

Abstract
A new fluorous 2-chloropyridinium hexafluorophosphate was prepared as a modified Mukaiyama condensation reagent, and it was applied in amide formation reactions. Good to excellent purities of amides were obtained after fluorous solid-phase extraction of reaction mixtures without additional chromatography.
Keywords: fluorous reagent, amide coupling, fluorous solid-phase extraction
Amide formation is one of the most common transformations in organic synthesis. As a part of our ongoing effort on the development of new fluorous reagents for high-speed organic synthesis,1 we recently initiated a project to develop chromatography-free, solution-phase amide formation protocols. Reactions involving fluorous reagents are usually conducted in common organic solvents, and the byproducts from fluorous reagents are removed by fluorous solid-phase extraction (F-SPE).2 Reactions with fluorous reagents have advantages of homogeneous reaction environment, while reactions with polymer-bound reagents are conducted under heterogeneous conditions where mode of agitation, quality of resin, and degree of swelling often affect the outcome of reactions.3
Since the Mukaiyama condensation reagent (N-methyl-2-chloropyridinium iodide) was first introduced in 1975 for esterification of carboxylic acids,4 various N-alkyl-2-halopyridinium salts have been used for coupling and dehydrating reactions.5 Here we report the synthesis of a new N-alkyl-2-chloropyridinium hexafluorophosphate containing a C9F19 fluorous tag, and its application as separation friendly Mukaiyama condensation reagent for amide formation reactions.6
Fluorous tagged 2-chloropyridinium hexafluorophosphate 3 was prepared from the corresponding benzyl alcohol 17 in 4 steps (Scheme 1). Pyridinium salt 3 is stable white powder that can be stored under air with no significant indication of decomposition over a year.
Scheme 1.
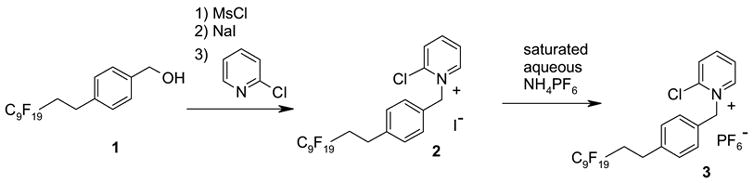
Preparation of fluorous pyridinium salt 3
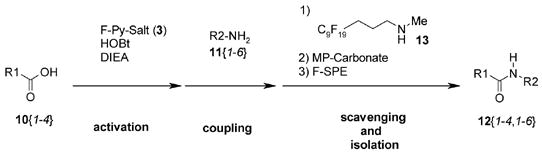
Because pyridinium salt 3 directly reacts with primary and secondary amines rapidly, we decided to activate carboxylic acid first by adding N,N-diisopropylethylamine (DIEA) to a mixture of carboxylic acid and pyridinium salt 3, and then to add an amine to form the corresponding amide (Scheme 2). LC-MS analysis of a mixture of N-methylindole-3-carboxylic acid 4, pyridinium salt 3, and DIEA showed three major peaks; two of which were assigned as carboxylic anhydride 5 and pyridone 6. The unidentified peak was presumably from acyloxypyridinium intermediate 7 since the peak disappeared immediately after the addition of dibenzylamine to give amide 8. Pyridone 6 was removed by F-SPE, and then resin-bound carbonate and trisamine (MP-Carbonate and MP-Trisamine)8 were used to scavenge the carboxylic anhydride 5 and the remaining carboxylic acid 4. This provided amide 8 in 51% yield.
Scheme 2.
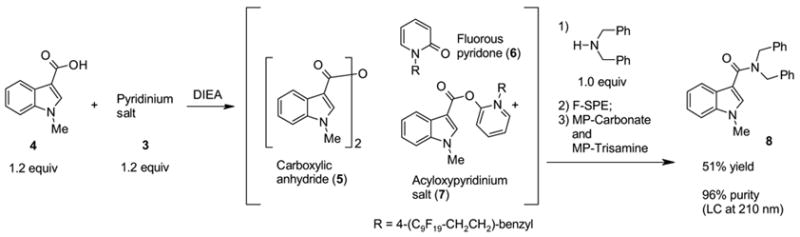
Amide formation without additive
In contrast, formation of the carboxylic anhydride 5 was completely suppressed in the presence of 1-hydroxybenzotriazole (HOBt). As shown in Scheme 3, DIEA was added to a mixture of carboxylic acid 4, pyridinium salt 3, and HOBt. LC-MS analysis of the mixture indicated that the corresponding OBt ester 9 formed within a few minutes after the addition of DIEA. Dibenzylamine was then added to form amide 8.9 To isolate amide 8, MP-Trisamine and MP-Carbonate were used to scavenge the remaining OBt ester 9 and HOBt, and then F-SPE was conducted to remove fluorous pyridone 6.
Scheme 3.
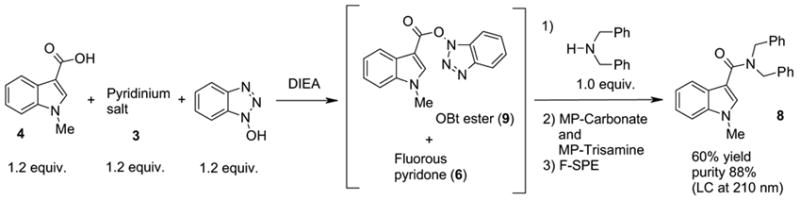
Amide formation in the presence of HOBt
To further examine this amide formation procedure with HOBt, we conducted a 4 x 6 matrix of reactions. The acids and the amines used for the demonstration are shown in Figure 1. In these 24 amide formations, a THF solution of each OBt ester was first prepared, and then it was split into 6 to react with each amine.10 In these reactions, to reduce the amount of polymer bound scavengers, a fluorous amine 13 (N-methyl-3-(perfluorononyl)propylamine) was used instead of MP-Trisamine to convert the excess OBt ester to the corresponding amide with a C9F19 tag so that the amide can be removed by F-SPE at the end. MP-Carbonate was then added to remove HOBt and to neutralize HCl and HPF6 salts of DIEA. Removal of the resin by filtration followed by F-SPE provided the desired coupling products. The yields and the purities of the products are summarized in Table 1.11 The yields are typically more than 80%; however, poor yields were obtained in several cases (entries 7, 9, and 23). Some of the low yields obtained are partly due to poor solubility of the amides in the loading solvent (DCM) for F-SPE.2b Two reactions that gave low yields were repeated (entries 7 and 9), and significantly higher yields of the products were obtained by using DMF as the loading solvent.
Figure 1.
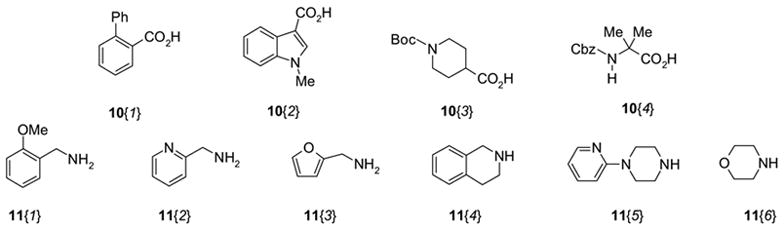
Acids and amines used for 4 × 6 matrix amide coupling reactions
Table 1.
4 × 6 amide coupling10
| entry | amide | yield (%) | purity (%)a | entry | amide | yield (%) | purity (%)a |
|---|---|---|---|---|---|---|---|
| 1 | 12{1,1} | 96 | >99b | 13 | 12{3,1} | 89 | >99 |
| 2 | 12{1,2} | 84 | 98 | 14 | 12{3,2} | 82 | 97 |
| 3 | 12{1,3} | 95 | >99 b | 15 | 12{3,3} | 98 | >99 b |
| 4 | 12{1,4} | 96 | 98 | 16 | 12{3,4} | 91 | 97 |
| 5 | 12{1,5} | 99 | >99 b | 17 | 12{3,5} | 93 | >99 b |
| 6 | 12{1,6} | 93 | >99 b | 18 | 12{3,6} | 98 | >99 b |
| 7 | 12{2,1} | 20 (99%c) | 98 | 19 | 12{4,1} | 94 | >99 b |
| 8 | 12{2,2} | 66 | >99 b | 20 | 12{4,2} | 87 | 75 |
| 9 | 12{2,3} | 40 (80%c) | >99 b | 21 | 12{4,3} | 88 | 86 |
| 10 | 12{2,4} | 95 | 96 | 22 | 12{4,4} | 86 | 96 |
| 11 | 12{2,5} | 93 | 99 | 23 | 12{4,5} | 46 | >99 b |
| 12 | 12{2,6} | 95 | 99 | 24 | 12{4,6} | 80 | >99 b |
measured by HPLC with an UV detector at 210 nm.
Single peak was detected by HPLC.
DMF was used as the loading solvent for F-SPE.
In summary, we have developed a new amide formation protocol by using fluorous pyridinium salt 3 and HOBt. The activation of carboxylic acids to the corresponding OBt esters typically finishes within 5 min. The protocol gave high purity products in general without chromatographic purifications.
Acknowledgments
We thank Professor Dennis Curran (University of Pittsburgh) for helpful discussions. This work was supported by the National Institutes of General Medical Sciences SBIR grant (2R44GM067326-02A1).
References and Notes
- 1.For general reviews on fluorous synthesis, see: Gladysz JA, Curran DP, Horvath IT, editors. Handbook of Fluorous Chemistry. Wiley-VCH; Weinheim: 2004. Zhang W. Chem Rev. 2004;104:2531. doi: 10.1021/cr030600r.Zhang W. Tetrahedron. 2003;59:4475.Pozzi G, Shepperson I. Coord Chem Rev. 2003;242:115.Tzschucke CC, Markert C, Bannwarth W, Roller S, Hebel A. Angew Chem Int Ed. 2002;41:3964. doi: 10.1002/1521-3773(20021104)41:21<3964::AID-ANIE3964>3.0.CO;2-3.Yoshida J, Itami K. Chem Rev. 2002;102:3693. doi: 10.1021/cr0103524.Dobbs AP, Kimberley MR. J Fluorine Chem. 2002;118:3.Curran DP. Synlett. 2001:1488.Curran DP. In: Stimulating Concepts in Chemistry. Stoddard F, Reinhoudt D, Shibasaki M, editors. Wiley-VCH; New York: 2000. pp. 25–37.Curran DP. Angew Chem, Int Ed. 1998;37:1175.
- 2.Curran DP. Separations with Fluorous Silica Gel and Related Materials. In: Gladysz JA, Curran DP, Horvath IT, editors. Handbook of Fluorous Chemistry. Wiley-VCH; Weinheim: 2004. pp. 101–127. [Google Scholar]; (b) for detailed F-SPE procedure, visit http://www.fluorous.com
- 3.(a) Barany G, Kempe M. The Context of Solid-Phase Synthesis. In: Czarnik AW, DeWitt SH, editors. A Practical Guide to Combinatorial Chemistry. American Chemical Society; Washington, DC: 1997. pp. 51–97. [Google Scholar]; (b) Yan B. Monitoring and Optimizing Organic Reactions Carried Out on Solid Support. In: Buchmeiser MR, editor. Polymeric Materials in Organic Synthesis and Catalysis. Wiley-VCH; Weinheim: 2003. pp. 503–526. [Google Scholar]
- 4.(a) Mukaiyama T, Usui M, Shimada E, Saigo K. Chem Lett. 1975:1045. [Google Scholar]; (b) Saigo K, Usui M, Kikuchi K, Shimada E, Mukaiyama T. Bull Chem Soc Jpn. 1977;50:1863. [Google Scholar]
- 5.(a) Mukaiyama T. Angew Chem, Int Ed Engl. 1979;18:707. [Google Scholar]; (b) Li P, Xu JC. Tetrahedron. 2000;56:8119. [Google Scholar]; (c) Convers E, Tye H, Whittaker M. Tetrahedron Lett. 2004;45:3401. [Google Scholar]; (d) Crosignani S, Gonzalez J, Swinnen D. Org Lett. 2004;6:4579. doi: 10.1021/ol0480372. [DOI] [PubMed] [Google Scholar]; (e) Donati D, Morelli C, Taddei M. Tetrahedron Lett. 2005;46:2817. [Google Scholar]
- 6.For other fluorous reagents for amide formation, see: Markowicz MW, Dembinski R. Synthesis. 2004:80.Palomo C, Aizpurua JM, Loinaz I, Fernandez-Berridi MJ, Irusta L. Org Lett. 2001;3:2361. doi: 10.1021/ol016165+.
- 7.Available from Fluorous Technologies, Inc. (http://www.fluorous.com)
- 8.(a) MP-Carbonate and MP-Trisamine are products of Argonaut Technologies, Inc http://www.argotech.com) (b) Four to five equiv of MP-Carbonate per each acid (HOBt, DIEA-HCl, and DIEA-HPF6) was used. (c) For use of MP-Carbonate in HOBt scavenging, see: Sauer DR, Kalvin D, Phelan KM. Org Lett. 2003;5:4721. doi: 10.1021/ol0358915.
- 9.The reaction time between OBt ester and an amine varies from within a minute to days depending on sterics and nucleophilicity of the amine used. Among the amines we tried, dibenzylamine was especially slow. The reaction mixture was stirred at room temperature for 3 d to obtain 60% yield of the amide 8. See also ref 8b.
- 10.Typical procedure: To a mixture of carboxylic acid (0.73 mmol), pyridinium salt (0.79 mmol), and HOBt (0.79 mmol) in THF (5.0 mL) was added DIEA (2.5 mmol) at 23 °C. After 5 min, total volume was adjusted to 6.0 mL by adding THF. After ½ h, 1.0 mL of the solution was added to an amine (0.1 mmol) in THF (1.0 mL). After 1 h, N-methyl-3-(perfluorononyl)propylamine (0.04 mmol) in THF (0.5 mL) was added to convert the excess OBt ester to fluorous tagged amide. After 16 h, MP-Carbonate (loading = 3.4 mmol/g, 0.45 g, 1.5 mmol) was added to remove HOBt and to neutralize HCl and HPF6 salts of DIEA, and the mixture was stirred vigorously for 3 h. The resin was filtered off, and was rinsed with THF (3 × 2 mL). The filtrate was concentrated, and the residue was dissolved in DCM (1 mL), and loaded onto a FluoroFlash SPE cartridge (3 g)7 that was conditioned with 80:20 MeOH-H2O.2b The amide was eluted with 80:20 MeOH-H2O (10 mL).
- 11.The products were characterized by 1H NMR and low resolution mass spectral analyses (LC-MS, APCI mode).