


Abstract
Analysis of the transcriptome by computational and experimental methods has established that sense–antisense transcriptional units are a common phenomenon. Although the regulatory potential of antisense transcripts has been experimentally verified in a number of studies, the biological importance of sense–antisense regulation of gene expression is still a matter of debate. Here, we report the identification of sequence features that are associated with antisense transcription. We show that the sequence composition of the first exon and the 5′end of the first intron of many human genes is similar to the sequence composition observed in promoter regions as measured by the density of known transcription regulatory motifs. Cloned intron-derived fragments were found to possess bidirectional promoter activity. In agreement with the reported abundance of antisense transcripts overlapping the 5′UTR, mapping of the 5′ends of antisense transcripts to the corresponding sense transcripts revealed that the first exon and the 5′end of the first intron are hotspots of antisense transcription as measured by the number of antisense transcription start sites per unit sequence. CpG dinucleotide suppression that is typically weak in non-methylated promoter regions is similarly weakened upstream as well as downstream of the first exon. In support of antisense transcripts playing a regulatory role, we find that 5′UTRs and first exons of genes with overlapping antisense transcripts are significantly longer than the genomic average. Interestingly, a similar size distribution of 5′UTRs and first exons is observed for genes silenced by CpG island methylation in human cancer.
INTRODUCTION
Over the past few years, computational as well as experimental evidence has firmly established that the transcribed fraction of mammalian genomes exceeds the fraction occupied by protein-coding genes by at least an order of magnitude and that many of these transcripts are transcribed in the antisense direction of known genes (1–15). These findings are supported by the widespread binding of different transcription factors to regions in the genome that are far removed from known promoters (16–19). In all of these studies, estimates on the number of antisense transcripts and transcription-factor-binding sites depend on specific biological systems under study where only subsets of genes are expressed. Computational approaches can alleviate this limitation by analyzing EST (expressed sequence tag) libraries of different origin (13). Nevertheless, general conclusions about the biological significance of antisense transcripts remain difficult to be drawn even though they have been found to be particularly abundant at the 5′ and 3′ends of genes, i.e. close to untranslated regions whose regulatory role has been documented in numerous studies (20).
A number of mechanisms that could be employed by antisense transcripts to regulate gene expression of corresponding sense transcripts have been discussed, including masking of regulatory motifs, RNA editing and RNA-interference-mediated mechanisms (21). In spite of their abundance and their enormous regulatory potential, a physiological role of mammalian antisense transcripts is widely accepted only in the regulation of imprinting and in X-chromosome dosage compensation (22). Therefore, the biological significance of most antisense transcripts is a matter of intense debate.
The identification of promoters of antisense transcripts will facilitate the design of experiments aimed at elucidating their regulatory potential and the signals that govern their expression. Here, we report that the distribution of transcription regulatory motifs along human genes closely resembles the distribution of 5′ends of antisense transcripts. Genome-wide searches for transcription regulatory motifs are known to produce large numbers of false-positive results (23). However, alignment of sequences at the transcription start site (TSS) shows that the density of transcription regulatory motifs immediately upstream of the TSS is much higher than the density observed in random alignments of genomic sequences and reflects the physiological role of these motifs. By aligning genes on exon–intron junctions, we measured the density of transcription regulatory motifs in the vicinity of exon–intron junctions in an attempt to identify regions of elevated density of transcription regulatory motifs downstream of the TSS. We show that the density of transcription regulatory motifs at the 5′end of the first intron of human genes is similar to the density observed in putative promoter regions and that cloned intron-derived fragments possess bidirectional promoter activity. Promoter regions are known to be unmethylated in vivo, leading to a significant weakening of CpG dinucleotide suppression in promoter regions as compared to other regions of the genome where methylation is common (24,25). We show that CpG dinucleotide suppression is similarly weakened upstream as well as downstream of the first exon, suggesting that the start of the first intron in many human genes is similarly unmethylated in vivo as the promoter region.
Antisense promoter activity at the 5′end of the first intron in vivo is supported by preferential mapping of antisense TSS in this region as well as within the first exon. In support of a functional role of antisense transcripts in gene regulation, our analysis of sense–antisense transcriptional units revealed that regions of sequence overlap are associated with longer 5′UTRs and first exons in the corresponding sense transcript as compared to the genomic average. A similar size distribution of 5′UTRs and first exons is observed for genes silenced by CpG island methylation in human cancer. Our results suggest that the 5′end of the first intron of human genes is a hotspot of antisense transcription and that antisense transcripts located at the 5′end of protein-coding genes are associated with regulatory functions that need to be explored in more detail.
RESULTS
With the aim of identifying hotspots of antisense transcription starting within the boundaries of known protein-coding genes, we estimated the number of antisense transcripts starting from known exons/introns of the corresponding protein-coding gene on the opposite strand. 5′ends of antisense transcripts were mapped to a non-redundant set of 18 008 RefSeq genes that was prepared as described in the Methods section. Antisense transcripts were identified according to the criteria defined by (11). In order to minimize the number of false positives, only antisense ESTs which are supported by an Aceview gene model were considered. Furthermore, when two or more antisense transcripts were found to originate from a sequence window of 200 bp, an antisense transcriptional starting region (ATSR) was defined and the starting point of antisense transcription was set to the starting position of the antisense transcript with the longest 5′end. Having defined ATSRs, we determined how many ATSRs were located in the first exon, first intron, second exon, second intron and so on of the corresponding sense transcripts. Figure 1A shows that the majority of ATSRs is located in the first intron, followed by the first exon, second intron and last intron of sense transcripts. Considering the number of bases occupied by first exons as compared to the sequence occupied by introns, these data suggest that antisense transcription starting from the first exon is strongly favored as compared to other exons/introns. Altogether, more than half of all identified ATSRs were located within exon1, intron1 and intron2, suggesting that ATSRs are observed primarily at the 5′end of genes.
Figure 1.

Distribution of ATSRs. (A) Pie chart representing the ATSR distribution per genomic element. The distribution was evaluated for RefSeq genes having at least three exons for which it was possible to distinguish unambiguously between first and last intron. Globally, we analyzed the distribution on 2671 RefSeq genes representing 94.3% of 2830 RefSeq genes having an ATSR. (B) Distribution of normalized ATSR distances. The distance between ATSR and the 5′end of the corresponding sense transcript was calculated and normalized to the length of sense RefSeq gene. (C) Distribution of ATSR distances relative to exon1–intron1 junction. Only ATSR mapping on first exon and first intron were considered. (D) Distribution of normalized ATSR distance from intron start on RefSeq introns. For ATSRs mapping on introns, the distance of the ATSR from the intron start was divided by intron length.
We tested this hypothesis by dividing genomic loci into 10 intervals of equal size and counted the number of ATSRs observed in each interval. As shown in Figure 1B, most ATSRs were located in the most 5′ interval. Considering the frequency of ATSRs in exon1 and intron1, we mapped ATSRs relative to the exon1–intron1 junction. A region covering 2000-bp upstream of the exon1–intron1 junction and 5000-bp downstream of the exon1–intron1 junction was analyzed for each locus. The 7000 bp under analysis were divided into 14 intervals of equal size (500 bp per interval) and the number of ATSRs in each interval was counted. Figure 1C illustrates that the interval with the largest number of ATSRs is represented by the first interval downstream of the exon1–intron1 junction followed by the first interval upstream of the exon1–intron1 junction and the second interval downstream of the exon1–intron1 junction. Altogether, the majority of ATSRs analyzed was found to be located within the first 1000 bp downstream of the exon1–intron1 junction. Considering that the first intron was found to give rise to the largest number of ATSRs (Figure 1A) and that the average length of the first intron (∼14 800 bp) is much larger than 1000 bp, these data suggested that the 5′end of the first intron is particularly rich in ATSRs.
In order to answer the question whether this observation is specific to intron1 or common to all introns, we analyzed the distribution of ATSRs along introns. Each intron containing an ATSR was divided into 10 equal sized intervals and the number of ATSRs within each interval was counted for ATSRs within a first intron, a second intron, a third intron, a fourth intron or a last intron. Figure 1D shows that the first interval of the first intron is particularly rich in ATSRs as compared to other introns. We conclude that ATSRs are preferentially located within first exons and/or the 5′end of the first intron. As a proof that the abundance of ATSRs in the first intron is not a mere consequence of the fact that first introns are larger than downstream introns, we calculated the probability of finding a given number of ATSRs in each exon/intron considering the number of bases that are represented by these genomic elements. The results of this calculation are shown in Table 1. The calculation was carried out separately for 18 008 non-redundant RefSeq genes as well as for ATSR genes having at least three exons (i.e. more than one intron). The total number of bases occupied by these genes in the genome and the number of bases occupied by exons and introns was calculated. Overrepresentation of ATSRs in a specific genomic element was estimated according to the hypergeometric distribution. The results indicate that ATSRs are strongly overrepresented in the first exon, first intron, last exon and, to a much lesser extent, in the last intron. All other genomic elements tested displayed a frequency of ATSRs compatible with a random distribution. These results confirm previous observations that antisense transcripts are frequently observed at the 5′ and 3′ends of protein-coding genes. However, they illustrate the genomic elements that are particularly prone to hosting ATSRs with exon1 and intron1 being the elements where ATSR overrepresentation is the strongest. It should also be noted that the P values calculated for intron1 are upper limits because the calculation assumes a uniform distribution of ATSRs along intron1 while we have observed that most ATSRs are actually located at the 5′end of the first intron (Figure 1D). Thus, we conclude that first exons and the 5′end of first introns of human genes are hotspots of antisense transcription.
Table 1.
ATSR overrepresentation in diverse genomic elements (exon–introns)
| Element | Natsr | FRACTIONatsr | N3bases | FRACTION3bases | NFULLbases | FRACTIONFULLbases | pval3 | pvalFULL |
|---|---|---|---|---|---|---|---|---|
| INTRON1 | 1222 | 0.34 | 77266164 | 0.2292 | 231868207 | 0.2310 | 6.48965E − 50 | 3.47121E − 48 |
| INTRON2 | 346 | 0.1 | 47512862 | 0.1410 | 140269449 | 0.1398 | 1.00000E + 00 | 1.00000E + 00 |
| INTRON_LAST | 277 | 0.08 | 20348242 | 0.0604 | 69393382 | 0.0691 | 4.65675E − 05 | 4.42930E − 02 |
| EXON1 | 303 | 0.08 | 775688 | 0.0023 | 4115038 | 0.0041 | 0.00000E + 00 | 2.89772E − 279 |
| INTRON3 | 186 | 0.05 | 32505134 | 0.0964 | 93731193 | 0.0934 | 1.00000E + 00 | 1.00000E + 00 |
| INTRON4 | 155 | 0.04 | 22193279 | 0.0658 | 65228174 | 0.0650 | 1.00000E + 00 | 1.00000E + 00 |
| INTRON5 | 106 | 0.03 | 17940288 | 0.0532 | 51523916 | 0.0513 | 1.00000E + 00 | 1.00000E + 00 |
| EXON_LAST | 125 | 0.03 | 3617144 | 0.0107 | 19179878 | 0.0191 | 1.67166E − 28 | 6.63873E − 10 |
| INTRON7 | 89 | 0.02 | 12556811 | 0.0373 | 34707870 | 0.0346 | 9.99992E − 01 | 9.99768E − 01 |
Overrepresentation of ATSRs considering the number of bases occupied by each genomic element was calculated based on the hypergeometric distribution as described in the Methods section. The calculation was carried out separately for 18 008 non-redundant RefGenes and for ATSR genes having at least three exons.
Natsr: number of ATSRs observed in genomic element (total ATSRs analyzed: 3619).
FRACTIONatsr: Natsr divided by 3619.
N3bases: number of bases occupied by genomic elements in ATSR genes with at least three exons (total number of bases occupied in the genome by ATSR genes with at least three exons: 337053089).
FRACTION3bases: N3bases divided by 337 053 089.
NFULLbases: number of bases occupied by genomic elements in 18 008 non-redundant RefGenes (total number of bases occupied in the genome by 18 008 non-redundant RefGenes: 1 003 561 228).
FRACTIONFULLbases: NFULLbases divided by 1003561228.
pval3: P value for ATSR genes with at least three exons.
pvalFULL: P value for 18 008 non-redundant RefGenes.
We were wondering whether the observed abundance of antisense transcripts starting from the first exon and the 5′end of the first intron is reflected by the distribution of transcription regulatory motifs along genomic loci. Therefore, we analyzed the distribution of consensus transcription regulatory motifs described by (26) as well as TRANSFAC (27) and JASPAR (28) repositories. Repeat masked sequences starting from the first exon and ending with the last exon for hg17 human RefSeq genes were downloaded from the UCSC genome browser and the location of consensus motifs was determined by a pattern matching approach. First, each locus was divided into 10 equal sized intervals and the number of matches for each consensus motif in each interval was counted. This analysis revealed that matches are particularly frequent in the first (5′end of genes) and the last (3′end of genes) interval (Figure 2A). In order to increase the resolution of this analysis, we determined the frequency of transcription regulatory motifs around exon–intron junctions. 1000-bp upstream and 1000-bp downstream of each exon–intron junction were analyzed. The 2000 bp under study were divided into 20 equal sized intervals and the number of matches in each interval was counted. The results of this analysis are shown in Figure 2B. The entire dataset was subjected to cluster analysis using Pearson correlation as distance measure. Two types of transcription regulatory motifs can be distinguished: GC-rich motifs that are particularly abundant upstream as well as downstream of the first exon, and GC-poor motifs that are underrepresented in these regions. In general, the frequency of transcription regulatory motifs upstream of the first exon (intervals to the left of P:E1, i.e. putative promoter regions) is very similar to the frequency observed downstream of the first exon (intervals to the right of E1:I1, i.e. 5′end of first introns).
Figure 2.


Sequence features associated with antisense transcription. (A) Distribution of transcription regulatory motifs along 18 008 non-redundant RefSeq loci. Consensus sites (5–10 mers) corresponding to transcription regulatory motifs reported in TRANSFAC, JASPAR, and Xie et al. (26) were mapped onto genomic loci starting from the beginning of exon1 up to the end of the last exon. Each motif position was normalized to locus length and assigned to one of ten intervals. The resulting matrix was subjected to hierarchical clustering using Pearson correlation as distance measure following median normalization of matrix rows. (B) Distribution of transcription regulatory motifs in the vicinity of exon–intron junctions. Transcription regulatory motifs located within 1000-bp upstream and downstream of exon–intron junctions were assigned to 1 of 20 intervals (each interval represents 100 bp). Each row of the resulting matrix was median normalized and subjected to hierarchical clustering using Pearson correlation as distance measure. The distribution of GC content of the motifs found in the two main clusters are shown to the right. (C) Bidirectional promoter activity of genomic fragments derived from the 5′end of the first intron of the indicated genes. Genomic fragments of ∼1000 bp were cloned into pGL3basic (Promega) in both orientations and basic promoter activity was determined in three different cell lines. (D) Relative density of transcription regulatory motifs in the vicinity of exon–intron junctions in ATSR genes as compared to non-ATSR genes. The number of transcription regulatory motifs located within 1000-bp upstream and downstream of exon–intron junctions was divided by the number of bases searched for ATSR genes and for non-ATSR genes. The motif density per unit sequence obtained for ATSR genes was divided by the motif density found in non-ATSR genes. The resulting matrix was subjected to hierarchical clustering using Pearson correlation as distance measure. The left panel displays the relative motif density including promoter and 3′ sequences. The right panel displays the relative motif densities around the exon–intron junctions E1:I1, E2:I2 and E3:I3. Ex:Ix = Exon x : Intron x junction. E-x:I-x = Exon x : Intron x junction counted from end of gene. Prom. = promoter. Dstr. = downstream.
The observed similarity of motif distributions upstream and downstream of the first exon led us to investigate the possibility that the 5′end of first introns may be associated with promoter activity and whether this promoter activity can explain the abundance of ATSRs at the exon1–intron1 junction shown in Figure 1C. Thus, we cloned intron1-derived fragments of ∼1000 bp from 15 randomly chosen ATSR loci with first introns longer than 1000 bp and cloned them in front of a luciferase reporter gene in both orientations (Figure 2C). Promoter activity of these fragments was tested in three different cell lines. We observed significant promoter activity in the sense direction for the majority of cloned fragments. Interestingly, however, for more than half of the fragments we also observed promoter activity in the antisense direction in at least one cell line. These data indicate that the 5′end of first introns can be associated with bidirectional promoter activity.
In order to obtain evidence that the observed abundance of transcription regulatory motifs in the first intron is indeed associated with antisense transcription, we studied the distribution of transcription regulatory motifs in ATSR-containing genes and compared it to the distribution in non-ATSR-containing genes. The number of observed motif matches in each of the 20 100-bp intervals surrounding the indicated exon–intron junctions was divided by the number of bases searched. The motif frequency per unit sequence obtained for ATSR-containing genes was then divided by the motif frequency per unit sequence observed in non-ATSR-containing genes. The results of this analysis are shown in Figure 2D. In this plot, red color indicates higher and green color indicates lower relative density of motifs in the indicated interval in ATSR- versus non-ATSR-containing genes. The complete dataset was subjected to cluster analysis, again using Pearson correlation as distance measure. If the distribution of regulatory motifs observed in Figure 2B is indeed associated with antisense transcription, cluster analysis of the data table representing the relative abundance of motifs in ATSR- versus non-ATSR-containing genes (Figure 2D) should separate GC-rich from GC-poor motifs as shown in Figure 2B. The quality of this separation can be judged from the color labels to the right of the data tables where yellow indicates GC-rich and red indicates GC-poor motifs. Run-length analysis of observing uninterrupted stretches of GC-poor and GC-rich motifs clustering together indicates that this separation is significant because the expected number of uninterrupted clusters of the observed size is <E-14. Interestingly, a very similar quality of separation of GC-poor and GC-rich motifs is achieved when only the sequences downstream of exon–intron junctions E1:I1, E2:I2 and E3:I3 are analyzed (See Figure 2D, right panel). We conclude that GC-rich motifs are more abundant downstream of the exon1–intron1 junction in ATSR-containing genes. These data suggest that CpG islands extending downstream of the start site of the sense transcript into the first exon and the first intron are frequently associated with antisense transcription.
In vivo, active promoters lack cytosine methylation at CpG sites while such methylation is frequently observed in non-promoter regions (24,25). Since methylated cytosine is prone to deamination-mediated C->T transition mutagenesis, CpG dinucleotides are strongly underrepresented in much of the genome. However, in non-methylated promoter regions, CpG suppression is much weaker. Therefore, we analyzed CpG dinucleotide suppression upstream and downstream of exons as an indirect measure of the methylation status in vivo of sequences in this region. The results of this analysis are shown in Figure 3. For each exon in ATSR genes, 1000 bases upstream and 1000 bases downstream were analyzed in 10 separate intervals of 100 bp each. The nucleotide composition in each interval was determined and the expected frequency of CpG dinucleotides was calculated based on the observed nucleotide frequencies. Then, the observed frequency of dinucleotides was divided by the expected frequency in each interval and plotted for exons one through five for CpG, GpC, and SpS (S = G or C) dinucleotides. The results indicate that CpG suppression is much weaker upstream of exon1 (i.e. putative promoter regions) as compared to the level of CpG suppression upstream of exons two through five. No significant deviation from the expected frequencies was observed for GpC and SpS dinucleotides, indicating that weakened suppression is CpG specific. Interestingly, we observed similarly weakened CpG suppression downstream of first exons but not downstream of other exons with GpC and SpS dinucleotides occurring at roughly the expected rates. These results suggest that sequences upstream as well as downstream of first exons are frequently non-methylated in vivo in ATSR genes. Since ATSRs are overrepresented in last introns and last exons, we also analyzed CpG suppression in these regions. However, no significant weakening of CpG suppression was detected (data not shown).
Figure 3.

CpG suppression upstream and downstream of exons. The number of nucleotides and dinucleotides up to 1000-bp upstream and up to 1000-bp downstream of exons one through five was determined. The expected number of dinucleotides was calculated from the base composition in each 100-bp interval. Plots show the observed to expected ratio for CpG, GpC and SpS (S = G or C) dinucleotides both upstream and downstream of the indicated exons for ATSR genes.
The results presented so far suggest that CpG islands extending into the first intron are associated in vivo with promoter activity that can be bidirectional and give rise to antisense transcripts. Thus, we analyzed the association of CpG island extension into the first intron with antisense transcription in more detail. This analysis is shown in Figure 4. First, we identified all RefSeq genes whose first exon is entirely embedded in a CpG island. Out of 18 008 RefSeq genes analyzed, 10 991 (61.0%) were found to belong to this class. Among the 2830 genes containing ATSRs, in 1962 (69.3%) genes the first exon was found embedded in a CpG island. Calculating the probability of observing this increase by chance according to the hypergeometric distribution indicates that this increase is highly significant (P = 1.12E–23). Furthermore, we analyzed the fraction of genes with CpG-island-embedded first exons overlapping with the RNA of antisense transcripts (i.e. genes with potential to form dsRNA hybrids if sense and antisense RNA are transcribed in the same cell). Among the 2830 genes containing an ATSR, 888 have the potential to form dsRNA hybrids. In 679 of these genes (76.4%), the first exon was found to be entirely embedded in a CpG island. Hypergeometric distribution suggests that this further increase from 69.3 to 76.4% is highly significant (P = 1.08E–8). We conclude that genes containing ATSRs are significantly enriched for genes whose first exon is entirely embedded in a CpG island and therefore within genomic regions that are known to be associated with bidirectional transcription (30).
Figure 4.

Sense–antisense exon1–exon overlap is preferentially observed in CpG islands which extend into the first intron. CpG islands analysis. In the set of 18 008 non-redundant RefSeq genes, we determined the presence of CpG islands in genomic sequences using method defined in (29). Alu repeats were excluded by filtering the sequences with RepeatMasker. We identified 10 991 genes (61.0%) characterized by the presence of a CpG island which includes the entire first exon (CpG islands that extend into the first intron). In this set of 10 991 genes, the fraction of ATSR genes (69.3%) and of ATSR genes with sense exon1–antisense exon overlap (76.4%) was determined.
Bidirectional transcription of promoters has been found to be a common phenomenon in the human genome and gene pairs arranged in a head-to-head fashion are often coordinately regulated (31). The vicinity of 5′ends of head-to-head gene pairs and the abundance of antisense transcripts originating at the 5′end of genes suggest that an antisense transcript of one head-to-head gene partner represents an alternative transcription start of the sense transcripts of the head-to-head counterpart, potentially providing an additional possible mechanism of co-regulation. Therefore, we examined human head-to-head genes for the presence of ATSRs and vice versa. We found that 18% (417/2318) of genes having a head-to-head partner starting within 5-kb upstream also contained ATSRs (see Figure 5). The overall frequency of ATSR-containing genes among the 18 008 RefSeq genes we analyzed was 13% (2380/18 008), suggesting that head-to-head genes are more frequently transcribed in the antisense direction than isolated genes. Vice versa, among the 2380 ATSR-containing genes, 826 genes (35%) were associated with a head-to-head partner. These results suggest that head-to-head configuration and antisense transcription are correlated.
Figure 5.

Antisense transcription and bidirectional gene pairs. (A) Fraction of head-to-head and antisense Aceview gene models. (B) Distribution of distances between the 5′end of Aceview gene models and the 5′end of the RefSeq transcripts. Only Aceview gene models whose 5′end is located within 5 kb of the RefSeq transcription start are shown (4270).
In order to obtain a detailed view of the relative frequencies of head-to-head versus antisense transcription as well as the relative distances of 5′ends of head-to-head and antisense transcripts, we analyzed Aceview gene models for genes having both head-to-head partners as well as ATSRs. The analysis was carried out by identifying for each antisense Aceview transcript the representative Aceview gene. Then we verified the existence of alternative isoforms of this gene whose transcription starts upstream of the 5′end of the ATSR gene (thus forming a head-to-head pair with the ATSR gene). Next, we calculated the distances of the 5′ends of these isoforms from the 5′end of the ATSR gene under study. This analysis was carried out for all 826 ATSR genes for which we identified the presence of both an antisense transcript and an alternative head-to-head isoform. In total, we found 7021 Aceview genes that were configured either in a head-to-head or an antisense orientation with the 826 ATSR genes studied. About 76% of the Aceview genes were arranged in a head-to-head fashion while the 5′ends of the remaining 24% of Aceview genes mapped within ATSR genes (i.e. are antisense transcripts) (Figure 5B). The distribution of the distances between the 5′ends of the ATSR gene and the corresponding Aceview genes shown in Figure 5A illustrates that the majority of Aceview genes starts within 1000-bp upstream (head-to-head) or downstream (antisense) of the annotated transcription start of the ATSR gene. Within this 2-kb interval around the start site of ATSR genes there are 1962 (60%) head-to-head transcripts and 1284 (40%) antisense transcripts forming a nearly symmetrical distribution centered at the 5′end of ATSR genes. These data suggest that the promoters of 826 head-to-head ATSR genes can be transcribed in a bidirectional fashion with start sites mapping both upstream as well as downstream of the major start site of the ATSR gene giving rise to head-to-head and antisense transcripts with nearly equal frequency. Evaluation of the biological significance of this finding and possible co-regulated expression of the genes involved has to await further studies.
From the biological point of view, an important question is whether preferential antisense transcription in the vicinity of start sites of sense transcripts is connected to biological function or whether it is a mere reflection of basic promoter activity of CpG islands that are known to be capable of being transcribed in a bidirectional fashion (30). Antisense transcripts overlapping with the sense transcripts at the 5′end could exert regulatory functions via the 5′UTR of sense transcripts. 5′UTRs in mammalian cells are not required for efficient initiation of translation (20). On the other hand, 5′UTRs with known regulatory functions are known to be longer than average 5′UTRs (20). Therefore, we tested the possibility that 5′UTRs and first exons in ATSR-containing genes might be longer than the genomic average. 5′UTR length was calculated according to the RefGene annotations available at the UCSC genome browser. The results of this analysis are shown in Figure 6. For genes without ATSR we observed a median 5′UTR length of 129 bases. For genes with ATSR and sense–antisense sequence overlap we found a median 5′UTR length of 178 and 206 bases, respectively. This increase in 5′UTR lengths was found to be highly significant according to Wilcoxon signed rank sum statistics. Since dsRNA-mediated mechanisms have been discussed as possible mediators of CpG island methylation in cancer, we also analyzed the 5′UTRs of genes which have been reported as silenced by CpG island methylation in human cancer. Interestingly, we observed a slight but significant increase in 5′UTR lengths also for methylated genes (see Supplementary Data for a comprehensive list of this gene set). A similar length distribution was observed for exon1 lengths. While for genes without ATSR we observed a median exon1 length of 187 bases, this value was found increased to 202 and 271 bases for genes with ATSR and exon–exon sequence overlap, respectively. Again, genes methylated in cancer were found to contain longer first exons than the genomic average (median 252 bases). All of these values were found to be highly significant according to Wilcoxon signed rank sum statistics. We interpret these data as indirect evidence for a functional relevance of antisense transcripts in the regulation of expression levels of their protein-coding counterparts. Furthermore, the observation of lengthened 5′UTRs and first exons in genes methylated in human cancer could indicate that antisense transcripts play an active role in controlling CpG island methylation.
Figure 6.

Analysis of 5′UTR and exon1 lengths. (A) 5′UTR lengths were determined according to UCSC genome browser annotations. Box plots show the distribution of 5′UTR lengths in non-ATSR genes, ATSR genes, ATSR genes with exon–exon overlap and in genes reported as CpG island methylated in human cancer. (B) Box plots show the distribution of exon1 lengths as annotated by the UCSC genome browser for non-ATSR genes, ATSR genes, ATSR genes with exon–exon overlap and for genes reported as CpG island methylated in human cancer.
DISCUSSION
Here, we show that ATSRs within human-protein-coding genes are preferentially located within the first exon and the 5′end of the first intron. CpG islands that have historically been associated with bidirectional promoter activity (30) are shown to extend into the first intron preferentially in genes with ATSRs. By cloning genomic fragments from the beginning of the first intron of randomly chosen genes, we show that bidirectional promoter activity is frequently observed in this region of protein-coding genes and that these regions show decreased suppression of CpG dinucleotides similar to promoter regions, indicating that they are less methylated in vivo than other portions of the genome. Although we cannot rule out that the observed abundance of ATSRs in these regions is a reflection of CpG-island-associated bidirectional background promoter activity (30), we provide a hint for a regulatory role of antisense transcripts: We show that 5′UTRs and first exons are significantly lengthened in ATSR genes, particularly in ATSR genes with sense–antisense overlap and a similar size distribution of exon1 and 5′UTR lengths has been observed for genes reported to undergo CpG island methylation in cancer.
Many researchers have reported computational identification of sense–antisense transcriptional units (7–15) and the number of identified antisense transcripts was found to be strongly dependent on the criteria employed for EST orientation. The criteria we applied during our searches for antisense transcripts are those described by (11). In contrast to previous studies, however, we focused on the identification of antisense transcripts whose 5′ends are mapping within protein-coding RefSeq genes with high-quality annotations of exon–intron boundaries. Thus, overlapping transcriptional units composed of poorly annotated ESTs have been ignored. We then analyzed the mapping of 5′ends of antisense transcripts in detail and combined the observed biases in the distribution of antisense 5′ends with searches for transcription regulatory motifs so as to identify hotspots of antisense transcription that are supported by a significant enrichment of transcription regulatory motifs.
Searches for transcription regulatory motifs along the genome are notoriously error prone (23). We nevertheless believe that our approach is able to identify physiologically meaningful results for two reasons: First, our searches are based on IUPAC consensus motifs rather than on position weight matrices (PWMs) that are highly sensitive to the specific cutoff used. Searches for consensus motifs are generally much more stringent than PWM searches. The binding sites listed in TRANSFAC (27) and JASPAR (28) repositories were converted to consensus motifs based on a PWM cutoff that corresponds to a false-positive rate of 5% (32). Second, we do not interpret binding sites found in single sequences. Rather, we align genes on exon–intron junctions in a way that is similar to aligning genes at the TSS so as to identify motifs that are overrepresented in promoter regions on a genome-wide scale. Following this approach, we studied the distribution of transcription regulatory motifs in the vicinity of exon–intron junctions and found that transcription regulatory motifs can be found downstream of the first exon with nearly the same frequency as they can be found upstream of it. The prediction that these regions are associated with promoter activity has been confirmed by cloning intron-derived fragments and testing them explicitly in luciferase assays where bidirectional promoter activity is frequently observed. Furthermore, we show that the sequences downstream of exon1 in ATSR genes show decreased levels of CpG dinucleotide suppression to an extent that is very similar to the loss of CpG dinucleotide suppression observed in promoter regions. It has recently directly been shown that active promoter regions are unmethylated and available for binding of transcription factors while most other portions of the genome display various levels of DNA methylation at CpG sites (25). Our observations suggest that the unmethylated portions of the promoter are often extended to sequences downstream of the first exon where they can give rise to antisense transcripts and/or alternative sense transcripts.
Recently, Carninci and co-workers (33) reported genome-wide identification of human promoters by mapping CAGE tags to genomic sequence. They found that use of alternative promoters is frequently observed and that promoters can be classified regarding the spread of TSS along the promoters. Promoters with the widest spread of TSS were found to be associated with CpG islands and their findings also indicate that CpG island promoters often support bidirectional transcription. We have observed that antisense transcription is frequently observed in genes that are organized in a head-to-head fashion. Here, the antisense transcript of a gene and the sense transcript of its head-to-head partner can be interpreted as resulting from the use of alternative TSS originating from the same, extended promoter region. Thus, co-regulation of head-to-head gene pairs may also involve antisense regulation in certain circumstances. It is also worthwhile mentioning that transcription factor binding to intronic sequences, particularly to the first intron, has been observed in numerous studies, with the first reports dating back more than 20 years (34,35). The intron-located regulatory motifs have often been interpreted as enhancer elements. However, it cannot be excluded that their influence on sense gene expression is mediated by antisense RNA and that these motifs function as bona fide proximal promoter elements during the initiation of antisense transcription.
The functional significance of ATSRs reported here has been explored by estimating the length of 5′UTRs and first exons in genes with and without ATSRs. 5′UTRs with regulatory significance have been shown previously to be longer than average (20) and we show that 5′UTRs and first exons of ATSR genes are significantly lengthened, particularly in genes where sense–antisense overlap is observed. This finding is supported by a recent report that detected lengthened 5′UTRs in genes with overlapping antisense transcripts in yeast (36). Since the first exons of ATSR-containing genes are often embedded in CpG islands, the 5′UTRs of these genes tend to be GC rich. On the other hand, longer UTRs have been associated previously with lower GC content, making the correlation reported here even more significant (20). Interestingly, we observed a similar lengthening of 5′UTRs and first exons in genes that have been reported as silenced by CpG island methylation in human cancer. Antisense transcripts have been shown to be involved in the regulation of chromatin modifications in imprinted genes (22). It is tempting to speculate that bidirectional transcription of CpG islands giving rise to antisense transcripts in genes methylated in cancer is of functional significance in regulating CpG island methylation in these genes and that antisense transcripts are the mediators of the ‘instructive mechanism of de novo methylation’ that has recently been identified (37).
METHODS
Computational identification of ATSRs
We performed genomic mapping of the 5′ends of antisense transcripts to corresponding sense transcripts with the aim of identifying hotspots of intragenic antisense TSSs. The sense transcripts reference dataset is represented by genomic alignments of RefSeq transcripts version 12, generated by UCSC genome browser. This dataset was filtered for redundancy: RefSeqs that produced high-quality alignments in multiple regions of the genome were discarded; for each gene, we selected only the longest RefSeq as representative sequence. This resulted in 18 008 non-redundant genomic alignments of RefSeqs transcripts (see Supplementary Data).
Starting from genomic alignments of 5′ESTs and GenBank mRNAs provided by UCSC genome browser, we selected correctly oriented antisense transcripts. Orientation was evaluated using criteria similar to those adopted by (11). Discrimination between 5′, 3′ and not annotated ESTs was performed using EMBL release 85, Dec 2005. In order to associate identified antisense 5′ESTs and GenBank mRNAs to a representative transcript, we identified Aceview genes (which are assembled by clustering these sequences [Aceview version August 2005, http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/]). When multiple Aceview antisense genes were mapped close to each other, they were assigned to distinct Antisense Transcription Starting Regions (ATSRs) if their 5′ends were more than 200-bp apart. Otherwise they were classified as being part of the same ATSR. A detailed account of GenBank mRNAs and ESTs analyzed and of the results of Aceview filtering is shown in Supplementary Data ‘TableGlobal.xls’.
Calculating overrepresentation of antisense transcripts in genomic elements shown in Table 1
We evaluated the enrichment of ATSRs in each of the most represented genomic elements. We calculated the total amount of bases belonging to each genomic element both for the 2671 RefSeq genes having an ATSRs and the number of exons greater than three (N3bases) and for the whole dataset of non-redundant RefSeqs (18 008) analyzed (NFULLbases). Significance of enrichment was assessed using cumulative hypergeometric distribution.
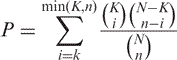 |
where K is the number of bases belonging to the considered genomic element; k is the number of ATSRs belonging to the considered genomic element; n is the total number of ATSRs and N is the total number of bases.
Run-length analysis of motif clusters
The expected number of GC-rich motifs clustering together without being interrupted by GC-poor motifs can be interpreted as the number of runs in n Bernoulli trials, where n is the sum of GC-rich and GC-poor motifs. The expected number of runs ≥R is given by NR = n × p (1 − p)R. Since the number of GC-rich motifs is the same as the number of GC-poor motifs, p = 0.5.
Consensus motifs
Motif searches were performed by matching IUPAC codes corresponding to known transcription regulatory motifs to the plus strand of repeat masked hg17 human genomic sequence. Searches were performed for each motif and its reverse complement counterpart. Transcription regulatory motifs were taken from the work of (26,38), as well as from repositories TRANSFAC (27) and JASPAR (28). Motifs reported as probabilistic models were converted to IUPAC consensus motifs in the following fashion: First, a weight matrix cutoff corresponding to a false-positive rate of 5% was determined as described by (32). Then, for each weight matrix, random motifs were generated using the nucleotide probabilities specified in the matrix as emission probabilities for each nucleotide in each motif position. After 500 motifs had been generated with a score beyond the 5% false-positive cutoff, a IUPAC consensus representing these 500 motifs was calculated. Since the consensus-generating algorithm is stochastic, the motifs generated can vary slightly from one run to the next. The full list of motifs is reported in the Supplementary Data.
Cell culture and luciferase assays
U-2 OS, T98G and HeLa cells were cultured in DMEM with 10% fetal bovine serum. Intron-derived genomic fragments were cloned from human genomic DNA (Roche) using Taq PCR core kit (Qiagen) with the primers specified in the Supplementary Data. Each primer contained a Nhe1 restriction site that was used to subclone the fragments into pGL3-basic luciferase reporter plasmid (Promega) in both orientations. Cells were grown in 12-well plates and transfected with the various luciferase reporter plasmids using FuGene6 reagent (Roche) according to the manufacturer's instructions. For normalization of transfection efficiency, cells were transfected with 5 ng of a renilla luciferase expressing vector. Luciferase assay was performed using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol.
Supplementary Material
ACKNOWLEDGEMENTS
The bioinformatics computation intensive tasks have been run on two HP multiprocessor Itanium servers granted to IFOM Bioinformatics Services by HP within the framework of the Integrity Scientific Program coordinated by Dr. Alessandro Guffanti. This work was supported by grants from AIRC (Italian Association of Cancer Research) and CARIPLO (Cassa di Risparmio della Regione Lombardia). Funding to pay the Open Access publication charge was provided by AIRC.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- 2.Rinn JL, Euskirchen G, Bertone P, Martone R, Luscombe NM, Hartman S, Harrison PM, Nelson FK, Miller P, et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003;17:529–540. doi: 10.1101/gad.1055203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 4.Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–342. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 6.Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 7.Lehner B, Williams G, Campbell RD, Sanderson CM. Antisense transcripts in the human genome. Trends Genet. 2002;18:63–65. doi: 10.1016/s0168-9525(02)02598-2. [DOI] [PubMed] [Google Scholar]
- 8.Shendure J, Church GM. Computational discovery of Sense–antisense transcription in the human and mouse genomes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-9-research0044. RESEARCH0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiyosawa H, Yamanaka I, Osato N, Kondo S, Hayashizaki Y. Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res. 2003;13:1324–1334. doi: 10.1101/gr.982903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, et al. Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 2003;21:379–386. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Sun M, Kent WJ, Huang X, Xie H, Wang W, Zhou G, Shi RZ, Rowley JD. Over 20% of human transcripts might form Sense–antisense pairs. Nucleic Acids Res. 2004;32:4812–4820. doi: 10.1093/nar/gkh818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 13.Ge X, Wu Q, Jung YC, Chen J, Wang SM. A large quantity of novel human antisense transcripts detected by LongSAGE. Bioinformatics. 2006;22:2475–2479. doi: 10.1093/bioinformatics/btl429. [DOI] [PubMed] [Google Scholar]
- 14.Li YY, Qin L, Guo ZM, Liu L, Xu H, Hao P, Su J, Shi Y, He WZ, et al. In silico discovery of human natural antisense transcripts. BMC Bioinformatics. 2006;7:18. doi: 10.1186/1471-2105-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Liu XS, Liu QR, Wei L. Genome-wide in silico identification and analysis of cis natural antisense transcripts (cis-NATs) in ten species. Nucleic Acids Res. 2006;34:3465–3475. doi: 10.1093/nar/gkl473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. U.S.A. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 18.Euskirchen G, Royce TE, Bertone P, Martone R, Rinn JL, Nelson FK, Sayward F, Luscombe NM, Miller P, et al. CREB binds to multiple loci on human chromosome 22. Mol. Cell. Biol. 2004;24:3804–3814. doi: 10.1128/MCB.24.9.3804-3814.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-3-reviews0004. REVIEWS0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavorgna G, Dahary D, Lehner B, Sorek R, Sanderson CM, Casari G. In search of antisense. Trends Biochem. Sci. 2004;29:88–94. doi: 10.1016/j.tibs.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 22.O'Neill MJ. The influence of non-coding RNAs on allele-specific gene expression in mammals. Hum. Mol. Genet. 2005;14:R113–R120. doi: 10.1093/hmg/ddi108. (Spec No 1) [DOI] [PubMed] [Google Scholar]
- 23.Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE. Human-mouse genome comparisons to locate regulatory sites. Nat. Genet. 2000;26:225–228. doi: 10.1038/79965. [DOI] [PubMed] [Google Scholar]
- 24.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 30.Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 31.Trinklein ND, Aldred SF, Hartman SJ, Schroeder DI, Otillar RP, Myers RM. An abundance of bidirectional promoters in the human genome. Genome Res. 2004;14:62–66. doi: 10.1101/gr.1982804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 34.Queen C, Baltimore D. Immunoglobulin gene transcription is activated by downstream sequence elements. Cell. 1983;33:741–748. doi: 10.1016/0092-8674(83)90016-8. [DOI] [PubMed] [Google Scholar]
- 35.Gillies SD, Morrison SL, Oi VT, Tonegawa S. A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell. 1983;33:717–728. doi: 10.1016/0092-8674(83)90014-4. [DOI] [PubMed] [Google Scholar]
- 36.David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM. A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keshet I, Schlesinger Y, Farkash S, Rand E, Hecht M, Segal E, Pikarski E, Young RA, Niveleau A, et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 2006;38:149–153. doi: 10.1038/ng1719. [DOI] [PubMed] [Google Scholar]
- 38.Vernell R, Helin K, Muller H. Identification of target genes of the p16INK4A-pRB-E2F pathway. J. Biol. Chem. 2003;278:46124–46137. doi: 10.1074/jbc.M304930200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.