google Scholar]
- 36.Juttler E, Potrovita I, Tarabin V, Prinz S, Dong-Si T, Fink G, Schwaninger M. The cannabinoid dexanabinol is an inhibitor of the nuclear factor-kappa B (NF-kappa B) Neuropharmacology. 2004;47:580–592. doi: 10.1016/j.neuropharm.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Proft T, Fraser JD. Bacterial superantigens. Clin Exp Immunol. 2003;133:299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwong MF. Have Cox2 inhibitors lived up to expectations? Best Pract Res Clin Gastroenterol. 2004;18(Suppl):13–16. doi: 10.1016/j.bpg.2004.06.006. [DOI] [PubMed] [Google Scholar]