


Abstract
The environmental carcinogen benzo[a]pyrene (BP) is metabolized to reactive diol epoxides that bind to cellular DNA by predominantly forming N2-guanine adducts (G*). Mutation hotspots for these adducts are frequently found in 5′- ··· GG ··· dinucleotide sequences, but their origins are poorly understood. Here we used high resolution NMR and molecular dynamics simulations to investigate differences in G* adduct conformations in 5′- ··· CG*GC ··· and 5′- ··· CGG* C··· sequence contexts in otherwise identical 12-mer duplexes. The BP rings are positioned 5′ along the modified strand in the minor groove in both cases. However, subtle orientational differences cause strong distinctions in structural distortions of the DNA duplexes, because the exocyclic amino groups of flanking guanines on both strands compete for space with the BP rings in the minor groove, acting as guideposts for placement of the BP. In the 5′- ··· CGG* C ··· case, the 5′-flanking G · C base pair is severely untwisted, concomitant with a bend deduced from electrophoretic mobility. In the 5′- ··· CG*GC ··· context, there is no untwisting, but there is significant destabilization of the 5′-flanking Watson–Crick base pair. The minor groove width opens near the lesion in both cases, but more for 5′- ··· CGG*C···. Differential sequence-dependent removal rates of this lesion result and may contribute to the mutation hotspot phenomenon.
INTRODUCTION
Polycyclic aromatic hydrocarbons (PAHs) are products of combustion of organic matter and are therefore ubiquitous in the environment, and have long been associated with induced human cancers (1,2). Benzo[a]pyrene (BP) is the most widely studied and representative compound of this class of chemical carcinogens (3). While BP is relatively inert, it is metabolically activated to highly reactive and genotoxic diol epoxide derivatives (4) that bind chemically to DNA and form mutagenic adducts (5). These DNA lesions have been correlated with the initiation of animal and human cancers (6). Among the most active and tumorigenic metabolites of BP is the (+)-7R,8S,9S,10R enantiomer of 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene ((+)-anti-BPDE); this metabolite binds predominantly to the exocyclic amino group of guanine in DNA to form the 10S (+)-trans-anti-[BP]-N2-dG adduct shown in Figure 1 (7). If DNA adducts are not excised by cellular repair mechanisms, they can persist until replication occurs and cause mutations. Multiple DNA mutations in critically important genes such as ras and p53 constitute genetic alterations that play key roles in the regulation of cell cycle control and cancer (1).
Figure 1.

(A) Chemical structure of the 10S (+)-trans-anti-[BP]-N2-dG adduct. Torsion angles α′ and β′ are defined as follows: α′, N1–C2–N2–C10(BP); β′, C2–N2–C10(BP)–C9(BP). The glycosidic torsion angle is denoted by χ, defined as O4′-C1′-N9-C4. (B) The sequences and numbering system of the 12-mer duplexes containing the 10S (+)-trans-anti-[BP]-N2-dG adduct. The lesion sites are marked G6* and G7*. The sequences are referred to as G6*G7 and G6G7*.
The diol epoxide (+)-anti-BPDE is highly mutagenic (8) and tumorigenic (9,10). Mutation hotspots associated with adduct formation derived from the reactions of BPDE with DNA are frequently found in runs of guanines, especially in GG sequence contexts (11–14). Examples of GG mutation hotspot phenomena include mutations in plasmids containing human c-Ha-ras1 sequences, reacted with anti-BPDE and transfected into mouse embryonic NIH 3T3 cells (15) in the coding region of the BPDE-modified HPRT gene in mammalian V-79 cells (14), and in the SupF gene of an E. coli plasmid (13). The mutational specificity and the mutation frequencies depend not only on base sequence contexts (5), but also on the nature of the host or the polymerases bypassing the lesion (16). Furthermore, in cellular environments, nucleotide excision repair of these bulky lesions may also depend on base sequence contexts and thus contribute to the mutation hotspot phenomena, by efficiently excising lesions in some sequence contexts but not in others. However, the structural basis of base sequence effects on mutagenesis and DNA repair is poorly understood.
Although the conformations of 10S (+)-trans-anti-[BP]-N2-dG adducts in double-stranded oligonucleotides have been studied by NMR methods (17,18), previous emphasis has been on understanding stereochemical rather than base sequence effects. In this work, we have used high resolution NMR methods to investigate the conformational properties of the mutagenic 10S (+)-trans-anti-[BP]-N2-dG adduct (Figure 1A) at either of the two guanines in a GG sequence context in a double-stranded oligonucleotide (Figure 1B). The central 6-mer sequence of this duplex contains the SupF mutation hotspot (13) 5′- ··· GCGGCC ··· with the BP lesion positioned either at the first (5′-side) or the second G (3′-side). In both sequence contexts, the BP rings are positioned 5′-directed along the modified strand in the minor groove (17,19), but with subtle differences in orientations which produce markedly different local duplex distortions. These distinctions arise from differential steric hindrance from the exocyclic amino groups of the neighboring undamaged guanine bases that compete with the bulky aromatic BP ring systems for space in the minor groove. This leads to significant differences in local helical untwisting, minor groove width opening near the lesion site, and weakening of local Watson–Crick base pairs. The unusual flexible bend observed in duplexes with the 10S (+)-trans-anti-[BP]-N2-dG adduct positioned only on the 3′-G in the 5′- ··· GG ··· sequence context (20) is consistent with severe local untwisting found only for this sequence. The pronounced differences in the local structural distortions affect the processing of the lesions in GG mutation hotspots by cellular enzymes.
MATERIALS AND METHODS
The methods of preparing of single-stranded G6*G7 and G6G7* sequences with the 10S (+)-trans-anti-[BP]-N2-dG lesions at G6* and G7*, respectively, have been described previously (21–23), and a summary is provided in Supplementary Data.
NMR measurements
The 2D nuclear overhauser effect spectroscopy (NOESY) spectra of both the G6*G7 and G6G7* duplexes (Figure 1B) in 2H2O phosphate buffer solution at 15°C were recorded at mixing times of 50, 100, 150 and 200 ms using a Bruker 800 MHz (for the G6*G7 sample) and a 600 MHz (for G6G7* sample) NMR spectrometers, equipped with cryoprobes, at the New York Structural Biology Center (NYSBC). The NOESY experiments in H2O at mixing times of 90, 175 and 250 ms at 5°C to visualize the imino protons were conducted using a Bruker 500 MHz NMR instrument at New York University. Full details are provided in Supplementary Data.
Molecular dynamics computations and intensity refinement
Starting models
Initial models were created from a high resolution NMR solution structure for the 10S (+)-trans-anti-[BP]-N2-dG adduct (17). We remodeled the base sequence to the two sequences listed in Figure 1B, using INSIGHT II 97.0 (Accelrys, Inc.).
Force field
MD simulations and intensity refinement against the NOE distances and volumes were carried out using SANDER in the AMBER 8.0 simulation package (University of San Francisco), the Cornell et al. force field (24), with the parm99.dat parameter set (25). Partial charges and all parameters for the 10S (+)-trans-anti-[BP]-N2-dG adduct employed in this work were computed previously (26).
Molecular dynamics computation protocols and best representative structures
Details of the molecular dynamics protocols and the computation of the best representative structure, using the cluster analyses option in MOIL-View (27) are provided in Supplementary Data.
NMR intensity refinement and structural analyses
The refinement started with the best representative structure of the final 1.5 ns unrestrained MD simulation. This structure was subjected to distance-restrained, and subsequently, intensity-restrained molecular dynamics computations in order to elucidate the solution structures of the 10S (+)-trans-anti-[BP]-N2-dG adduct in the G6*G7 and G6G7* sequence contexts. The SANDER module in AMBER 8.0 was employed. Details of the structural analyses are provided in Supplementary Data.
RESULTS
Exchangeable proton spectra
The G6*G7 duplex
The exchangeable 1D proton NMR spectrum (12.0–14.0 ppm) of the G6*G7 duplex (sequence context shown in Figure 1B) in H2O buffer solution (pH = 6.8) at 5°C is plotted in Figure 2A. The imino protons were assigned via their NOE connectivities to the amino protons of the hydrogen bond partner and the flanking base pairs, as described in earlier publications (17,28–31). The observed distinguishable imino proton resonances indicate a single adduct conformation. An expanded contour plot of the NOESY spectrum in H2O solution determined at 250 ms mixing time at 5°C is shown in Figure 2B. NOEs between deoxyguanosine imino and cytosine amino protons are characteristic of a G · C Watson–Crick basepair (12.4–13.1 ppm), while NOEs between thymidine imino and adenine H2 protons are characteristic of an A · T Watson–Crick base pair (13.4–13.9 ppm). The observed NOE pattern establishes Watson–Crick pairing for G · C pairs (peaks labeled A to C) and A · T pairs (peaks labeled E and F), including the G6* · C19 base pair, in the 12-mer duplex. However, somewhat weaker NOE cross-peaks are observed at the lesioned G6* · C19 base pair and the 5′-flanking C5* · G20 base pair. Imino–imino proton sequential connectivities can be traced throughout the duplex except at G20(NH1)–G6*(NH1), G7(NH1)–G17(NH1) and G17(NH1)–G16(NH1). Some of these connectivities cannot be observed, presumably because they are too close to the diagonal, while imino protons at the terminal base pairs are difficult to observe due to fraying effects. None of the imino proton resonances are upfield shifted (Figure 2A), which indicates that the aromatic BP ring system is not intercalated between adjacent base pairs in the duplex (28,29) (Figure 2C). An observable BP(H8)–G6*(H1) cross-peak is also observed in this region of the spectrum (labeled D in Figure 2B).
Figure 2.

NMR spectra of the G6*G7 (A–C) and G6G7* (D–F) duplexes. (A) G6*G7 imino proton spectrum (12.0–14.0 ppm) in H2O buffer solution at 5°C. The imino proton assignments are also shown. (B) G6*G7 expanded NOESY (250 ms mixing time) contour plot in H2O buffer solution at 5°C. The NOE connectivities between imino protons of all guanines and amino protons of cytosines across the G · C base pairs and thymidine imino to adenine H2 protons in a T · A base pair. Cross-peaks A–H are assigned as follow: A1: G6*(H1)–C19(NH2 non-bonded H atom); A2: G6*(H1)–C19(NH2-bonded H); A3: G6*(H1)–G6*(NH2); B1: G20(H1)–C5(NH2-non-bonded H); B2: G20(H1)–C5(NH2-bonded H); C1: G7(H1)–C18(NH2-non-bonded H); C2: G7(H1)–C18(NH2-bonded H); D: BP(H8)–G6*(H1); E: T3(H3)–A22(H2); F: T23(H3)–A2(H2); G: T14(H3)–A11(H2); H: T10(H3)–A15(H2). (C) G6*G7 NOE 250 ms mixing time contour 2D plot showing distance connectivities between imino-to-imino protons in the duplex. The labels are, I: G13(H1)–T14(H3); J: G4(H1)–T3(H3); K: G6*(H1)–G7(H1); L: G4(H1)–G20(H1); M: T3(H3)–T23(H3); N: G16(H1)–T10(H3). (D) G6G7* Imino proton spectrum (12.0–14.0 ppm) in H2O buffer solution at 5°C. (E) G6G7* expanded NOESY (250 ms mixing time) 2D contour plot in H2O buffer solution at 5°C. The NOE connectivities between imino protons of all guanines and amino protons of cytosines across the G · C base pairs and thymidine imino to adenine H2 protons in T · A base pairs are shown. Cross-peaks A–H are assigned as follows: A1: G7*(NH)–C18(NH2 non-bonded H); A2: G7*(NH)–C18(NH2-bonded H); A3: G7*(NH)–G7*(NH2); B1: G6(NH)–C19(NH2 non-bonded H); B2: G6(NH)–C19(NH2-bonded H); C1: G17(NH)–C8(NH2 non-bonded H); C2: G17(NH)–C8 (NH2-bonded H); D: BP(H8)–G7*(H1); E: T3(H3)–A22(H2); F: T14(H3)–A11(H2); G: T23(H3)–A2(H2); H: T8(H3)–A15(H2). (F) G6G7* NOE 250 ms mixing time 2D contour plot showing distance connectivities between imino-to-imino protons in the G6G7* duplex. Peak labels I–N are defined as I: T3(H3)–T23(H3); J: G16(H1)–T10(H3); K: G4(H1)–T3(H3); L: G6(H1)–G20(H1); M: G16(H1)–G17(H1); N: G4(H1)–G20(H1).
The G6G7* duplex
The 1D imino proton NMR spectrum (12.0–14.0 ppm) in H2O buffer of the G6G7* duplex, together with the assignments, is depicted in Figure 2D. The imino proton resonances are distinguishable and establish the presence of a single conformation in this duplex. An expanded contour plot of the NOESY spectrum (250 ms mixing time) in H2O buffer at 5°C is shown in Figure 2E. Characteristic interstrand Watson–Crick NOE cross-peaks are observed between guanine imino and cytosine amino protons of G · C base pairs (peaks labeled A to C), and thymine imino and adenine H2 protons (peaks labeled E to H). Although Watson–Crick hydrogen bonding is observed at the modified base pair (peak A in Figure 2E) and the two flanking base pairs G6 · C19 (peak B) and C8 · G17 (peak C), the NOE at the lesion site (G7* · C18), is relatively weak (peak A) when compared to the NOE connectivities between other base pairs. This implies that hydrogen bonding at the lesion site is weakened. A similar conclusion can be drawn for the 5′-flanking G6 · C19 base pair where hydrogen bonding is also diminished. On the other hand, the amino–imino NOE cross-peak for the C8 · G17 pair is quite strong (peak C), most likely reflecting a unidirectional orientation of the BP ring system. Imino–imino proton sequential assignments can be traced for the entire duplex except at the G6(NH1)-G7*(NH1), G7*(NH1)-G17(NH1) and the terminal base pairs. None of the imino proton resonances are upfield shifted (Figure 2D) suggesting that the aromatic BP ring system is not intercalated between adjacent base pairs in this duplex.
Temperature dependence of imino proton resonances in G6*G7 and G6G7* duplexes
Imino proton chemical shifts and linewidth broadening of the central three base pair segments around the lesion as a function of temperature show interesting differences between the two adducts (Figure S1, Supplementary Data). Increases in linewidths of the imino protons reflect enhanced solvent exposure, and thus increased exchange rates with solvent water molecules. These phenomena are therefore indicators of the relative stability of the local duplex environment (32). As the temperature is increased from 5 to 20°C, there is a pronounced broadening of the imino proton resonances of G6*, G7 and G20; the latter two bases belong to base pairs flanking G6* · C19 in the G6*G7 duplex. However, in the case of the G6G7* duplex, the changes in the linewidths of imino protons of G7*, and neighboring bases G6 and G17 are less pronounced (Figure S1A, Supplementary Data).
Upfield chemical shift changes of imino protons are attributed to a premelting conformational transition associated with increased duplex opening rates (32). We have examined the chemical shift values of the imino protons at the lesion site and the two adjacent G · C base pairs as a function of temperature (Figure S1B, Supplementary Data). In the case of the G6G7* duplex, rather small and monotonous upfield chemical shifts of the imino proton resonances are observed at the site of the G7* lesion and the two flanking base pairs as the temperature increases from 5 to 45°C (Figure S1B, Supplementary Data). However, in the G6*G7 duplex, pronounced upfield chemical shift changes occur at the 5′-side of G6*, i.e. at the C5 · G20 base pair and, to a lesser extent, at the G7 · C18 base pair flanking the lesion on both sides in the G6*G7 duplex (Figure S1B, Supplementary Data). All three base pairs, C5 · G20, G6* · C19, and G7 · C18, appear to premelt at temperatures of ∼20–30°C, well below the global melting point, Tm, of either of the two duplexes determined from measurements of the UV absorbance as a function of temperature (Figure S1C, Supplementary Data). When the absorbance is monitored at the DNA absorption maximum of 260 nm, the melting curves are cooperative and the Tm values are 57 ± 1 and 55 ± 1°C for the G6G7* and G6*G7 duplexes, respectively. When the temperature-dependent absorbance is monitored at 346 nm, corresponding to the absorption maximum of the aromatic BP rings, the local premelting of the duplexes in the immediate vicinity of the lesions can be monitored. The local Tm is 50 ± 1°C in both duplexes, which is significantly lower than the global melting point for the entire duplex measured at 260 nm. It is evident that the region of the duplex in the immediate vicinity of the lesions is destabilized by the 10S (+)-trans-anti-[BP]-N2-dG adducts, causing local premelting of the double-stranded DNA.
The pronounced upfield shift changes of the G20(H1), G6*(H1) and G7(H1) imino protons in the G6*G7 duplex suggest that significant local changes occur at temperatures much lower than the global Tm, i.e. between 20 and 30°C. Changes in local duplex structure associated with increased opening rates occur at these relatively low temperatures. The lack of similar changes in linewidths and chemical shifts of the analogous base pairs in the G6G7* duplex, indicates that the G6 · C19, G7* · C18, and C8 · G17 are more stable in the same 20–30°C temperature interval (Figure S1A and B, Supplementary Data).
Non-exchangeable proton spectra
The G6*G7 duplex
A portion of the 2D NOESY contour plot (mixing time 250 ms) of the G6*G7 duplex in 2H2O buffer solution (pH = 6.8) at 15°C is plotted in Figure 3A. The characteristic NOE patterns between base protons and their own and 5′-flanking base sugar H1′ protons for a right-handed B-DNA duplex (33) are traced for the C5–G6*–G7–C8 segment of the modified strand (solid lines) and for the G17–C18–C19–G20–C21–A22 segment of the non-modified strand (broken lines). Internucleotide base to sugar NOEs could not be identified unequivocally in the G20–C21 segment of the complementary strand because of severe overlap of the C19(H6), G20(H8) and T23(H6) base protons. We tentatively assigned the G20(H1′) and G20(H3′) sugar protons at 4.18 and 3.52 ppm, respectively. These protons exhibit NOE cross-peaks to the G20(H2′) and G20(H2″) sugar protons. Large upfield shifts of the base (6.77 ppm) and sugar H1′ (4.55 ppm) proton of C21, as well as similar upfield shifts for all other sugar protons of G20 and C21 (Figure 3B), indicate that the aromatic BP ring system of G6* is in contact with the G20–C21 sugar rings, and is therefore positioned in the minor groove. These results also indicate that the aromatic BP ring system is oriented toward the 5′-end of the modified strand in the G6*G7 duplex. The aromatic BP protons, assigned on the basis of comparisons of NOESY and TOCSY patterns for these protons and their chemical shifts, are shown in Table 1.
Figure 3.

(A) G6*G7 expanded NOESY (250 ms mixing time) 2D contour plot in 2H2O buffer at 15°C using a Bruker 800 MHz spectrometer. NOE connectivities are shown between a base (purine H8 and pyrimidine H6) proton and their own and 5′-flanking sugar H1′ protons from dC5 to dC8 on the modified strand (solid arrows) and from dG17 to dA22 on the non-modified strand (broken arrows). The NOEs between the 2.45 Å fixed distance H6 and H5 of the cytosine are designated by crosses. Internucleotide base to sugar NOEs, at the non-modified strand, were not identified clearly for G20 → C21 as a result of the overlap of the C19–H6, G20–H8 and T23–H6 protons. There is a tentative assignment of the G20(H1′) and G20(H3′) sugar protons at 4.18 and 3.52 ppm, respectively through NOEs between theses protons and the G20(H2′) and G20(H2″) sugar protons. A cross-peak between the BP(H11)–G6*(H1′) is observed (square box). (B) Graphical representations of the chemical shift perturbations in the G6*G7 duplex relative to the unmodified control 12-mer duplex. Positive values indicate upfield shifts, whereas negative values indicate downfield shifts. (C) NOE cross-peaks between C21(H4′) and BP(H1–H3–H4–H2) protons and between G20(H4′) and BP(H6–H4/5) protons.
Table 1.
Comparison of experimental distance restraints with those observed for the NMR energy-minimized structures of the G6*G7 and G6G7* duplexesa
| BP proton | G6*G7 | G6G7* | ||||||
|---|---|---|---|---|---|---|---|---|
| Restraints | Restraints | |||||||
| Interproton distances (Å) | Interproton distances (Å) | |||||||
| BP chemical shift (ppm) | Experimental bounds | Observed | BP chemical shift (ppm) | Experimental bounds | Observed | |||
| H1 | 8.3 | C21H4′ | 2.8–3.6 | 3.5 (0.3) | 8.1 | G20H4′ | 2.0–4.0 | 3.9 (0.4) |
| H2 | 8.1 | C21H4′ | 2.7–3.4 | 2.9 (0.3) | 8.3 | G20H4′ | 2.7–3.6 | 2.9 (0.3) |
| H3 | 8.3 | C21H4′ | 4.2–4.6 | 3.6 (0.4) | 8.2 | G20H4′ | 2.8–5.1 | 3.4 (0.3) |
| H4 | 8.2 | C21H4′ | 3.7–6.5 | 5.1 (0.3) | 8.3 | – | – | – |
| G20H4′ | 3.5–4.1 | 4.1 (0.2) | ||||||
| H5 | 8.2 | G20H4′ | 3.6–3.4 | 2.6 (0.2) | 8.3 | C19H4′ | 2.1–3.6 | 2.6 (0.2) |
| C19H3′ | 3.8–7.0 | 5.3 (0.3) | ||||||
| H6 | 8.4 | G20H4′ | 3.2–4.4 | 2.9 (0.2) | 8.3 | C18H2″ | 4.6–8.1 | 7.5 (0.3) |
| C18H1′ | 4.4–4.9 | 4.9 (0.2) | ||||||
| H7 | 5.0 | – | – | – | 5.0 | – | – | – |
| H8 | 4.5 | G7H1′ | 4.0–4.7 | 3.9 (0.3) | 4.6 | C8H1′ | 3.2–4.3 | 4.2 (0.3) |
| H9 | 4.5 | G7H1′ | 2.4–3.0 | 2.2 (0.2) | 4.5 | C8H1′ | 2.8–3.1 | 2.7 (0.4) |
| C8H6 | 3.9–5.2 | 4.8 (0.2) | ||||||
| G7H8 | 4.6–7.6 | 6.8 (0.2) | ||||||
| G7H1′ | 3.1–3.9 | 3.8 (0.2) | ||||||
| H10 | 6.2 | G7H1′ | 3.8–5.2 | 4.4 (0.3) | 6.1 | G7H1′ | 3.5–3.8 | 3.8 (0.2) |
| G6H1′ | 3.2–4.0 | 3.7 (0.1) | ||||||
| H11 | 8.4 | G6H1′ | 4.0–4.4 | 4.1 (0.3) | 8.2 | G7H1′ | 3.9–5.2 | 4.8 (0.3) |
| H12 | 8.3 | – | – | – | 8.3 | – | – | – |
aStandard deviations are given in parentheses.
The G6G7* duplex
Representative 2D NOESY spectra of the G6G7* duplex (mixing time 250 ms) in 2H2O buffer solution (pH = 6.8) at 15°C are shown in Figure 4A. The NOE connectivities between base protons and their own and 5′-flanking H1′ sugar protons are traced for the C5–G6–G7*–C8 segment of the modified strand (solid lines), and for the G17–C18–C19–G20–C21–A22 segment of the unmodified strand (dashed lines). All connectivities are present, confirming that the G6G7* duplex is consistent with a B-DNA helix conformation. The upfield shift observed for the C19 base proton (6.71 ppm) and its sugar H1′ proton (3.87 ppm) indicate that the aromatic BP ring stacks mainly over the sugar ring of the C19 residue of the G6 · C19 base pair flanking the lesion G7* on the 5′-side of the modified strand. The BP aromatic ring system extends less toward the G20 sugar protons, since smaller upfield chemical shifts are observed for this base than for C19 (Figure 4B). The aromatic BP protons were assigned on the basis of comparisons of NOESY and TOCSY patterns for the indicated protons, and their chemical shift values are shown in Table 1.
Figure 4.

(A) G6G7* expanded NOESY (250 ms mixing time) 2D contour plot in 2H2O buffer at 15°C using a Bruker 600 MHz spectrometer with a cryoprobe. The NOE connectivities between base protons (purine H8 and pyrimidine H6) and their own and 5′-flanking sugar H1′ protons from C5 to C8 on the modified strand (solid arrows), and from G17 to A22 on the unmodified complementary strand (broken arrows) are shown in the figure. The NOEs between the 2.45 Å fixed-distance H6 and H5 of the cytosine are designated by crosses. Connectivities can be followed on both strands without disruption. Cross-peaks between BP–BP and BP–DNA protons are also shown. The upfield shift of the base and sugar protons at C19 and the corresponding cross-peaks between the BP protons with the C19 and G20 protons suggest that the BP rings is in the minor groove with its major axis pointing toward the 5′-end of the modified strand. (B) Graphical representations of the chemical shift perturbations in the G6G7* duplex relative to the unmodified control 12-mer duplex. Positive values indicate upfield shifts, whereas negative values indicate downfield shifts. (C) NOE connectivities G7*(H1′)–BP(H9), BP(H9)–BP(H10), BP(H8)–BP(H10), BP(H9)–C8(H1′) and BP(H8)–C8(H1′).
Comparisons of nucleic acid chemical shifts in the G6*G7 and G6G7* duplexes
In order to compare the effects of the aromatic BP rings on chemical shifts of DNA proton resonances, plots of chemical shifts, relative to those of the unmodified control duplexes, are presented in Figures 3B and 4B. The relative chemical shifts for sugar protons H1′, H2′, H2″ and H3′, and H6/H8 base protons are generally unperturbed except in the vicinity of the lesion (C5–G7), and up to three bases on the 5′-side of the lesion in the complementary strand. These observations are consistent with a 5′-orientation of the BP moiety in both the G6*G7 and G6G7* duplexes, with the aromatic ring system contacting mostly sugar protons of the complementary strand in the minor groove (17). However, there are significant differences. In the case of the G6G7* duplex, the largest upfield shifts, caused by aromatic ring current effects associated with the BP rings, are observed at the sugar protons of the 5′-adjacent base C19 (Figure 4B). However, in the case of G6*G7, the effects of the BP rings extend to protons on the 5′-flanking G20 and C21 in the complementary strand, up to two bases from the modification site (G6*) (Figure 3B). These results suggest that the long axes of the BP aromatic ring systems in the minor grooves are oriented differently in the G6*G7 and G6G7* duplexes.
BP–DNA proton NOE cross-peaks
We have identified and assigned all BP aliphatic and aromatic protons in both 12-mer duplexes. A total of 12 NOE cross-peaks between the BP rings and DNA protons were identified for the G6*G7 sample. Several of the NOEs between minor groove sugar protons and the BP rings are shown in Figure 3A and C for the G6*G7 duplex. The intermolecular NOE connectivities involve protons on both the modified and the unmodified strands. In the case of the G6G7* duplex, a total of 14 intermolecular NOE cross-peaks between the BP rings and DNA protons were identified. Several of these intermolecular NOEs are shown in Figure 4A and C, and involve sugar and BP aromatic ring protons. A list of all of the BP–DNA intermolecular NOE cross-peaks for the G6*G7 and the G6G7* duplexes are summarized in Table 1. These intermolecular connectivities provide further evidence that the BP aromatic ring systems are positioned in the minor groove in both duplexes and are directed toward the 5′-side of the modified strand. The results are fully consistent with the chemical shift data (Figures 3B and 4B) showing that the BP aromatic ring system stacks predominantly over the G20 and C21 sugar rings in the G6*G7 duplex, and over the C19 sugar ring in the case of the G6G7* duplex.
Determination of the conformations of the 10S (+)-trans-anti-[BP]-N2-dG adducts in the G6*G7 and G6G7* duplexes
NMR distance and intensity-restrained molecular dynamics computations were employed to define the solution structures of the 10S (+)-trans-anti-[BP]-N2-dG adducts in the G6*G7 and G6G7* sequence contexts at the 12-mer duplex level. Details concerning the initial models and molecular dynamics computation protocols for unrestrained and distance and intensity-restrained MD simulations are given in the Materials and Methods section. The restraints and refinement statistics are listed in Table 2.
Table 2.
NMR refinement statistics for the 10S (+)-trans-anti-[BP]-N2-dG adduct in the G6*G7 and G6G7* sequence contexts
| A. NMR distance restraints | G6*G7 | G6G7* | ||
| Total non-exchangeable distance restraints | 144 | 246 | ||
| Hydrogen bond restraints | 32 | 32 | ||
| Total number of carcinogen intramolecular distance restraints | 9 | 12 | ||
| Total number of carcinogen intermolecular distance restraints | 12 | 14 | ||
| B. Structural statistics of the adduct NMR violations | ||||
| Number >0.2 Å | 0 | 0 | ||
| Maximum violations (Å) | 0.1 | 0.1 | ||
| The sixth root R factor Rx for different mixing timesa | ||||
| Mixing time (ms) | 60 | 135 | 175 | 250 |
| G6*G7 | 0 | 0.12 | 0.12 | 0.17 |
| G6G7* | 0 | 0.17 | 0.13 | 0.12 |
a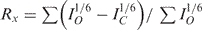 .
.
Solution structures
Stick views of five superpositioned intensity-refined structures of the 10S (+)-trans-anti-[BP]-N2-dG adducts in G6*G7 and G6G7* sequence contexts are shown in Figure 5. The views shown are into the minor grooves of the G6*G7 (Figure 5A) and the G6G7* duplexes (Figure 5B). In both cases, the BP moiety is in the B-DNA minor groove, 5′-directed along the modified strand. However, there are subtle differences in the orientations of the aromatic BP rings within the minor groove of the DNA. Figure 6 shows the origin of this phenomenon. In each case, the BP rings are positioned to avoid collisions with adjacent exocyclic amino groups of the flanking guanine bases that are protruding into the minor groove. In the case of the G6*G7 duplex, the relevant amino groups are those belonging to G7 on the 3′-side of the lesion G6* and positioned on the same strand, and G20 on the complementary strand. These two amino groups are in contact with the aliphatic ring and the aromatic ring system of BP, respectively (Figure 6A). In G6G7*, the relevant amino groups are those belonging to G6 on the 5′-side to the lesion G7* and positioned on the same strand, and G17 on the complementary strand. These two amino groups are also in contact with the aromatic ring and the aliphatic ring system of BP, respectively. The difference between the two cases is that the amino group of G6 flanking the modified guanine G7* is on the same strand and on the 5′-side of G7* (Figure 6B), while in the G6*G7 duplex the 5′-flanking amino group is part of G20 on the complementary strand (Figure 6A). These variations in positions of the amino groups relative to the modified guanines produce steric hindrance differences; subtle orientational differences result, manifested by distinctions in the distribution of the torsion angles at the carcinogen–DNA linkage site α′, β′ and the glycosidic torsion angles χ (Figure 7A and Table 3). As a result, the G6*G7 and G6G7* duplexes have distinct structural properties.
Figure 5.

View into the minor groove of superpositioned intensity-refined structures of the (A) G6*G7 and (B) G6G7* sequence contexts at the 12-mer duplex level. The structures shown are the five best representative conformations for each sequence context from the final 1 ps of unrestrained MD simulation after intensity refinement. The BP rings are in yellow, the modified base guanine and its partner cytosine are in cyan, and the rest of the DNA is in white except for the phosphorus atoms, which are colored red.
Figure 6.

Effects of exocyclic amino groups of guanines on the positioning of the 10S (+)-trans-anti-[BP]-N2-dG adduct in (A) G6*G7, (B) G6G7* and (C) CG*C (17,26) sequence contexts in double-stranded DNA. Only the central 7-mers are shown. The BP moiety and relevant guanine amino groups are in CPK representation. The color scheme is the same as in Figure 5. In addition, relevant amino group nitrogen atoms are shown in purple. Hydrogen atoms in the DNA duplexes, except in the relevant amino groups, are not displayed.
Figure 7.

Differences in structural parameters in G6*G7 and G6G7* duplexes. (A) Distributions of the carcinogen–DNA linkage site and glycosidic torsion angles α′, β′ and χ for the G6*G7 (red), and G6G7* (blue) duplexes. The analysis was based on each frame of the final 1 ps unrestrained MD simulations after intensity refinement. The indicated values are ensemble averages. (B) Minor groove widths of the modified duplexes. The definitions of the minor groove widths for the G6G7 12-mer duplexes is illustrated above the figure. The minor groove widths are the distance between phosphate groups P5 and P24, P6 and P23, P7 and P22, etc. The values shown were obtained after subtracting 5.8 Å from the pairwise distances to account for the van der Waals radius of the P atoms (34). The minor groove widths of the unmodified control were obtained from an unrestrained 3.0 ns simulation of the unmodified sequence (Cai et al., unpublished data), using the last 1.5 ns of production. (C) Computed ensemble average of Twist angles for the G6G7* and the G6G7* duplexes. The numbering scheme for the nucleotide base pair steps is that C1 · G24 to A2 · T23 is step 1, A2 · T23 to T3 · A22 is step 2, … and so on. The minor groove widths and Twist angles were calculated using MD Toolchest (35).
Table 3.
Carcinogen–DNA linkage site and glycosidic bond torsion angles
| Torsion angles | G6*G7 | G6G7* | CG*Ca |
|---|---|---|---|
| α′ | 173° ± 8° | 175° ± 7° | 176° ± 8° |
| ß′ | 269° ± 10° | 279° ± 8° | 263° ± 13° |
| χ | 248° ± 9° | 266° ± 7° | 253° ± 14° |
Most notably, there are significant differences in the Twist angles flanking the modified guanines in the G6*G7 and G6G7* duplexes (Figure 7B). In the G6*G7 duplex, the Twist angle varies within the narrow range of 29–32° from base pair A2 · T23 to base pair T10 · A15. However, in the case of the G6G7* duplex, there is a significantly larger Twist (40°) at the modified base pair G7* · C18, and a strikingly lower Twist angle of only 13° at the 5′-flanking base pair G6 · C19 (Figure 7B). These results indicate that there is a significant impact of guanine G6 flanking the modified guanine G7* on the 5′-side, since the base pair G7* · C18 is over-twisted while the 5′-flanking unmodified G6 · C19 base pair is severely undertwisted. Undertwisting is accompanied by increased Roll (Figure S2, Supplementary Data). In addition, there are differences in the minor groove dimensions near the lesion (Figure 7C), with average values of 10.6 Å for P7–P22 (defined in Figure 7C) in the G6*G7 sequence context, and 12.3 Å for P8–P21 in the G6G7* duplex.
The amino groups governing the position of the BP moiety in the minor groove facilitate extra hydrogen bonding between the hydroxyl groups of the BP benzylic ring and O2 atoms of cytosines in the immediate vicinity of the lesion; this is seen in both the G6*G7 and the G6G7* sequence contexts in the computed structures (Figure S3, Supplementary Data). These cytosines are either partner bases to the modified guanines, or their 3′ side-flanking neighbors. In the case of G6*G7, there is one hydrogen bond between the HO7 and O2 of C19, the partner to the modified G6*. (Figure S3A, Supplementary Data). In the G6G7* case, there are two hydrogen bonds: one between the HO7 of the benzylic ring and O2 of C18, the partner base to the modified guanine, G7*; the other is between the HO8 and O2 of C8, on the 3′-side of G7* (Figure S3B, Supplementary Data). These intermolecular hydrogen bonds between the benzylic ring and cytosines further anchor the BP moiety in the position governed by the guanine amino groups. Thus the differential orientation of the BP in the different sequence contexts is dictated by the competing amino groups in the minor groove, with possible additional stability derived from hydrogen-bonding interactions of the hydroxyl groups on the BP benzylic ring with keto groups of neighboring cytosines.
DISCUSSION
Steric competition in minor groove produces sequence-governed conformational differences
The present study provides detailed insights into the structures of two BP–DNA adducts that have guanine bases flanking the modified G*, on either the 3′-side or the 5′-side. The exocyclic amino groups on the modified and complementary strands in the immediate vicinity of the adducts protrude into the minor groove and strongly affect the structural properties of the 10S (+)-trans-anti-[BP]-N2-dG adduct conformations in the minor groove. Like the structures in the CG*C sequence context studied previously by Cosman et al. (17) and in a TG*C sequence investigated by Fountain and Krugh (19), the G6*G7 and G6G7* duplexes exhibit 5′-directed BP minor groove conformations. However, the orientations of the BP rings are subtly different (Figure 5). In both the G6*G7 and G6G7* duplexes, the exocyclic amino groups on the 3′-side contact the aliphatic ring of BP, while the amino group on the 5′-side contacts the aromatic ring system. Yet, different steric hindrance effects by the unmodified guanine amino groups combine to produce distinct positioning of the BP moiety in each case. This is evident from the intermolecular NOE patterns (Table 1). The large upfield chemical shifts due to stacking of the BP rings over the sugar protons of the two bases G20 and C21 in the G6*G7 duplex (Figure 3B), and predominantly over one complementary base C19 only, in the G6G7* duplex (Figure 4B), is evidence for the different orientations of the BP rings in these duplexes. These differences are illustrated schematically in Figure S4, Supplementary Data. The chemical shifts of the aliphatic and aromatic BP ring protons are not sensitive to their interactions with the different sugar protons in the G6*G7 and G6G7* duplexes, which is also consistent with minor groove conformations (17,19) (Figure S5, Supplementary Data). Intercalative structures are characterized by significant upfield shifts of the BP aromatic ring protons due to base stacking interactions (36).
We compared the conformations of the BP residues in the G6*G7 and G6G7* duplexes with the structure of the same 10S (+)-trans-anti-[BP]-N2-dG adduct in a CG*C double-stranded sequence context, previously studied by NMR (17), using an MD-derived ensemble which reproduced well the NOE-derived distance restraints (26) and employed the same force field as in the current work. There are subtle differences in the orientation of the BP rings in the minor groove in all three sequence contexts G6*G7, G6G7* and CG*C, with attendant small differences in χ, α′ and β′ torsion angles (defined in Figure 1A) (Figure 7A and Table 3). The combination of subtle differences in these three torsion angles together reflects the subtly different positioning of the pyrenyl ring system in the three sequences (Figure 6), because the steric hindrance effects differ for each case. G6G7* is most distinct because it uniquely contains a 5′-flanking amino group on the same strand.
Specifically, in the G6G7* duplex, the amino group of G6 on the 5′-side of G7* sterically forces the BP rings towards the complementary strand (Figure 6B). In addition, the bulky amino group of G17, the partner base of C8, is in van der Waals contact with the benzylic ring of BP and thus also affects the positioning of the BP aromatic ring system in the minor groove. In the case of the G6*G7 duplex, the amino group of G7 which is 3′ to G6*, as well as the amino group of base G20 which is the Watson–Crick partner of C5, must be avoided by the bulky benzylic ring (Figure 6A). In the case of the CG*C duplex, the pyrenyl and benzylic rings are again positioned to avoid collisions with the amino groups of G16 and G18 in the complementary strand, which are the Watson–Crick partners of C5 and C7, respectively (Figure 6C). In all cases, the BP rings are 5′-directed along the modified strand, the torsion angles α′ β′ remaining in the low energy domains (α′ = 180 ± 40°, β′ = 270 ± 40°) computed for this 10S (+)-trans-anti-[BP]-N2-dG adduct (37) and observed in all NMR solution structures in which the cytosine partner to the lesion is present (18), and also in a crystal structure within a replicative polymerase (38). Therefore, modest flexibility in the α′, β′ torsion angles within the allowed domains (37) permits the subtle differential accommodation of the bulky BP rings in the minor groove.
Sequence-governed duplex distortions
It has been shown that oligonucleotide duplexes with 10S (+)-trans-anti-[BP]-N2-dG adducts in the G6G7* sequence context exhibit a bend at the site of the lesion that are characterized by remarkably diminished electrophoretic mobilities relative to the same duplexes but with a G6*G7 sequence context (20). The most striking difference in the G6*G7 and G6G7* duplexes is the remarkable untwisting in the latter case (Figure 7B). There is a striking decrease in the Twist angle of the G6 · C19 base pair to 13° relative to that of the adjacent modified G7* · C18 base pair whose Twist angle is somewhat above normal (40°). The Twist angles are about 30° in the same region in the G6*G7 duplex (Figure 7B). This large untwisting effect in the G6G7* duplex is consistent with a bend at the lesion site, which markedly decreases the electrophoretic mobilities of such duplexes with 10S (+)-trans-anti-[BP]-N2-dG adducts in 5′- ··· GG*··· sequence contexts (20). Untwisting has been shown to correlate with increased Roll (seen also in our G6G7* case, Figure S2, Supplementary Data) and hence DNA bending (39–41). In contrast, the electrophoretic mobilities are normal in the 5′- ··· GG*··· sequence context (20). The bend in the G6G7* duplex is attributed to the steric hindrance by the exocyclic amino group in the minor groove at G6, on the same strand, which causes the untwisting needed to accommodate BP, with the attendant bend. In support of this interpretation, it was found that the replacement of the 5′-flanking G in GG* duplexes by inosine, lacking the amino group, abolishes the unusual electrophoretic mobilities (Ruan,Q. and Geacintov,N.E., unpublished data). However, in the CG*C sequence context, no significant effect on electrophoretic mobility (20) and untwisting (26) is observed.
Another difference we observe between the G6*G7 and G6G7* duplexes is the marked local helix destabilization in only the former case, which exhibits low-temperature (20–30°C) perturbation of Watson–Crick base pairing. While these differences are important in the base pair opening local dynamics around the lesion sites, the effect on the overall cooperative melting of the two duplexes is small (Figure S1D, Supplementary Data).
Another structural difference we find is widening of the minor groove centered about the lesion site, which is greater in the G6G7* case (Figure 7C). We find that an amino group of a guanine base flanking the 10S (+)-trans-anti-[BP]-N2-dG adduct on the 5′-side causes a greater distortion of the minor groove and prevents the bulky BP rings from aligning as well within the normal groove contours. However, the strain caused by the greater groove opening does not strongly affect the thermal stabilities at the modified base pair nor the flanking base pairs in G6G7* duplexes. The space filling views in Figure S6, Supplementary Data, show the overall structures of the G6*G7 and G6G7* duplexes and the differences in groove widths.
In sum, the steric effects of the amino groups differ depending on their exact position, and produce different structural distortions. In the G6G7* case, the steric hindrance produces prominent untwisting with attendant bending. In the G6*G7 case, the necessary accommodation of the BP rings to the amino groups causes instead local helix destabilization. Local minor groove width opening centered about the lesion site occurs in both cases, but is somewhat greater for G6G7*. In all cases, these amino groups act as guideposts for the placement of the BP in the minor groove.
Biological implications
The flanking guanine bases strongly affect the structural characteristics of the 10S (+)-trans-anti-[BP]-N2-dG lesions in double-stranded DNA. The G6G7* duplex is characterized by a wider minor groove centered about the lesion site than the G6*G7 duplex, but the base pairing at the lesion site and flanking base pairs is more stable in the former than the latter. Local untwisting with concomitant bending occurs only in the G6G7* duplex. Differences in distortions caused by the exocyclic amino groups of guanines surrounding these lesions on either side may be distinguished by DNA repair enzymes that are known to probe the structural integrity of the damaged DNA (42). Indeed, recent experiments in our laboratory have shown that human nucleotide excision repair enzymes in cell-free extracts exhibit distinct differences in excising sequences with the 10S (+)-trans-anti-[BP]-N2-dG adduct positioned at either one or the other G in GG sequence contexts. The dual incisions are more efficient in the G6*G7 than the G6G7* sequence context by a factor of ∼2 (manuscript in preparation). While this difference is not large, the lesions that are more slowly repaired in cellular environments and in tissues are more likely to survive until DNA replication occurs. Finally, sequence effects can also manifest themselves in DNA replication. The relevance of the minor groove orientation of the 10S (+)-trans-anti-[BP]-N2-dG lesion to polymerase structure and function has recently been shown by crystallographic analyses of a replicative DNA polymerase containing the 10S (+)-trans-anti-[BP]-N2-dG adduct (38).
SUPPLEMENTARY DATA
Supplementary Data is available at NAR online.
ACKNOWLEDGEMENTS
This work was supported by NIH grants CA 099194 (NEG), CA 28038 (SB) and CA 046533 (DJP). Components of this work were conducted in the Shared Instrumentation Facility at NYU that was constructed with support from Research Facilities Improvement Grant C06 RR-16572 from the National Center for Research Resources, NIH. The acquisition of the 500 MHz spectrometer was supported by NSF Grant MRI 0116222. NMR resources were also used at the New York Structural Biology Center (NYSBC) where Professors D.J. Patel and N.E. Geacintov are affiliate faculty. The Center is a STAR site supported by the New York State Office of Science, Technology and Academic Research and NMR resources are supported by NIH P41 GM66354. The NMR experimental work was carried out by F.A.R., Y.C. performed the computational modeling, C.L. participated in interpretation of the NMR data, Y.T. and A.K. prepared and purified the modified oligonucleotides, S.A. provided the diol epoxides, D.J.P. provided guidance in NMR data interpretation, S.B. guided the modeling work and N.E.G. provided overall direction of the project. Funding to pay the Open Access publication charge was provided by NIH grant CA 099194.
Conflict of interest statement. None declared.
REFERENCES
- 1.Luch A. Nature and nurture—lessons from chemical carcinogenesis. Nat. Rev. Cancer. 2005;5:113–125. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 2.Kriek E, Rojas M, Alexandrov K, Bartsch H. Polycyclic aromatic hydrocarbon-DNA adducts in humans: relevance as biomarkers for exposure and cancer risk. Mutat. Res. 1998;400:215–231. doi: 10.1016/s0027-5107(98)00065-7. [DOI] [PubMed] [Google Scholar]
- 3.Phillips DH. Fifty years of benzo[a]pyrene. Nature. 1983;303:468–472. doi: 10.1038/303468a0. [DOI] [PubMed] [Google Scholar]
- 4.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer. Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 5.Kozack R, Seo KY, Jelinsky SA, Loechler EL. Toward an understanding of the role of DNA adduct conformation in defining mutagenic mechanism based on studies of the major adduct (formed at N(2)-dG) of the potent environmental carcinogen, benzo[a]pyrene. Mutat. Res. 2000;450:41–59. doi: 10.1016/s0027-5107(00)00015-4. [DOI] [PubMed] [Google Scholar]
- 6.Hemminki K. DNA adducts, mutations and cancer. Carcinogenesis. 1993;14:2007–2012. doi: 10.1093/carcin/14.10.2007. [DOI] [PubMed] [Google Scholar]
- 7.Cheng SC, Hilton BD, Roman JM, Dipple A. DNA adducts from carcinogenic and noncarcinogenic enantiomers of benzo[a]pyrene dihydrodiol epoxide. Chem. Res. Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 8.Jelinsky SA, Liu T, Geacintov NE, Loechler EL. The major, N2-Gua adduct of the (+)-anti-benzo[a]pyrene diol epoxide is capable of inducing G- ->A and G- ->C, in addition to G- ->T, mutations. Biochemistry. 1995;34:13545–13553. doi: 10.1021/bi00041a034. [DOI] [PubMed] [Google Scholar]
- 9.Buening MK, Wislocki PG, Levin W, Yagi H, Thakker DR, Akagi H, Koreeda M, Jerina DM, Conney AH. Tumorigenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides in newborn mice: exceptional activity of (+)-7beta,8alpha-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10- tetrahydrobenzo[a]pyrene. Proc. Natl. Acad. Sci. USA. 1978;75:5358–5361. doi: 10.1073/pnas.75.11.5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slaga TJ, Bracken WJ, Gleason G, Levin W, Yagi H, Jerina DM, Conney AH. Marked differences in the skin tumor-initiating activities of the optical enantiomers of the diastereomeric benzo(a)pyrene 7,8-diol-9,10- epoxides. Cancer Res. 1979;39:67–71. [PubMed] [Google Scholar]
- 11.Mattes WB, Hartley JA, Kohn KW, Matheson DW. GC-rich regions in genomes as targets for DNA alkylation. Carcinogenesis. 1988;9:2065–2072. doi: 10.1093/carcin/9.11.2065. [DOI] [PubMed] [Google Scholar]
- 12.Yang JL, Chen RH, Maher VM, McCormick JJ. Kinds and location of mutations induced by (+/−)-7 beta,8 alpha-dihydroxy-9 alpha,10 alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in the coding region of the hypoxanthine (guanine) phosphoribosyltransferase gene in diploid human fibroblasts. Carcinogenesis. 1991;12:71–75. doi: 10.1093/carcin/12.1.71. [DOI] [PubMed] [Google Scholar]
- 13.Hanrahan CJ, Bacolod MD, Vyas RR, Liu T, Geacintov NE, Loechler EL, Basu AK. Sequence specific mutagenesis of the major (+)-anti-benzo[a]pyrene diol epoxide-DNA adduct at a mutational hot spot in vitro and in Escherichia coli cells. Chem. Res. Toxicol. 1997;10:369–377. doi: 10.1021/tx9601925. [DOI] [PubMed] [Google Scholar]
- 14.Wei SJ, Chang RL, Hennig E, Cui XX, Merkler KA, Wong CQ, Yagi H, Jerina DM, Conney AH. Mutagenic selectivity at the HPRT locus in V-79 cells: comparison of mutations caused by bay-region benzo[a]pyrene 7,8-diol-9,-10-epoxide enantiomers with high and low carcinogenic activity. Carcinogenesis. 1994;15:1729–1735. doi: 10.1093/carcin/15.8.1729. [DOI] [PubMed] [Google Scholar]
- 15.Vousden KH, Bos JL, Marshall CJ, Phillips DH. Mutations activating human c-Ha-ras1 protooncogene (HRAS1) induced by chemical carcinogens and depurination. Proc. Natl. Acad. Sci. USA. 1986;83:1222–1226. doi: 10.1073/pnas.83.5.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo KY, Nagalingam A, Miri S, Yin J, Chandani S, Kolbanovskiy A, Shastry A, Loechler EL. Mirror image stereoisomers of the major benzo[a]pyrene N2-dG adduct are bypassed by different lesion-bypass DNA polymerases in E. coli. DNA Repair (Amst) 2006;5:515–522. doi: 10.1016/j.dnarep.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, Margulis LA, Live D, Geacintov NE, et al. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc. Natl. Acad. Sci. USA. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. NMR solution structures of stereoisometric covalent polycyclic aromatic carcinogen-DNA adduct: principles, patterns, and diversity. Chem. Res. Toxicol. 1997;10:111–146. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 19.Fountain MA, Krugh TR. Structural characterization of a (+)-trans-anti-benzo[a]pyrene-DNA adduct using NMR, restrained energy minimization, and molecular dynamics. Biochemistry. 1995;34:3152–3161. doi: 10.1021/bi00010a004. [DOI] [PubMed] [Google Scholar]
- 20.Liu T, Xu J, Tsao H, Li B, Xu R, Yang C, Amin S, Moriya M, Geacintov NE. Base sequence-dependent bends in site-specific benzo[a]pyrene diol epoxide-modified oligonucleotide duplexes. Chem. Res. Toxicol. 1996;9:255–261. doi: 10.1021/tx9501086. [DOI] [PubMed] [Google Scholar]
- 21.Geacintov NE, Cosman M, Mao B, Alfano A, Ibanez V, Harvey RG. Spectroscopic characteristics and site I/site II classification of cis and trans benzo[a]pyrene diolepoxide enantiomer-guanosine adducts in oligonucleotides and polynucleotides. Carcinogenesis. 1991;12:2099–2108. doi: 10.1093/carcin/12.11.2099. [DOI] [PubMed] [Google Scholar]
- 22.Mao B, Xu J, Li B, Margulis LA, Smirnov S, Ya NQ, Courtney SH, Geacintov NE. Synthesis and characterization of covalent adducts derived from the binding of benzo[a]pyrene diol expoxide to a -GGG- sequence in a deoxyoligonucleotide. Carcinogenesis. 1995;16:357–365. doi: 10.1093/carcin/16.2.357. [DOI] [PubMed] [Google Scholar]
- 23.Cosman M, Ibanez V, Geacintov NE, Harvey RG. Preparation and isolation of adducts in high yield derived from the binding of two benzo[a]pyrene-7,8-dihydroxy-9,10-oxide stereoisomers to the oligonucleotide d(ATATGTATA) Carcinogenesis. 1990;11:1667–1672. doi: 10.1093/carcin/11.9.1667. [DOI] [PubMed] [Google Scholar]
- 24.Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the multimolecule and multiconformational Resp methodology to biopolymers - charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995;16:1357–1377. [Google Scholar]
- 25.Cheatham TE, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 26.Yan S, Wu M, Patel DJ, Geacintov NE, Broyde S. Simulating structural and thermodynamic properties of carcinogen-damaged DNA. Biophys. J. 2003;84:2137–2148. doi: 10.1016/S0006-3495(03)75020-6. (Correction for TABLE 5 in press Biophys. J., January 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simmerling C, Elber R, Zhang J. In: Modeling of Biomolecular Structure and Mechanisms. Pullman A, editor. Netherlands: Kluwer; 1995. pp. 241–265. [Google Scholar]
- 28.Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, Harvey R, Geacintov NE, Broyde S, et al. Solution conformation of the (+)-cis-anti-[BP]dG adduct in a DNA duplex: intercalation of the covalently attached benzo[a]pyrenyl ring into the helix and displacement of the modified deoxyguanosine. Biochemistry. 1993;32:4145–4155. doi: 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- 29.Cosman M, Fiala R, Hingerty BE, Amin S, Geacintov NE, Broyde S, Patel DJ. Solution conformation of the (+)-cis-anti-[BP]dG adduct opposite a deletion site in a DNA duplex: intercalation of the covalently attached benzo[a]pyrene into the helix with base displacement of the modified deoxyguanosine into the minor groove. Biochemistry. 1994;33:11518–11527. doi: 10.1021/bi00204a014. [DOI] [PubMed] [Google Scholar]
- 30.Cosman M, Fiala R, Hingerty BE, Amin S, Geacintov NE, Broyde S, Patel DJ. Solution conformation of the (+)-trans-anti-[BP]dG adduct opposite a deletion site in a DNA duplex: intercalation of the covalently attached benzo[a]pyrene into the helix with base displacement of the modified deoxyguanosine into the major groove. Biochemistry. 1994;33:11507–11517. doi: 10.1021/bi00204a013. [DOI] [PubMed] [Google Scholar]
- 31.de los Santos C, Cosman M, Hingerty BE, Ibanez V, Margulis LA, Geacintov NE, Broyde S, Patel DJ. Influence of benzo[a]pyrene diol epoxide chirality on solution conformations of DNA covalent adducts: the (−)-trans-anti-[BP]G.C adduct structure and comparison with the (+)-trans-anti-[BP]G.C enantiomer. Biochemistry. 1992;31:5245–5252. doi: 10.1021/bi00138a002. [DOI] [PubMed] [Google Scholar]
- 32.Patel DJ, Pardi A, Itakura K. DNA conformation, dynamics, and interactions in solution. Science. 1982;216:581–590. doi: 10.1126/science.6280281. [DOI] [PubMed] [Google Scholar]
- 33.Hare DR, Wemmer DE, Chou SH, Drobny G, Reid BR. Assignment of the non-exchangeable proton resonances of d(C-G-C-G-A-A-T-T-C-G-C-G) using two-dimensional nuclear magnetic resonance methods. J. Mol. Biol. 1983;171:319–336. doi: 10.1016/0022-2836(83)90096-7. [DOI] [PubMed] [Google Scholar]
- 34.Fratini AV, Kopka ML, Drew HR, Dickerson RE. Reversible bending and helix geometry in a B-DNA dodecamer: CGCGAATTBrCGCG. J. Biol. Chem. 1982;257:14686–14707. [PubMed] [Google Scholar]
- 35.Ravishanker G, Swaminathan S, Beveridge DL, Lavery R, Sklenar H. Conformational and helicoidal analysis of 30 PS of molecular dynamics on the d(CGCGAATTCGCG) double helix: “curves”, dials and windows. J. Biomol. Struct. Dyn. 1989;6:669–699. doi: 10.1080/07391102.1989.10507729. [DOI] [PubMed] [Google Scholar]
- 36.Zhang N, Lin C, Huang X, Kolbanovskiy A, Hingerty BE, Amin S, Broyde S, Geacintov NE, Patel DJ. Methylation of cytosine at C5 in a CpG sequence context causes a conformational switch of a benzo[a]pyrene diol epoxide-N2-guanine adduct in DNA from a minor groove alignment to intercalation with base displacement. J. Mol. Biol. 2005;346:951–965. doi: 10.1016/j.jmb.2004.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie XM, Geacintov NE, Broyde S. Stereochemical origin of opposite orientations in DNA adducts derived from enantiomeric anti-benzo[a]pyrene diol epoxides with different tumorigenic potentials. Biochemistry. 1999;38:2956–2968. doi: 10.1021/bi9825605. [DOI] [PubMed] [Google Scholar]
- 38.Hsu GW, Huang X, Luneva NP, Geacintov NE, Beese LS. Structure of a high fidelity DNA polymerase bound to a benzo[a]pyrene adduct that blocks replication. J. Biol. Chem. 2005;280:3764–3770. doi: 10.1074/jbc.M411276200. [DOI] [PubMed] [Google Scholar]
- 39.Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorin AA, Zhurkin VB, Olson WK. B-DNA twisting correlates with base-pair morphology. J. Mol. Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 41.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.