

Abstract
Thio-ether bonds in the cysteinyl side-chain of peptides, formed with the most commonly used cysteine blocking reagent iodoacetamide, after conversion to sulfoxide, releases a neutral fragment mass in a low energy MS/MS experiment in the gas-phase of the mass spectrometer [Steen et al. J Am Soc Mass Spectrom. 2001, 12, 228–32.]. In this study, we show that the neutral loss fragments produced from the mono-oxidized thio-ether bonds (sulfoxide) in peptides, formed by alkyl halide or double-bond containing cysteine blocking reagents are different under low energy MS/MS conditions. We have evaluated the low energy fragmentation patterns of mono-oxidized modified peptides with different cysteine blocking reagents, such as iodoacetamide, 3-maleimidopropionic acid, and 4-vinylpyridine using FTICR-MS. We propose that the mechanisms of gas-phase fragmentation of mono-oxidized thio-ether bonds in the side-chain of peptides, formed by iodoacetamide and double-bond containing cysteine blocking reagents, maleimide and vinylpyridine, are different due to the availability of acidic β-hydrogens in these compounds. Moreover, we also investigated the fragmentation characteristics of mono-oxidized thio-ether bonds within the peptide sequence in order to develop novel mass-spectrometry identifiable chemical cross-linkers. This methionine type of oxidized thio-ether bond within the peptide sequence did not show anticipated low energy fragmentation. Electron capture dissociation (ECD) of the side-chain thio-ether-bond containing oxidized peptides was also studied. ECD spectra of the oxidized peptides showed a greater extent of peptide backbone cleavage, as compared to CID spectra. This fragmentation information is critical to researchers for accurate data analysis of this undesired modification in proteomics research, as well as other methods that may utilize sulfoxide derivatives.
Introduction
In conventional or bottom-up proteomics research, proteins are commonly digested with trypsin and analyzed by mass spectrometry [1]. Prior to digestion, the disulfide bonds in the proteins are first cleaved with either DTT or β-mercaptoethanol and their reformation must be blocked using different cysteine blocking reagents. Several cysteine-blocking reagents exist; the most common are iodoacetamide, N-ethylmaleimide, acrylamide, and 4-vinylpyridine [2, 3, 4]. These methods that use iodoacetamide rely on an alkyl halide to react with the sulfhydryl group of cysteine. The other three blocking reagents, maleimide, acrylamide, and vinylpyridine utilize double bonds to react with the sulfhydryl group of cysteine by the Michael addition type mechanism [5]. All of these blocking reagents form thio-ether bonds in the cysteinyl side-chain of proteins. Steen et al. reported that these thio-ether bonds can be oxidized in current protein profiling techniques, such as tryptic digestion and gel electrophoresis [6]. The oxidized thio-ether bond formed with iodoacetamide in the cysteinyl side-chain of the peptide, loses a neutral fragment mass in the gas phase under low energy MS/MS conditions. Steen et al. exploited this low energy fragmentation characteristic of mono-oxidized thio-ethers to profile phosphorylation by β-elimination and Michael addition chemical strategy [7]. It was reported that this characteristic neutral loss was generated by the formation of dehydroalanine in a cysteinyl residue after releasing a sulfenic acid fragment (RSOH, where R stands for added tag mass from the blocking reagent) from the modified peptide in a low energy MS/MS experiment.
This study was carried out to investigate new features for novel low energy mass spectrometry cleavable cross linkers where bonds can be selectively dissociate with low energy MS/MS in the gas-phase environment of a mass spectrometer [8, 9]. Low energy fragmentation characteristics of oxidized thio-ether bonds have been reported [6,7]. While incorporating this bond to develop novel mass spectrometry cleavable PIR-cross linkers, we have observed different low energy fragmentation behavior for mono-oxidized thio-ether bonds formed from different sulfhydryl reactive functional groups.
In this manuscript, we show that the characteristic low energy fragment loss for mono-oxidized thio-ether bonds in peptides is different if they are formed with double bonds-containing cysteine blocking reagents. These results show that the gas-phase fragmentation pathways for mono-oxidized thio-ether bond in the side-chain of the peptide are different for different sulfhydryl reactive blocking reagents. We propose that due to the availability of additional acidic β-hydrogens in the double-bond-containing cysteine blocking reagents, the mono-oxidized thio-ether residues fragment differently in the gas phase environment of a mass spectrometer. We have also incorporated a methionine-like thio-ether bond within a peptide sequence to validate the feasibility of constructing a novel mass spectrometry cleavable cross-linker. The low energy MS/MS of peptide containing thio-ether bond within the sequence was compared with its oxidized counterpart. The internal oxidized thio-ether containing peptide did not show any low energy fragmentation. The ECD spectra of this modification also show different fragmentation patterns as compared with low energy CID spectra.
Experimental
Iodoacetamide, 3-maleimidopropionic acid and 4-vinylpyridine were purchased from Sigma-Aldrich (St Loius, Mo, USA). The 30% H2O2 solution was also purchased from Sigma-Aldrich. Cysteinyl peptides, SEVAHRFKC and CHWKQNDEQM were synthesized in WSU peptide synthesis facility with an Applied Biosystems 431 A peptide synthesizer. The sulfhydryl group of cysteine in the peptides was blocked with different sulfhydryl reactive reagents in presence of the NH4HCO3 or PBS buffer. The selective mono oxidation of sulfur in the thio-ether was accomplished by reacting the peptide in a 3 % H2O2 solution for 15 min. Desalting was accomplished with ZipTip (Millipore, Billerica, MA) and SepPack C18 (Water, Milford, MA, USA) purification.
All FTICR-MS spectra were obtained with a Bruker Daltonics 7T APEX Q-FTICR mass spectrometer by direct infusion of samples with a nano ESI source made with fused silica capillary (360 um o.d and 20 um i.d). The capillary tip was etched with 49% HF. The FTICR mass spectra were processed with the software, ICR-2LS, developed by Pacific Northwest National Laboratory (PNNL) [10]. CID Mass spectra were calibrated externally utilizing standard peptides, Substance P (RPKPQQFFGLM), Bradykinin (RPPGFSRFR), and the peptide SEVAHRFKC. The electrospray solution was MeOH/H2O/AcOH (49:49:2) unless otherwise mentioned. Electron capture dissociation was performed using a heated hollow cathode dispenser located outside the ICR cell to obtain the MS/MS data. The cathode dispenser was heated gradually to 1.8A–1.9A. Side kick trapping voltage [11] was maintained between +6V to −6V. Electrons used for ECD were accelerated with 3V. The electron injection time was 200 ms. ECD spectra were calibrated externally with a standard peptide, Substance P.
Synthesis of the internal thio-ether bond-containing peptide
The peptide containing an internal thio-ether bond was synthesized using an ABI 431A peptide synthesizer (Applied Biosystems, Foster City, CA, USA) utilizing typical Fmoc peptide synthesis cycles except the thio-ether bond was introduced by the reaction with 2-amino-ethanethiol (Aldrich, St. Loius, Mo) with bromoacetic acid (Aldrich, St. Loius, Mo, USA). Bromoacetic acid was coupled to the alanine residue of the peptide, ALVG, using standard Fmoc cycles. Next, solid cysteamine HCL (2-amino-ethanethiol) was added directly to the reaction vessel that contains NMP solvent, 0.1ml of 1.0 M Dimethylaminopyridine/N,N Dimethylformamide (Applied Biosystems, Foster city, CA) and 0.1 ml water. This condition was used to increase the reaction rate by deprotonating the thiol group. The primary amine group of the cysteamine attached peptide was further coupled with phenylalanine residue of a small peptide, LVF, using same Fmoc chemistry. The final product was dried under vacuum. ESI-FTICR-MS analysis reveals the purity of the peptide product was about 50%. No further purification was done on the peptide product. Selective oxidation of sulfur in the thio-ether bond was achieved by reacting the peptide product with 3% H2O2 solution for 15 min.
Results and discussions
Our study was carried out with two peptides which have cysteine residues situated at the N-terminal and C-terminal sites of peptides. We first carried out experiment with a cysteine-containing peptide (SEVAHRFKC). The peptide was reacted with iodoacetamide to form a carbamidomethyl modification on the sulfhydryl group of cysteine, a common procedure in proteomics research to block free thiols. After selective oxidation of the thio-ether bond, desalting and vacuum drying, the sample was reconstituted in MeOH:H2O:AcOH (49:49:2) for ESI-FTICR-MS analysis. To observe the fragmentation characteristics of oxidized and modified peptides in FTICR-MS, the solutions were sprayed with a nanoelectrospray source with 20 μL/h flow rate. Ions at m/z 575.2794 correspond to the expected [M+2H]2+ ion of the oxidized iodoacetamide-labeled peptide and were isolated in the quadrupole (Figure 1, A) (Table 1). The ions were then fragmented in the hexapole by increasing the collisional trap voltage by three increments (Figure 1, B, C, D). At −6V the characteristic low energy signature ion at m/z 521.7770, appeared with high intensity in the spectra after the predicted neutral loss of RSOH fragment (R = 58.0292 Da) (Figure 1, B). The expected accurate mass of this neutral fragment, RSOH (C2H5NO2S) was 107.0042 Da. We observed the neutral loss of 107.0048 Da from the low energy MS/MS experiment (m/z 575.2794 − m/z 521.7770 = 53.5024x2 = 107.0048 Da) in FTICR-MS with a mass error of 0.6 mDa (Table 1). At −9V this ion, m/z 521. 7770, appeared in the spectrum with high intensity. No significant peptide backbone fragments were generated at this collisional trap voltage other than the fragment generated after loss of RSOH from the side-chain of the modified peptide. Increasing the voltage settings (−12V) increased the intensity of m/z 521.7770 (Figure 1, D). This confirms the low energy fragmentation characteristic of a side-chain mono-oxidized iodoacetamide labeled peptide as previously reported by Steen et al., using a q-TOF instrument [6].
Figure 1.
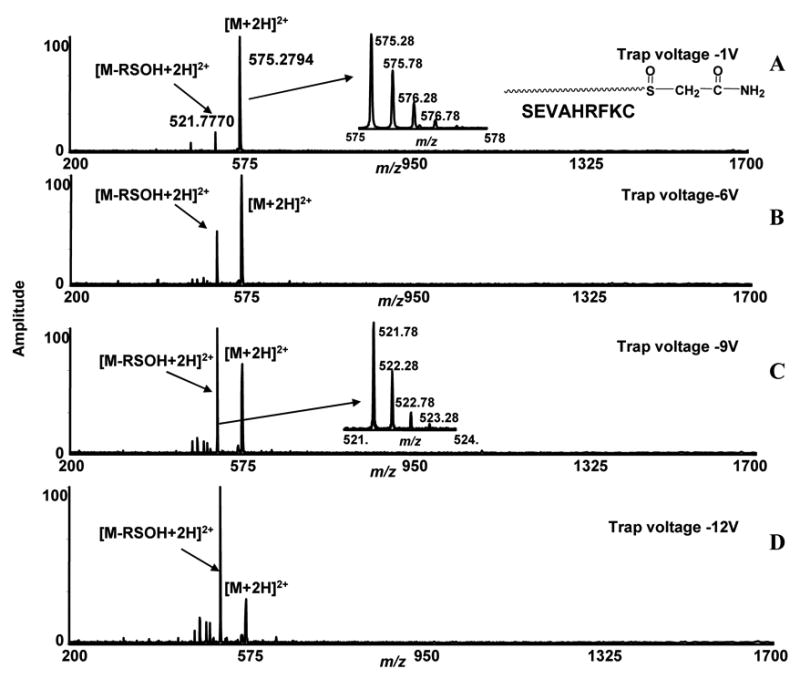
Low energy ESI-FTICR-MS/MS spectra of a cysteine-containing peptide (SEVAHRFKC) labeled with iodoacetamide. The sulfur in the side-chain thio-ether residue is converted to sulfoxide by oxidation. The ions at m/z 575.2794, correspond to [M+2H]2+ ions of oxidized modified peptides and were isolated in the quadrupole and subjected to different collisional trap voltage settings. Mass spectra shown were acquired with −1V, −6V, −9V, and −12V (A, B, C, D).
Table 1.
List of m/z values and calculated neutral and observed masses.
| Neutral Masses in Da | Expected m/z | Observed m/z | Observed Mass | Delta Mass in Da | Mass Errors in mDa | |
|---|---|---|---|---|---|---|
| Peptide-1 SEVAHRFKC | 1075.5232 | 538.7689 (+2) | 538.7688 (+2) | 1E-04 | 0.1 | |
| Peptide-2 CHWKQNDEQM | 1317.5229 | 659.7688 (+2) | 659.7681 (+2) | 7E-04 | 0.7 | |
| Peptide-2 (Ox) CHWKQNDEQM | 1333.5178 | ------------------ | ------------------ | ------------ | ------------ | ------------ |
| Iodoacetamide (IA) [added mass] | 58.0292 | ------------------ | ------------------ | ------------ | ------------ | |
| 3-maleimidopropionic acid (MP) | 169.0375 | ------------------ | ------------------ | ------------ | ------------ | ------------ |
| 4-vinylpyridine (VP) | 105.0578 | ------------------ | ------------------ | ------------ | ------------ | ------------ |
| Peptide-1+IA+[O] | 1148.5395 | 1149.5468 (+1)
575.2771 (+2) |
575.2794(+2) | ------------ | 2.3E-03 | 2.3 |
| Peptide-1+MP+[O] | 1260.5556 | 1261.5629 (+1)
631.2851 (+2) |
631.2848 (+2) | ------------ | 3E-04 | 0.3 |
| Peptide-1+VP+[O] | 1196.5759 | 1197.5832 (+1)
599.2953 (+1) |
599.2939 (+2) | 1.4E-03 | 1.4 | |
| Peptide-2 (Ox)+MP+[O] | 1518.5502 | 1519.5575 (+1)
760.2824 (+2) |
760.2837 (+2) | 1.3E-03 | 1.3 | |
| RSOH (IA) | 107.0041 | ------------------ | ------------------ | 107.0048 | 7E-04 | 0.7 |
| R+ H2O (MP) | 187.0480 | ------------------ | ------------------ | 187.0470
187.0484 |
1E-03
4E-04 |
1.0
0.4 |
| R+ H2O (VP) | 123.0683 | ------------------ | ------------------ | 123.0698 | 1.5E-03 | 1.5 |
To determine if this characteristic sulfenic acid fragment loss (RSOH) from the side-chain mono-oxidized thio-ether residues in peptides is common among other cysteine blocking reagents, we studied fragmentation characteristics with two other commonly used reagents. We first studied the fragmentation characteristics of the same cysteinyl peptide (SEVAHRFKC) modified with a maleimide group. Maleimide contains an unsaturated bond which reacts with the sulfhydryl group of cysteine by a Michael addition type reaction mechanism. We reacted the peptide SEVAHRFKC with 3-maleimidopropionic acid under the same conditions we utilized for iodoacetamide reaction. The sulfur in the newly formed thio-ether bond was then oxidized as previously described. The ions observed at m/z = 631.2848, correspond to [M+2H]2+ ions of the oxidized maleimide-modified peptide and were isolated in the quadrupole and subjected to increased collisional trap voltage (Figure 2, A, B, C) (Table 1). Two fragment ions appeared in the spectra at m/z 537.7613 and m/z 521.7758. The intensity of the ion at m/z 537.7613 increased with increasing collisional trap voltage (Figure 2, B, C). The expected low energy fragment ion (i. e. loss of RSOH) at m/z 521.7758 appeared in the spectra at a very low intensity using low collisional trap voltages; and did not increase with increased voltages. The ions at m/z 537.7613 appeared as the dominant peak at −15 V due to the fragmentation of the molecular peak but no significant peptide backbone cleavage was observed. These data show that the low energy fragmentation characteristics of iodoacetamide and maleimide-labeled mono-oxidized thio-ether residues in the same peptide sequence are different. In this case, low energy fragment ions correspond to a neutral fragment loss of R + H2O from the oxidized modified peptide (R = MW of 3-maleimidopropionic acid) were observed. The accurate mass for the loss of ROH2 is 187.0480 Da. We observed the neutral loss of 187.0470 Da in low energy ESI-FTICR-MS/MS analysis with an error of 1 mDa (Table 1).
Figure 2.
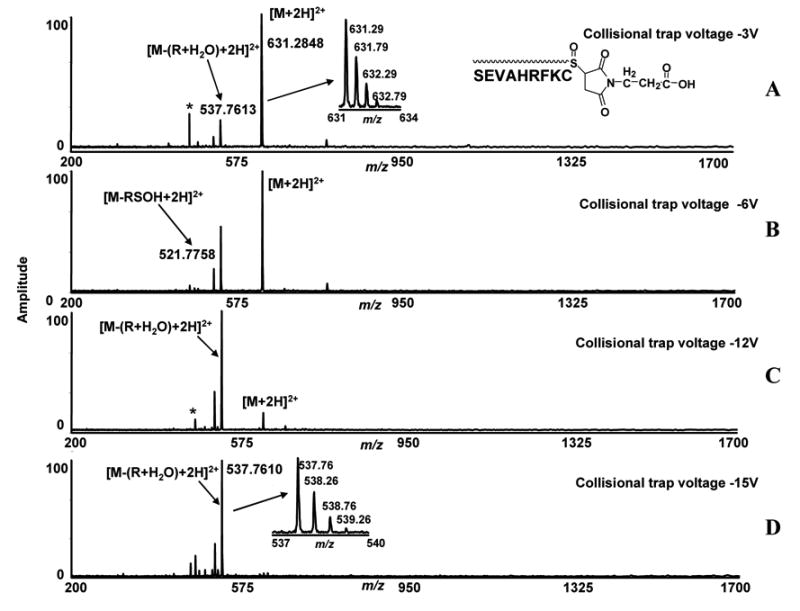
Low energy ESI-FTICR-MS/MS spectra of cysteine-containing peptide (SEVAHRFKC) labeled with 3-maleimidopropionic acid. The sulfur in the side-chain thio-ether residue is converted to sulfoxide by oxidation. The ions at m/z 631.2848 correspond to [M+2H]2+ ions of oxidized modified peptides and were isolated in the quadrupole and subjected to different collisional trap voltage settings. Mass spectra shown were acquired with −3V, −6V, −12V and −15V (A, B, C, D).
To see if this neutral loss is specific for maleimide modified peptides, we reacted the same peptide with another cysteine labeling reagent, 4-vinylpyridine. The 4-vinylpyridine has the same double bond functional group, but has asymmetry around the double bond. The observed ions at m/z 599.2939 which correspond to [M+2H]2+ ions of oxidized vinylpyridine-modified peptides, were isolated in the quadrupole and the trap voltage was increased as previously described (Figure 3) (Table 1). Two fragment ions appeared in the spectra, showing the same fragmentation as we observed for the maleimide labeled peptide. The ions at m/z 537.7590, which correspond to neutral loss of R + H2O, were observed in the spectra and the intensity of these ions increased with increasing collisional trap voltages (Figure 3, B, C, D). The characteristic ions, generated after neutral loss of R + H2O, were also observed at +1 charge state in the spectra (m/z 1074.5105 Da). The ions at m/z 521.7590, which correspond to the loss of the expected neutral fragment mass from the peptide (loss of RSOH) were also observed in the spectra but did not show intensity changes after setting different voltages in the collisional trap. These data showed that both maleimide and vinylpyridine have similar low energy fragmentation characteristics for side-chain mono-oxidized thio-ether residues in peptides, which are the neutral loss of the R + H2O fragment, not the loss of RSOH as observed with iodoacetamide-modified peptides. The accurate neutral mass for R + H2O of vinylpyridine labeled peptide is 123.0683 Da. We observed the neutral loss of 123.0698 Da with an error of 1.5 mDa (Table 1).
Figure 3.
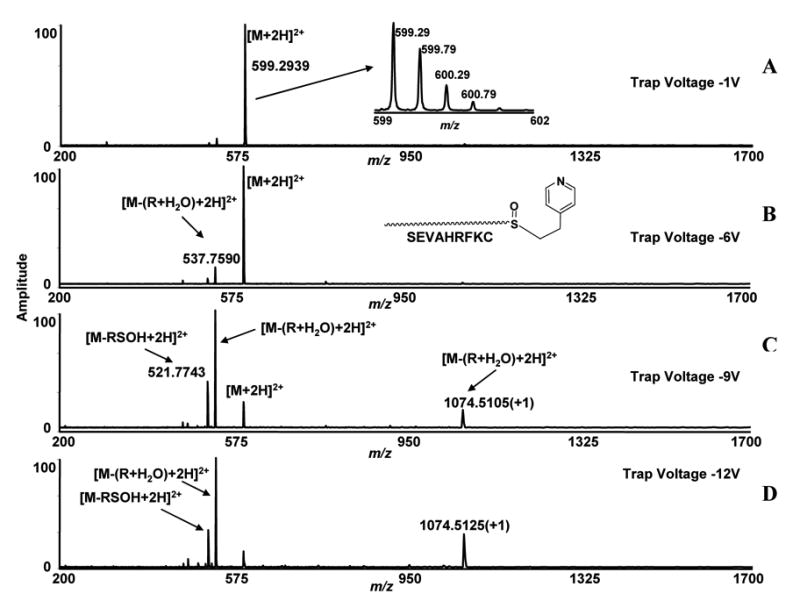
Low energy ESI-FTICR-MS/MS spectra of cysteine-containing peptides (SEVAHRFKC) labeled with 4-vinylpyridine. The sulfur in the side-chain thio-ether residue is converted to sulfoxide by oxidation. The ions at m/z 599.2939 correspond to [M+2H]2+ ions of oxidized modified peptides and were isolated in the quadrupole and subjected to various collisional trap voltage settings. Mass spectra shown were acquired with −1V, −6V, −9V and −12V (A, B, C, D).
To confirm these characteristic losses with other peptide sequences, we reacted the peptide CHWKQNDEQM with 3-maleimidopropionic acid. The newly formed thio-ether bond was selectively oxidized to sulfoxide. The cysteine side-chains blocked with iodoacetamide and 3-maleimidopropionic acid were confirmed from the MS/MS and ECD spectra of un-oxidized iodoacetamide and maleimide labeled peptides. ECD study of unoxidized iodoacetamide derivatized peptide was reported by several authors [12] [13]. The ECD and CID of corresponding unoxidized iodoacetamide-and maleimide-labeled peptides confirmed the labeling on the cysteinyl side-chain of the peptides. The methionine residue in the peptide CHWKQNDEQM was found to be mono-oxidized after these modification reactions (Supplementary data 1,2). The ions at m/z 760.2837, which corresponds to [M+2H]2+ ions of modified and oxidized peptides, were isolated in the quadrupole and fragmented in the hexapole [Figure 4, A–D] (Table 1). The fragmentation spectra showed the same neutral loss fragment behavior that we observed with other peptides labeled with maleimide and vinylpyridine. The ions at m/z 666.7595 (+2) correspond to the neutral loss of R + H2O from the methionine oxidized vinylpyridine modified peptides. A competing water loss fragment was also observed from the oxidized peptide. Thus, neutral loss fragments observed under low energy CID conditions with peptides modified by double-bond containing cysteine reactive reagents, maleimide and vinylpyridine were primarily R + H2O.
Figure 4.
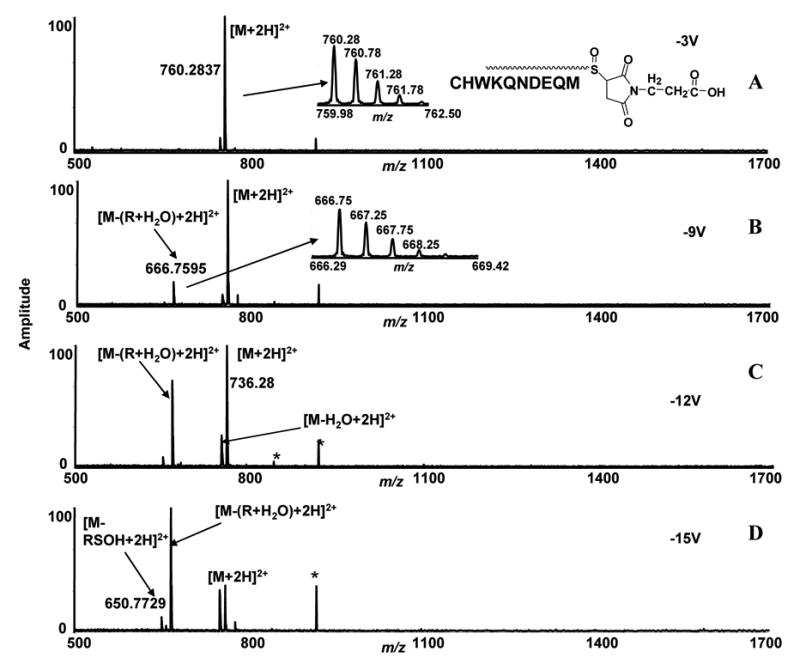
Low energy ESI-FTICR-MS/MS spectra of cysteine containing peptides (CHWKQNDEQM) labeled with 3-maleimidopropionic acid. The sulfur in the side-chain thio-ether residue is converted to sulfoxide by oxidation. The ions at m/z 760.2837 correspond to [M+2H]2+ ions of oxidized modified peptides and were isolated in the quadrupole and subjected to different collisional trap voltage settings. Mass spectra shown were acquired with −3V, −9V, −12V, and −15V (A, B, C, D). *denotes noise peaks.
The side-chains of cysteine residues of peptides SEVAHRFKC, were labeled with different cysteine blocking reagents and they showed distinct low energy fragmentation in the gas-phase in a mass spectrometer. To determine, if a sulfoxide group within a peptide sequence would show similar low energy fragmentation characteristics, we introduced a thio-ether bond similar to the side-chain of methionine within a peptide sequence. The low energy fragmentation characteristic of mono-oxidized methionine side-chain residue (methionine sulfoxide) was also reported [14] [15] [16]. Our studies of mono-oxidized thio-ether bonds of methionine side-chains also showed low energy fragmentation characteristics, but did not show the same labile characteristics as we observed for the side-chains of mono-oxidized thio-ether bonds of cysteine in peptides (Supplementary data 3). To compare the fragmentation characteristics of the internal thio-ether bond-containing peptide, we first selectively oxidized sulfur to sulfoxide. The MS/MS of both peptides, oxidized and non-oxidized, were obtained using −15 V collisional trap voltage (Figure 5, A, B)). Both oxidized and non-oxidized peptides showed similar fragmentation characteristics. Several ‘b’ ions were observed without any selective cleavage in the thio-ether bond. These data suggest that within peptide sequence, the methionine-like thio-ether residues fragment differently than the oxidized thio-ether residue in the amino acid side-chain.
Figure 5.
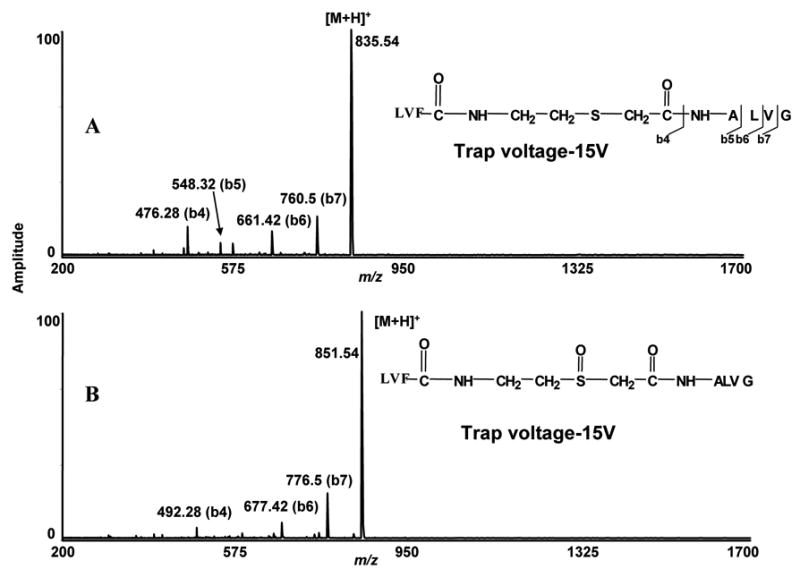
ESI-FTICR-MS/MS spectra of peptide containing either oxidized or non-oxidized thio-ether bonds within the sequence. A) ESI-FTICR-MS/MS spectra of a peptide containing non-oxidized thio-ether bond within the backbone of the sequence. At −15V trap voltage few b ions were generated. No specific fragmentation occurred at non oxidized thio-ether bond. B) ESI-FTICR-MS/MS spectra of peptide containing mono-oxidized thio-ether bond inside the sequence. At −15V trap voltage, a similar fragmentation pattern was observed with the oxidized peptide.
To compare the ECD of these modified peptides with that of CID, we first looked at the ECD of oxidized cysteinyl peptide, SEVAHRFKC, blocked with iodoacetamide. We isolated [M+2H]2+ ions in the quadrupole and low energy electrons were irradiated with conditions mentioned previously in the experimental section. We observed three distinct ‘c’ ions, c6, c7, and c8 in the spectrum (Figure 6). Since no such fragments were observed with the addition of the modification mass, these data strongly suggest the modification exists on the C-terminal cysteine, as one might expect for sulfhydryl-reactive compounds. Ions observed at m/z = 1149.57 correspond to [M+H]+ ion of this modified peptide and were detected after electron irradiation. Ions at m/z = 1091.57 were observed in the spectrum and correspond to the loss of iodoacetamide tag from the modified and oxidized peptide. Both ions which correspond to [M- (R + H2O)] and [M-RSOH] were observed in this ECD spectrum.
Figure 6.
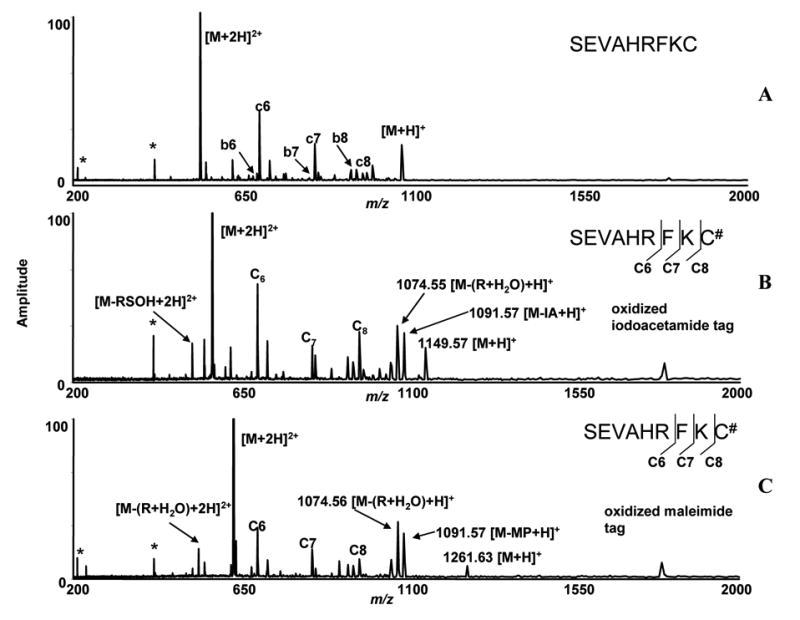
ECD-FTICR mass spectra of the peptide (SEVAHRFKC) reacted with iodoacetamide and 3-maleimidopropionic acid. The sulfur atoms in thio-ether bonds were converted to sulfoxide (See Figure 1 and 2 for CID mass spectra). A) ECD spectrum of unmodified peptide. B) ECD spectrum of mono-oxidized iodoacetamide labeled peptide. C) ECD spectrum of mono-oxidized maleimide labeled peptide. # denotes modification and * denotes noise peaks.
The ECD of the maleimide-labeled oxidized cysteinyl peptide and iodoacetamide labeled oxidized peptide SEVAHRFKC, showed similar fragmentation behavior. The m/z 631.28 corresponds to the [M+2H]2+ of ions of oxidized modified peptides and the ions were isolated in the quadrupole and trapped in the ICR cell, and were irradiated with electrons for 150–200 ms. Three detected ‘c’ ions (c6, c7, c8), were also observed for iodoacetamide-labeled oxidized peptides. The ions at m/z 1074.56 and m/z 537.78, which correspond to the loss of ROH2 molecule from the oxidized modified peptide, were also observed. Ions at m/z = 1091.57, which corresponds to loss of maleimide tag from the oxidized peptides, were observed in the spectrum.
The ECD spectra of both oxidized iodoacetamide and maleimide-labeled mono-oxidized peptides showed similar fragmentation patterns in FTICR-MS. These data showed much more backbone cleavage than was observed in the CID spectra. The difference in the ECD spectra of iodoacetamide-labeled and maleimide-labeled oxidized peptide is in the observation of low energy signature ions. In iodoacetamide labeled oxidize peptides, we observed both [M-RSOH] and [M- (R + H2O)] ions where as in maleimide labeled peptides, we observed only [M- (R + H2O)] ions.
Gas-phase fragmentation of aliphatic and alicyclic sulfoxides and sulphones with electron impact ionization has been reported [17]. A mechanism of gas-phase β-elimination of oxidized thio-ether bonds in peptide side-chain was proposed by Steen et al [6]. The abstraction of an acidic β-hydrogen from the peptide backbone by the oxygen of thio-ether residues results in the migration of a hydrogen and the loss of the iodoacetamide tag as sulfenic acid. This mechanism also results in formation of a double bond at the position of the cysteinyl side-chain of the peptide. For peptides labeled with maleimide and vinylpyridine, two additional acidic β-hydrogens (alpha to carbonyl group, pKa~25) are available in these groups besides the acidic β-hydrogen in the peptide backbone. The abstraction of β-hydrogens from these compounds generates a sulfenic-acid containing peptide. These mechanisms are illustrated with the maleimide group in Figure 7. Extreme instability of sulfenic acid in the gas-phase was reported by Lacombe et al [18]. The mass spectra of methane and ethanesulfenic acid was investigated by Turecek et al [19]. The CID mass spectrum of methanesulfenic acid showed the loss of H2O from this molecule [19]. In our case, the sulfenic acid-containing peptides readily lost water molecules in the gas-phase to generate more stable sulfide, which results in the appearance of [M- (R + H2O)] ion in the spectra following the loss of the maleimide tag and a water molecule. In another pathway, the abstraction of a β-hydrogen from the peptide backbone causes the migration of sulfenic acid in the tag and the loss of RSOH fragment from the peptide. Both pathways were observed for maleimide and vinylpyridine but the abstraction of a β-hydrogen from the maleimide and vinylpyridine rather than the peptide backbone appears more favorable. For peptides containing a mono-oxidized thio-ether residue within the sequence, two β-hydrogens are present near the sulfoxide group. Abstraction of these hydrogens (pKa~ 45–50) and formation of an unsaturated bond is not favorable. Therefore, we did not observe characteristics low energy fragmentation behavior for this internal oxidized thio-ether containing peptide.
Figure 7.

Proposed fragmentation pathways for the loss of RSOH and ROH2 from the mono oxidized thio-ether residues from the two double bond containing cysteine reactive reagents; maleimide and vinylpyridine. Fragmentation pathways are shown with the 3-maleimidopropionic acid group.
Conclusions
In this study, we report low energy neutral fragmentation of side-chain mono-oxidized thio-ether bonds in peptides and differences observed with various cysteine-blocking reagents. The mono-oxidized thio-ether bonds formed with commonly used iodoacetamide or double-bond-containing cysteine reactive reagents, showed different neutral fragment loss in the gas-phase environment of the mass spectrometer. We also report that these mono-oxidized thio-ether bonds showed low energy fragmentation characteristics that appear to depend upon the availability of acidic β-hydrogens. On the other hand, the methionine-like mono-oxidized thio-ether bond within the peptide sequence did not show any low energy fragmentation behavior. The ECD spectra of this peptide showed more backbone cleavage and sequence info than observed in the corresponding CID spectra. We believe this information is very important for proteomics research because alkyl halides and Michael acceptors containing acidic hydrogens are commonly used functional groups to block cysteines in proteins. Therefore, fragmentation characteristics of this undesired modification must be understood and implemented in the search algorithms for accurate data analysis in proteomics research. Some time ago, thio-ether modifications were also discovered in the internal sequence of some peptide antibiotics, called lantibiotics [20], as is more recently discussed [21]. We feel the fragmentation study of the oxidized thio-ether in the internal sequence may also assist in that lantibiotic research area. Finally, better understanding of the low energy fragmentation behavior of this sulfoxide group can aid development of new class of mass spectrometry cleavable chemical reagents.
Supplementary Material
Acknowledgments
This research was supported by the Office of Science (BER), U.S. Department of Energy, Grant No. DE-FG02-04ER63924 and The Murdock Charitable trust. The authors thank Gordon A. Anderson for the data analysis tools used for this research.
Abbreviations
- CID
Collision Induced Dissociation
- ECD
Electron Capture Dissociation
- FTICR-MS
Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
- IA
Iodoacetamide
- MS/MS
Tandem Mass Spectrometry
- MP
3-Maleimidopropionic Acid
- q-TOF
Quadrupole – Time of Flight
- VP
4-vinylpyridine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bogdanov B, Smith RD. Proteomics by FTICR Mass Spectrometry: Top Down and Bottom Up. Mass Spectrom Rev. 2005;24:168–200. doi: 10.1002/mas.20015. [DOI] [PubMed] [Google Scholar]
- 2.Sechi S, Chait BT. Modification of Cysteine Residues by Alkylation. A Tool in Peptide Mapping and Protein Identification. Anal Chem. 1998;70:5150–8. doi: 10.1021/ac9806005. [DOI] [PubMed] [Google Scholar]
- 3.Schilling B, Yoo CB, Collins CJ, Gibson BW. Determining cysteine oxidation status using differential alkylation. Int J Mass Spectrom. 2004;236:117–127. [Google Scholar]
- 4.Fullmer CS. Identification of Cysteine-Containing Peptides in Protein Digests by High-Performance Liquid Chromatography. Anal Biochem. 1984;142:336–9. doi: 10.1016/0003-2697(84)90473-1. [DOI] [PubMed] [Google Scholar]
- 5.Perlmutter P. Conjugate Addition Reactions in Organic Synthesis. Pergamon Press; Oxford, England: 1992. [Google Scholar]
- 6.Steen H, Mann M. Similarity between Condensed Phase and Gas Phase Chemistry: Fragmentation of Peptides Containing Oxidized Cysteine Residues and Its Implications for Proteomics. J Am Soc Mass Spectrom. 2001;12:228–32. doi: 10.1016/S1044-0305(00)00219-1. [DOI] [PubMed] [Google Scholar]
- 7.Steen H, Mann M. A New Derivatization Strategy for the Analysis of Phosphopeptides by Precursor Ion Scanning in Positive Ion Mode. J Am Soc Mass Spectrom. 2002;13:996–1003. doi: 10.1016/S1044-0305(02)00415-4. [DOI] [PubMed] [Google Scholar]
- 8.Anderson GA, Bruce JE, editors. ICR-2LS 1995. Pacific Northwest National Laboratory; Richland, WA: 1995. [Google Scholar]
- 9.Tang X, Munske GR, Siems WF, Bruce JE. Mass Spectrometry Identifiable Cross-Linking Strategy for Studying Protein-Protein Interactions. Anal Chem. 2005;77:311–8. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- 10.Chowdhury SM, Munske GR, Tang X, Bruce JE. CAD, ECD, of Several Mass Spectrometry-Identifiable Chemical Cross-Linkers. Anal Chem. 2006 doi: 10.1021/ac060789h. accepted. [DOI] [PubMed] [Google Scholar]
- 11.Caravatti P, Allemann M. The Infinity Cell : A New Trapped-Ion Cell with Radiofrequency Covered Trapping Electrodes for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Org Mass Spectrom. 1991;26:514–518. [Google Scholar]
- 12.Mormann M, Ma ek B, De Peredo AG, Hofsteenge J, Peter-Katalini J. Structural Studies on Protein O-fucosylation by Electron Capture Dissociation. Int J Mass Spectrom. 2004;234:11–21. [Google Scholar]
- 13.Adamson JT, Håkansson K. Infrared Multiphoton Dissociation and Electron Capture Dissociation of High-Mannose type glycopeptides. J Proteome Res. 2006;5:493–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 14.Galeva NA, Esch SW, Williams TD, Markille LM, Squier TC. Rapid Method for Quantifying the Extent of Methionine Oxidation in Intact Calmodulin. J Am Soc Mass Spectrom. 2005;16:1470–80. doi: 10.1016/j.jasms.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Reid GE, Roberts KD, Kapp EA, Simpson RI. Statistical Mechanistic Approaches to Understanding the Gas-Phase Fragmentation Behavior of Methionine Sulfoxide Containing Peptides. J Proteome Res. 2004;3:751–9. doi: 10.1021/pr0499646. [DOI] [PubMed] [Google Scholar]
- 16.Guan Z, Yates NA, Bakhtiar R. Detection and Characterization of Methionine Oxidation in Peptides by Collision-Induced Dissociation and Electron Capture Dissociation. J Am Soc Mass Spectrom. 2003;14:605–13. doi: 10.1016/S1044-0305(03)00201-0. [DOI] [PubMed] [Google Scholar]
- 17.Smakman R, Boer DJT. The Mass Spectra of Some Aliphatic and Alicyclic Sulphoxides and Sulphones. Organic Mass Spectrom. 1970;3:1561–88. [Google Scholar]
- 18.Lacombe S, Banchereau MLE, Simon M, Pfister-Guillouzo G. Sulfenic Acids in the Gas Phase: A photoelectron Study. J Am Chem Soc. 1996;118:1131–38. [Google Scholar]
- 19.Turecek F, Drinkwater DE, McLafferty FW. Gas-Phase Chemistry of CH3SOH, -CH2+SHOH, CH3SO., and .CH2SOH by Neutralization-Reionization Mass Spectrometry. J Am Chem Soc. 1989;111:7696–7701. [Google Scholar]
- 20.Gross E, Morell JL. Structure of Niasin. J Am Chem Soc. 1971;93:4634–4635. doi: 10.1021/ja00747a073. [DOI] [PubMed] [Google Scholar]
- 21.Xie L, Van Der Donk WA. Post-Translational Modifications During Lantibiotic Biosynthesis. Curr Opin Chem Biol. 2004;8:498–507. doi: 10.1016/j.cbpa.2004.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.