

Abstract
A new synthetic protocol for sclerotigenin-type benzodiazepine-quinazolinone library scaffold is introduced. A fluorous benzyl protecting group is used for synthesis of 4-benzodiazepine-2,5-dione intermediate and also as a phase tag for fluorous solid-phase extraction (F-SPE).
Keywords: sclerotigenin; benzodiazepine-quinazolinone; 1,4-benzodiazepine-2,5-dione; fluorous synthesis; solid-phase extraction
Sclerotigenin was isolated from the sclerotia of Penicillium sclerotigenum and has shown promising antiinsectan activity.1 It is the simplest member of the benzodiazepine-quinazolinone natural alkaloid family. Other members in this family such as circumdatins A–G isolated from terrestrial fungus Aspergillus ochraceus2 and benzomalvins A–C isolated from fungus Penicillium sp also possess interesting biological activities.3
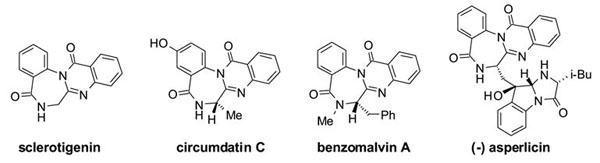
Privileged 1,4-benzodiazepine-2,5-dione ring systems are the key intermediates for synthesis of benzodiazepine-quinazolinone alkaloids.4 As part of our continuous effort on the development of fluorous synthetic protocols, we have employed a series of fluorous protecting groups for library synthesis.5,6 Reported here is a new approach to synthesize benzodiazepinedione scaffold using fluorous benzyl as a protecting group and also as a phase tag for fluorous solid-phase extraction (F-SPE).7 Further derivatization of benzodiazepinediones leads to formation of sclerotigenin ring skeleton.
Taking the advantage that numbers of conventional solution-phase and solid-phase synthetic methods for benzodiazepine have been reported in literature,8 we adopted Ellman’s solid-phase method for fluorous synthesis (Scheme 1).9 Fluorous benzaldehyde 1 prepared by reaction of a hydroxybenzaldehyde with a fluorous alcohol was used as the starting material. Compound 2 was produced by reductive amination of 1 with an amino ester. Compound 2 was reacted with an anthranilic acid in the presence of 1-ethyl-3-3-dimethylaminopropyl)carbodiimide (EDCI) and N-methylpyrrolidine (NMP). The 1,4-benzodiazepine-2,5-dione ring formation was accomplished by base-promoted cyclization of 3. Compounds 2, 3, and 4 generated in this reaction sequence were purified by simple workup or F-SPE with FluoroFlash® cartridges.10 In F-SPE, the first wash with 80:20 MeOH–H2O eluted the non-fluorous components. The desired fluorous compound was eluted with 100% MeOH. A total of nine analogs of compound 4 with substitution variations (R1 and R2) were prepared.11
Scheme 1.
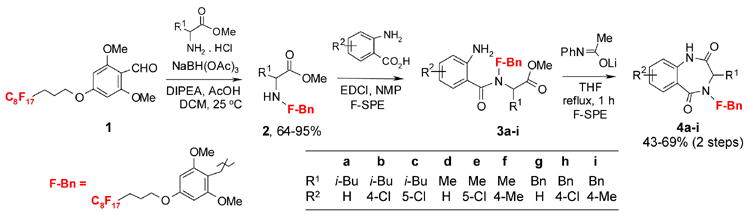
Fluorous synthesis of benzodiazepinedione 4
With nine different benzodiazepinediones 4 in hand, we then conducted parallel synthesis to construct the quinazolinone ring skeleton (Scheme 2).1c Compound 4 was acylated with 2-nitrobenzoyl chloride in the presence of t-BuN=P(NMe2)3 as a base to give compound 5 (Table 1). If substituted 2-nitrobenzoyl chloride was employed for acylation, the third diversity point (R3) could be introduced. Compounds 5 were purified by automated RapidTrace F-SPE.12 The nitro group of 5 was reduced with zinc dust in acetic acid under sonication conditions. Resulted amino group simultaneously underwent cyclization to form quinazolinone ring of 6. The parallel sonication reactions of 5 gave the reduction/cyclization products 6 in a broad range of yield (21–73%). Since some reactions had low yields, F-SPE was not sufficient for purification. Reverse-phase chromatography was applied to purify compounds 6. The capability to purify fluorous compounds by non-fluorous technique is a useful option. It could be a difficult task in solid-phase synthesis to separate resin-bound impurities. At the last step, F-benzyl tag of compounds 6 was removed by treated with 90:5:5 TFA-H2O-dimethylsulfide (DMS) under microwave radiation, followed by F-SPE on RapidTrace® workstation to give the final product 7 with the sclerotigenin ring skeleton.13
Scheme 2.
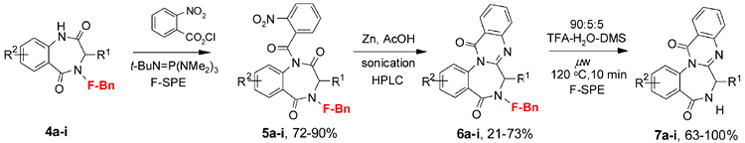
Parallel synthesis of nine benzodiazepine-quinazolinones 7
Table 1.
Yields for analogs of compounds 5, 6, and 7
| a | b | c | d | e | f | g | h | i | |
|---|---|---|---|---|---|---|---|---|---|
| R1 | i-Bu | i-Bu | i-Bu | Me | Me | Me | Bn | Bn | Bn |
| R2 | H | 4-Cl | 5-Cl | H | 5-Cl | 4-Me | H | 4-Cl | 4-Me |
| 5a–i | 82% | 80% | 90% | 75% | 94% | 90% | 75% | 72% | 81% |
| 6a–i | 44% | 50% | 67% | 21% | 51% | 62% | 70% | 65% | 73% |
| 7a–i | 83% | 86% | 91% | 91% | 63% | 71% | 100% | 89% | 97% |
In summary, we have developed a new approach for the synthesis of fluorous 1,4-benzodiazepine-2,5-diones. The key intermediates can be readily converted to sclerotigenin ring skeleton. The new method which produces the library scaffold with substitution variation coupled with the simple F-SPE separation is an alternative way for solution-phase parallel synthesis of benzodiazepine-quinazolinone analogs.
Acknowledgments
This work was supported by the National Institutes of General Medical Sciences SBIR grant (2R44GM067326-02A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a) Penhoat M, Bohn P, Dupas G, Papamicael C, Marsais F, Levacher V. Tetrahedron Asymm. 2006;17:281–286. [Google Scholar]; b) Liu JF, Kaselj M, Isome Y, Chapnick J, Zhang B, Bi G, Yohannes D, Yu L, Baldino CM. J Org Chem. 2005;70:10488–10493. doi: 10.1021/jo051876x. [DOI] [PubMed] [Google Scholar]; c) Grieder A, Thomas AW. Synthesis. 2003:1707–1711. [Google Scholar]; d) Snider BB, Busuyek MV. Tetrahedron. 2001;57:3301–3307. [Google Scholar]; e) He F, Foxman BM, Snider BB. J Am Chem Soc. 1998;120:6417–6418. [Google Scholar]
- 2.Rahbaek L, Breinholt J. J Nat Prod. 1999;62:904–905. doi: 10.1021/np980495u. [DOI] [PubMed] [Google Scholar]
- 3.a) Sun HH, Barrow CJ, Sedlock DM, Gillum AM, Cooper R. J Antibiotics. 1994;47:515–522. doi: 10.7164/antibiotics.47.515. [DOI] [PubMed] [Google Scholar]; b) Sugimori T, Okawa T, Eguchi S, Kakehi A, Yashima E, Okamoto Y. Tetrahedron. 1998;54:7997–8008. [Google Scholar]
- 4.Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 5.a) Zhang W, Lu Y, Chen CH-T, Zeng L, Kassel DB. J Comb Chem 2006. 8:687–695. doi: 10.1021/cc060061e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang W, Lu Y, Chen CH-T, Curran DP, Geib S. Eur J Org Chem. 2006:2055–2059. doi: 10.1002/ejoc.200600077. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang W, Lu Y, Geib S. Org Lett. 2005;7:2269–2272. doi: 10.1021/ol0507773. [DOI] [PubMed] [Google Scholar]; d) Zhang W, Chen CHT.Tetrahedron Lett 2005461807–1810.18079977 [Google Scholar]; e) Lu Y, Zhang W. Mol Diversity. 2005;9:91–98. doi: 10.1007/s11030-005-1293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Nagashima T, Zhang W. J Comb Chem. 2004;6:942–949. doi: 10.1021/cc049885r. [DOI] [PubMed] [Google Scholar]; g) Lu Y, Zhang W. QSAR Comb Sci. 2004;23:827–835. doi: 10.1901/jaba.2004.23-827. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Zhang W, Tempest P. Tetrahedron Lett. 2004;45:6757–6760. [Google Scholar]; i) Zhang W, Lu Y. Org Lett. 2003;5:2555–2558. doi: 10.1021/ol034854a. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Chen CHT, Zhang W. Org Lett. 2003;5:1015–1017. doi: 10.1021/ol0274864. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Zhang W. Org Lett. 2003;5:1011–1014. doi: 10.1021/ol027469e. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Zhang W, Luo Z, Chen CH-T, Curran DP. J Am Chem Soc. 2002;124:10443–10450. doi: 10.1021/ja026947d. [DOI] [PubMed] [Google Scholar]
- 6.Selected reviews on fluorous synthesis. Curran DP. Aldrichemica Acta. 2006;39:3–9.Curran DP. In: Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Wiley-VCH: Weinheim; 2004. pp. 101–127.Zhang W. Chem Rev. 2004;104:2531–2556. doi: 10.1021/cr030600r.Zhang W. Curr Opin Drug Discov Develop. 2004;7:784–797.Zhang W. Tetrahedron. 2003;59:4475–4489.Curran DP. Angew Chem Int Ed Eng. 1998;37:1174–1196. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P.
- 7.For reviews on F-SPE, see Zhang W, Curran DP. Tetrahedron. 2006;62:11837–11865. doi: 10.1016/j.tet.2006.08.051.Curran DP. In: Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Wiley-VCH; Weinheim: 2004. pp. 101–127.Curran DP. Synlett. 2001:1488–496.. See also Zhang W, Lu Y, Nagashima T. J Comb Chem. 2005;7:893–897. doi: 10.1021/cc050061z.
- 8.For a recent review on solid-phase synthesis of benzodiazepines, see Kamal A, Reddy KL, Devaiah V, Shankaraiah N, Reddy DR. Mini-Rev Med Chem. 2006;6:53–68. doi: 10.2174/138955706775197875.
- 9.Boojamra CG, Burow KM, Thompson LA, Ellman JA. J Org Chem. 1997;62:1240–1256. [Google Scholar]
- 10.FluoroFlash® SPE cartridges are available from Fluorous Technologies, Inc. (www.fluorous.com)
- 11.A general procedure for the synthesis of compounds 2 and 4. To a solution of leucine methyl ester hydrochloride (7.6 g, 42 mmol), 2,6-dimethoxy-4-[3-(perfluorooctyl)propyloxy]benzaldehyde 1 (26 g, 40 mmol), and N,N-diisopropylethylamine (7 mL, 0.04 mol) in CH2Cl2 (0.3 L) was added 4 Å molecular sieves (3 g) at 23 °C. NaBH(OAc)3 (13 g, 60 mmol) was added after 4 h, then water was added after additional 3 h. The CH2Cl2 layer was washed with aq. NH4Cl and brine. After most of the solvent was removed using a rotary evaporator, the residue was passed through a pad of silica gel (50 mL). The product was eluted with hexanes–EtOAc (1:1, 300 mL). The concentrated product was further triturated with hexanes–Et2O to give the desired compound 2 (R1 = i-Bu, 3.9 g, 95% yield). 1H NMR (270 MHz, CDCl3) δ 6.08 (s, 2H), 4.02 (t, 2H, J = 5.8 Hz), 3.78 (s, 6H), 3.59 (s, 3H), 3.26 (t, 1H, J = 7.1 Hz), 2.45–2.00 (m, 5H), 1.82–1.35 (m, 3 H), 0.88 (d, 3H, J = 6.5 Hz), 0.81 (d, 3H, J = 6.4 Hz). 13C NMR (67.5 MHz, CDCl3) δ 176.4, 159.3, 108.9, 90.7, 66.3, 59.0, 55.5, 51.3, 42.8, 39.9, 28.2, 27.9, 27.5, 24.8, 22.6, 22.0, 20.5. LC-MS (APCI+) m/z 772 [M+1]+. To a solution of 2 (R1 = i-Bu, 4.3 g, 5.6 mmol) in N-methylpyrrolidine (30 mL), 4-chloroanthranilic acid (1.9 g, 11 mmol) and EDCI-HCl (2.1 g, 11 mmol) were added as solid at 23 °C. The same amounts of the acid and EDCI-HCl were added after 2 h and 4 h. One day after the final addition, the reaction mixture was diluted with DMSO (300 mL), and was loaded onto an F-SPE cartridge (50 g), and the flask was rinsed with DMSO (100 mL), and was loaded to the silica gel. The nonfluorous components were eluted with MeCN–H2O (1:1, 300 mL, and 4:1, 200 mL), and then most of the solvent was drained from the cartridge. The amide coupling product was eluted with MeCN (0.4 L). The MeCN solution was concentrated in a rotary evaporator, and the residue was treated with a solution of lithium acetanilide (0.33 M in THF, 30 mL). The mixture was refluxed for 1 h. After cooling, AcOH (0.6 mL) was added, and the solvent was removed in a rotary evaporator. MeOH (30 mL) was added to the residue, and it was heated until the solvent started to boil. The mixture was left at 23 °C for 1 d, and product 4b (R1 = i-Bu, R2 = 4-Cl) was collected as a solid by filtration (3.5 g, 69% yield based on the amount of 2). 1H NMR (270 MHz, CDCl3) δ 9.55 (s, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.16 (dd, J = 7.1, 1.9 Hz 1,), 6.90 (d, J = 1.8 Hz, 1H), 6.09 (s, 2H), 5.23 (d, J = 13.8 Hz, 1H), 4.56 (d, J = 13.8 Hz, 1H), 4.15–3.85 (m, 3H), 3.75 (s, 6H), 2.45–2.00 (m, 4H), 1.60–1.45 (m, 1H), 1.35–1.15 (m, 2H), 0.80 (d, J = 6.4 Hz, 3H), 0.73 (d, J = 6.6 Hz, 3H). 13C NMR (67.5 Hz, CDCl3) δ 173.5, 165.1, 160.6, 160.1, 137.7, 136.2, 133.3, 125.5, 124.6, 119.4, 104.1, 90.6, 66.3, 59.5, 55.5, 42.0, 38.3, 27.9 (t, J = 22 Hz), 25.2, 22.3, 22.1, 20.5. LC-MS (APCI +) m/z 893 [M+1]+.
- 12.Zhang W, Lu Y. J Comb Chem. 2006;8:890–896. doi: 10.1021/cc0601130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.A general procedure for the synthesis of compounds 5, 6, and 7. To a solution of 4 in CH2Cl2 was added t-butylimino-tris(dimethylamino)phosphorane (10 equiv) and 2-nitrobenzoic acid (2 equiv). The reaction mixture was stirred for 10 min and then concentrated in a rotary evaporator. The residue was dissolved in DMF (1 mL) and purified on RapidTrace® SPE workstation with 2 g cartridges to afford 5 in 72–90% yield. A solution of 5 in acetic acid (1 mL) was added Zn dust (20 equiv) and sonicated at room temperature for 2 h. The Zn was filtered and the filtrate was diluted with EtOAc and washed with NaHCO3 and brine. The EtOAc solution was dried and concentrated in a rotary evaporator. The residue was dissolved in MeCN and purified by C18 HPLC to afford 6 in 21–73% yields. A solution of 6 in TFA-H2O-DMS (90:5:5) was stirred for 3 days before being concentrated in a rotary evaporator. The residue was dissolved in DMF (1 mL) and purified by RapidTrace® SPE workstation to afford 7 in 63–100% yields. Analytical date for compound 7b (R1 = i-Bu, R2 = 4-Cl): 1H NMR (275 Hz, CDCl3) δ 0.90 (d, J = 6.5 Hz, 3H), 1.00 (d, J = 6.5 Hz, 3H), 1.80–2.05 (m, 2H), 2.05–2.25 (m, 1H), 4.10–4.35 (m, 1H), 6.66 (d, J = 6.2 Hz, 1H), 7.45–7.59 (m, 2H), 7.67 (d, J = 1.9 Hz, 1H), 7.70–7.85 (m, 2H), 7.91 (d, J = 8.4 Hz, 1H), 8.31 (dd, J = 1.4, 8.0 Hz, 1H); 13C NMR (67.5 Hz, CDCl3) δ 22.0, 23.1, 24.3, 38.0, 52.4, 121.3, 127.5, 127.8, 127.9, 128.7, 128.9, 129.5, 131.0, 134.3, 135.2, 137.4, 146.0, 154.1, 161.5, 167.1; LCMS (APCI+) 368 [M+1]+.