

Abstract
Tumors secrete proangiogenic factors to induce the ingrowth of blood vessels from the surrounding stroma, the end targets of which are vascular endothelial cells. The homeobox gene Gax inhibits angiogenesis and induces p21WAF1/CIP1 expression in vascular endothelial cells. In order to elucidate the mechanism through which Gax activates p21WAF1/CIP1 expression, we constructed Gax cDNAs with deletions of either the N-terminal domain, the homeodomain, or the C-terminal domain, and then assessed these constructs for their ability to activate p21WAF1/CIP1. There was an absolute requirement for the homeodomain, while deleting the C-terminal domain decreased, but did not abolish, transactivation of the p21WAF1/CIP1 promoter by Gax. Deleting the N-terminal domain did abolish transactivation. Next, we performed chromatin immunoprecipitation and found approximately 15 kb upstream of the p21WAF1/CIP1 ATG an ATTA-containing Gax binding site (designated A6) with a sequence similar to that of other homeodomain binding sites. Gax was able to bind to A6 in a homeodomain-dependent manner and thereby activate the expression of a reporter gene coupled to this sequence, and this activation was abolished by mutating specific residues in this sequence. Based on the sequence of A6, we were then able to locate other ATTA-containing sequences that also bound Gax and activated transcription in reporter constructs. Finally, we found that the ability of these Gax deletions to induce G0/G1 arrest correlates with their ability to transactivate the p21WAF1/CIP1 promoter. We conclude that Gax activates p21WAF1/CIP1 through multiple upstream AT-rich sequences. Given the multiple biological activities of Gax in regulating EC function, identification of a putative Gax binding site will allow study of how Gax activates or represses other downstream targets to inhibit angiogenesis.
INTRODUCTION
Angiogenesis is critical to the growth, invasion, and metastasis of human tumors because the diffusion of oxygen and nutrients is limited to less than 1 mm in aqueous solution (1). Key to the process of angiogenesis is the vascular endothelial cell (EC) (2). In health ECs respond to a balance between pro- and antiangiogenic factors secreted by various cell types to maintain blood vessel homeostasis (3,4). Tumors hijack this process by secreting proangiogenic factors in order to supply themselves with oxygen and nutrients, a transition known as the “angiogenic switch” (3,4). Because targeting angiogenesis has developed into a promising avenue of research and treatment for malignancies (5), understanding the transcriptional regulation of the angiogenic phenotype in ECs has become particularly important.
Although the extracellular signals and downstream signaling pathways activated by pro- and antiangiogenic factors have been topics of intense study (6–9), less is known about the transcriptional regulators that lead to the upregulation and downregulation of batteries of genes necessary for the angiogenic phenotype. Because of their ubiquitous nature and their importance in regulating morphogenesis, organogenesis, cell proliferation and migration, and tumor formation, we considered it likely that homeodomain proteins (10–14) are involved in the transcriptional regulation of angiogenesis. Indeed several homeobox genes have now been so implicated (15–26). For example, HOXD3 expression activates the angiogenic phenotype in vascular ECs through the activation of urine plasminogen activator and integrins αVβ3 and α5α1 (15,16), and HOXA5, a HOX cluster gene implicated in regulating p53 expression in human breast cancer (27–29) inhibits angiogenesis by downregulating VEGFR1 and ephrin A1 in vascular ECs (26).
Recently, we reported that a homeobox gene Gax (whose mouse homolog is Meox-2) strongly influences EC phenotype (30–33). Originally described in vascular smooth muscle cells (VSMCs) (32,34–38), in the adult Gax is expressed primarily, but not exclusively, in the cardiovascular system, kidney (32,39,40), and placenta (41,42). Pointing to a role in regulating VSMC proliferation and phenotype is its pattern of regulation, in which mitogenic signals results in a rapid downregulation of Gax expression, while growth arrest signals induce a slower upregulation (32). Given its association with mesoderm (31,43), we looked for Gax expression in vascular ECs, observing a similar pattern of expression to what has been observed in VSMCs (20,25), with mitogenic and proangiogenic signals rapidly downregulating Gax expression (25). Moreover, Gax also inhibits nuclear factor-κB (NF-κB) signaling in vascular ECs (25), and inhibition of NF-κB activity is antiangiogenic in several systems (44–49). Finally, Gax expression also inhibits angiogenesis in both in vitro and in vivo models (20,25).
In addition to the inhibition of NF-κB signaling, a potentially important mechanism through which Gax could inhibit tumor-induced angiogenesis is the inhibition of cell-cycle progression by activating the cyclin kinase inhibitor p21WAF1/CIP1 (20,37). In vascular ECs, Gax also inhibits proliferation and trans-activates the p21WAF1/CIP1 (20). However, the sequences in the p21WAF1/CIP1 promoter and the likely mechanisms through which Gax accomplishes cell cycle arrest in vascular ECs have not yet been elucidated. In this study, we dissected the Gax protein and identified its homeodomain and polyhistidine (CAX, or opa) repeat as important for DNA binding and trans-activation, respectively. We also identified at least two upstream sequences to which Gax binds and thereby activates p21WAF1/CIP1 expression. These findings suggest that a major component of the mechanism by which Gax inhibits angiogenesis is its ability to induce cell cycle arrest in ECs by directly activating p21WAF1/CIP1 expression and thus inhibiting the early, proliferative stage of angiogenesis. Gax may thus represent a potentially promising target for the antiangiogenic therapy of human tumors.
EXPERIMENTAL PROCEDURES
Cells and cell culture
Human umbilical vein endothelial cells (HUVECs) and EGM-2 medium were obtained from BioWhittaker (Walkersville, MD). HUVECs cultured according to the manufacturer’s instructions.
Expression and reporter constructs
Gax deletions were constructed using polymerase chain reaction. First, a full length (aa1-aa303) human Gax (hugax) cDNA clone (32,33) was isolated from HUVEC total RNA using reverse transcriptase polymerase chain reaction (RT-PCR) with appropriate primers. Next, the N-terminal fragment of Gax, hugax-NT (aa1-aa187), hugaxΔCT (aa1-aa245) and an N-terminal deletion fragment of Gax, hugaxΔNT (aa188-aa303), were generated using PCR with appropriate primers. Constructs in which either the Gax homeodomain was deleted (hugaxΔHD, Δaa188-aa245) or the Gax CAX (opa) repeat was deleted (hugaxΔCAX) were produced by overlap PCR. All cDNA deletion constructs contained EcoRI and XhoI restrict enzyme sites and were initially cloned into the pCR-Blunt II-TOPO vector (Invitrogen, CA), after which they were inserted into pcDNA3.1 expression vector (Invitrogen, CA) in frame with a Flag tag at the N-terminal end of the peptides. Mammalian retroviruses were similarly constructed as derivatives of LZRSpBMN-Z in which the lacZ gene had been excised to make LZRSΔ (50). All PCR-amplified cDNA constructs were sequenced completely and protein expression verified by Western blot.
The construction of the Gax exression vector pCGN-Gax and the replication-deficient adenoviral vectors expressing the rat and human homologs of Gax (Ad.hGax and Ad.rGax, respectively), all conjugated to the Δ-hemagluttinin (HA) epitope, has been described (37). The control replication-deficient adenoviral vector expressing green fluorescent protein (Ad.GFP) was a kind gift of Dr. Daniel Medina (The Cancer Institute of New Jersey, New Brunswick, NJ). The p21WAF1/CIP1 promoter-Luciferase plasmid was also a kind gift from Dr. Kenneth Walsh (Boston University). It contains 2.4 kb of p21WAF1/CIP1 upstream sequence beginning at nucleotide 11 in the cDNA and is the same construct used in (20,37) and described in detail in (51). Finally, the shRNA gene silencing vectors targeted at Gax (Ad.shMeox2/Gax) and GFP (Ad.shGFP, control) were a kind gift of Berislav Zlokovic (52).
Gene expression and promoter assays
Transfections were carried out using Trans-IT® Jurkat Transfection Reagent (Mirus Bio Corporation, Madison, WI) according to a modification of the manufacturer’s instructions. In general, a 1 μl:l μg. ratio of transfection reagent to plasmid was used, after which cells were incubated 16 to 24 hours and then harvested for experiments.
Protein expression
Whole cell extracts from treated HUVECs were electrophoresed through 12% SDS-polyacrylamide gels and transferred to polyvinylidene diflouride membranes. The membranes were blocked with PBS plus 5% nonfat dry milk and 0.1% Tween 20 before being incubated with the appropriate dilution of primary antibody (mouse monoclonal anti-Flag, mouse monoclonal anti-Δ-tubulin, mouse monoclonal anti-p21WAF1/CIP1, and mouse monoclonal anti-p53, Sigma MO) in blocking solution, or polyclonal rabbit anti-Gax antibody (40). Blots were washed with blocking solution and incubated with secondary antibody, either goat anti-mouse IgG or goat anti-rabbit IgG (Pierce Biotechnology, Inc., Rockford, IL) and then washed again with blocking solution. Bands were visualized by chemiluminescence using the ECL-Plus reagent (Amersham, Piscataway, NJ) and quantified using densitometry, with each band being normalized to α-tubulin.
Chromatin immunoprecipitation (ChIP)
ChIP assays utilizing HUVECs expressing Gax and Gax truncates were carried out as follows. HUVECs were infected with either LZRSΔ vector, LZRSΔ-flag-hugax, and LZRSΔ-hugax for 2 days, and then incubated in fresh EGM-2 media (Cambrex, MD) overnight. Next, formaledehyde (37%) was added directly to tissue culture media to a final concentration of 1% with gentle shaking for 10 minutes at room temperature to crosslink the protein-DNA complexes, after which a final concentration of 0.125 M of glycine was added and the cells further incubated for 5 minutes to stop the crosslinking reaction. For the remaining steps of the protein isolation, all buffers used to isolate the proteins contained PMSF and protease inhibitor cocktail. Cells were rinsed twice with cold PBS, harvested by gentle scraping, and pelleted by centrifugation at 2,000 rpm for 4 minutes at 4°C, after which the pellets were washed once with 1X PBS. The cell pellets were resuspended in 300 μl of cell lysis buffer and incubated on ice for 10 minutes to release the nuclei. Following that, nuclei were pelleted by centrifugation for 5 minutes at 5,000 rpm at 4°C. Lysis buffer (300 μl) was added to the pelleted nuclei, and the mixture incubated on ice for 10 minutes to lyse the nuclei and release the chromatin.
Chromatin samples were sonicated on ice to an average length of 600 bp and then pelleted by centrifugation for 10 minutes at 14,000 rpm at 4°C. The supernatant was transferred to a new tube and precleared by adding 30 μl of blocked protein A beads (Sigma, MO) to 1 ml of IP supernatant with gentle shaking for 30 min at 4°C. After preclearing, the supernatant was recovered after pelleting the beads by centrifugation. Target protein-DNA complexes were immunoprecipitated by adding 30 μl of blocked anti-FLAG antibody beads (Sigma, St. Louis, MO) to each sample, followed by incubation at 4° C overnight. To the IP input control was added 10 μl of blocked protein G beads. The antibody-protein-DNA complex samples were collected by centrifugation for 2 min 14,000 rpm at 4°C. Pellets were washed with 1X dialysis buffer twice and IP wash buffer four times at room temperature. Immunoprecipitated antibody-protein-DNA complexes were eluted with 250 μl of freshly prepared IP elution buffer (1% SDS, 0.1M NaHCO3). To reverse crosslinking, 5 M NaCl was added to each combined eluate to a final concentration of 0.3 M, followed by heating at 65°C for 5 hours. Finally, DNA was eluted into 50 μl of H2O using a DNA gel extraction kit (Qiagen, Valencia, CA).
To detect the enrichment of chromatin sequences in the immunoprecipitate due to Gax binding to the chromatin upstream of p21CIP1/WAF1, we chose ChIP primers to amplify approximately 200 bp fragments at 1–2 kb intervals beginning near the p21WAF1/CIP1 start codon and continuing upstream 15 kb (Table 1). Purified DNA from each ChIP assay (0.5 to 1 μl) was subjected to PCR under the following conditions: initial denaturation at 94° C for 2 min; then 35 cycles at 94 °C for 30 seconds (denaturation), 60° C for 30 seconds (annealing), 72° C for 45 seconds (extension). PCR products were subjected to gel electrophoresis through 2% agarose gels. After initial identification of a putative Gax DNA binding site, we verified that this binding depended upon the homeodomain by performing ChIP on HUVECs transduced with the Gax deletion constructs. Finally, repeat ChIP experiments were carried out to detect the binding of endogenous Gax to the p21WAF1/CIP1 promoter using previously generated polyclonal rabbit antibodies to the Gax protein (40).
TABLE 1.
Primers for the initial ChIP assay
| Region | Primer sequence (5′ to 3′) | |
|---|---|---|
| A | Forward | CCC AGC AGA TAC AGG GTT GT |
| Reverse | CTT GTC CTT GCC TTT GCT TC | |
| B | Forward | GGT TAT CCT GCG TGT GAC CT |
| Reverse | TTT GTA GTT GCC TCC CCT TG | |
| C | Forward | CTT CAA GGC AGT GGG AGA AG |
| Reverse | GAT TGT GGC TAA ACC CCA GA | |
| D | Forward | CTC TCC AAT TCC CTC CTT CC |
| Reverse | AGA AGC ACC TGG AGC ACC TA | |
| E | Forward | TTC CCT CTC CGA AAG CTA CA |
| Reverse | CAG CTC CAA GAT GCT TTT CC | |
| F | Forward | AGC TTT CAC CCC CAG AAA CT |
| Reverse | CCC TTC AGG AGA GGG AAA AC | |
| G | Forward | CAC CTT TCA CCA TTC CCC TA |
| Reverse | GCA GCC CAA GGA CAA AAT AG | |
| H | Forward | ACC CCA GGT AAA CCT TAG CC |
| Reverse | AGT TTG CAA CCA TGC ACT TG | |
| I | Forward | GGT CAG GGG TGT GAG GTA GA |
| Reverse | TGT GGC TCC AAA ATG ACA AA | |
Gene promoter assays
To verify regulation of p21WAF1/CIP1 transcription by genomic fragment A in which positive binding for Gax protein in the ChIP assay was observed, 1.5 kb fragments containing p21WAF1/CIP1 ChIP-A and p21WAF1/CIP1 ChIP-C were cloned and inserted into pGL3. Co-transfection assays of reporter plasmid DNA and pCDNA3.1-flag-hugax and pCDNA3.1-flag-hugaxΔHD were performed the same as described above in order to determine which domains of the Gax protein are important for regulation of p21WAF1/CIP1 transcription. luciferase reporter plasmid inserted fragment p21WAF1/CIP1
Promoter activities were measured using constructs with the relevant regulatory sequences placed upstream of Luciferase, using a plasmid containing Luciferase from Renilla reniformus under the control of the SV40 promoter (pRL-SV) as a normalization control for transfection efficiency. Firefly and Renilla Luciferase activities were measured using the Dual Luciferase Assay Kit (Promega, Madison, WI), and firefly Luciferase activity from the p21WAF1/CIP1-Luciferase promoter construct normalized to constitutive Renilla Luciferase activity. For each experiment, HUVECS at approximately 80% confluence in 6-well plates were transfected with differing amounts of plasmid as described in individual experiments. HUVECs were then incubated with transfection reagents for three hours, and then refreshed with fresh EBM (Cambrex, MD) and supplements (Cambrex, MD) overnight. Empty pCDNA3.1-Flag vector was used to equalize the total plasmid DNA transfected for each well.
Quantitative reverse transcriptase real time polymerase chain reaction (QRT-PCR)
After each ChIP assay, the resuspended chromatin immunoprecipitate and flow-through were subjected to quantitative real time PCR utilizing TaqMan probes (53) to determine whether the immunoprecipitate was enriched for the p21WAF1/CIP1 upstream chromatin sequences of interest. QRT-PCR was carried out using a Cepheid SmartCycler thermocycler, with the associated SmartCycler v.2.0 software used to analyze the data and determine the threshold count (Ct) for each reaction. The fluorophore used was Sybr Green, and the sequences of the primers and probes were the same as those used in the initial ChIP reaction described above. Real time PCR cycles started with an initial 1.5 minute denaturation step at 95° C, followed by 30 to 40 cycles of denaturation at 95° C for 10 seconds; annealing at 56° for 20 seconds; and extension at 72° C for 30 seconds. Each sample was run in triplicate and Ct determined for the target gene. To normalize the signal for each ChIP target and identify which targets were enriched in the chromatin by Gax expression, immunoprecipitate and flow-through target gene levels were normalized to β-actin sequence levels using the ΔΔCt method (54), using described previously (55,56), and the result presented as a ratio to chromatin-bound sequence/unbound. Differences in the target/α-actin ratio were evaluated using one-way ANOVA, followed by the Bonferonni post-test.
Electrophoretic mobility shift and supershift assays
To explore the possible direct binding sites of Gax on p21WAF1/CIP1 genomic DNA, the p21WAF1/CIP1 ChIP positive binding fragment A was analyzed by probes approximately 30 bp long as listed in Table 2. 100 ng purified PCR products were end-labeled using γ-32P-ATP and T4 Kinase. Resulting labeled probes were then purified on G-50 Micro-columns (Amersham). Binding reactions were then carried out in 1x binding buffer (50 mM Tris pH 7.5, 25 mM NaCl, 3.5 μM MgCl2, 0.5 μM EDTA, 5% Glycerol, 0.05% NP-40,and 0.25mg/ml BSA), 5 μM DTT, 50 ng/μl poly(dI-dC), using 50,000 cpm labeled probe and either 0, 100 ng, and 200 ng protein in 20 μl reaction mixture at room temperature for 20 min. Binding complexes were separated from unbound probe on a non-denaturing 4% acrylamide gel. Gels were dried for 45 min and exposed to film at −80°C. For the supershift assay, the binding reaction was as follows: 1x binding buffer (50 μM Tris pH 7.5, 25 μM NaCl, 3.5 μM MgCl2, 0.5 μM EDTA, 5% Glycerol, 0.05% NP-40, and 0.25 mg/ml BSA), 5 μM DTT, 50 ng/μl poly(dI-dC), 100–500 ng protein, 2 μl of anti-Flag in a total volume of 20 μl. Reactions were incubated on ice for 20 min., after which of labeled probe (50,000 cpm) was added and the mixture incubated at room temperature for 10 minutes prior to gel electrophoresis.
TABLE 2.
Probe sequences for identification of the Gax binding site within Sequence A
| Sequence Name | Sequence |
|---|---|
| A1 | CAA TGA TTC CTC CCA GCA GAT ACA GGG TT |
| A2 | GTT AGA AAC CAC TGA GGA TAG GGA AGA GGG |
| A3 | AGT GAC ATC TCC TCC CTG GCT CTG AAC TTG |
| A4 | GCT CTA GTG TGG CCA CTA GCA TTT GGG GCT T |
| A5 | GGG GGT TGC AGC TGT CGC ATT CCA CAG TGG |
| A6 | CCC CGA TGG CAT TAC AAT TAC AGA TGA CAC T |
| A7 | TAG AGC CAC CCT AGG GAA GCA AAG GCA AG |
| A7 | AGA TAC AGG GTT GTT AGA AAC CA |
| A8 | TAG GGA AGA GGG AGT GAC ATC TC |
| A9 | GGC TCT GAA CTT GGC TCT AGT GTG GC |
| A10 | CAT TTG GGG CTT GGG GGT TGC AGC TGT |
| A11 | CCA CAG TGG CCC CGA TGG CAT T |
| A12 | TAC AGA TGA CAC TTA GAG CCA CCC TA |
Flow cytometry/cell cycle analysis
Flow cytometry and cell cycle analysis were performed using HUVECs as previously described (37). In brief, sparsely plated randomly cycling HUVECs were transfected with pCDNA3.1-Gax or its truncates, incubated overnight in normal growth medium supplemented with 5 ng/ml VEGF. Cells were then harvested and resuspended in cold PBS. Approximately 1 x 106 cells were fixed with 3 ml of −20°C cold absolute ethanol for 1 hour at 4°C, washed twice, and then incubated with 1 ml of 50 μg/ml propidium iodide (PI) staining solution supplemented with 50 μl of 10 μg/ml of RNase A for 3 hours at 4°C. Cells were pelleted and washed twice in PBS before flow analysis on a Beckman-Coulter Cytomics FC500 flow cytometer (Fullerton, CA).
RESULTS
Activation of p21WAF1/CIP1 expression by Gax requires the Gax homeodomain and the N-terminal domain
To test which domains of the Gax protein are important for the activation of p21WAF1/CIP1 expression by Gax, we constructed multiple Gax truncates, inserted them into expression vectors and tagged with Flag at the N-terminus. These constructs (Figure 1A) included: Flag-hugax (full length); Flag-hugaxNT (N-terminal domain lacking the homeodomain and C-terminal domain; Flag-hugaxΔCT (deletion of C-terminal domain; Flag-hugaxΔNT (deletion of N-terminal domain); Flag-hugaxΔHD (deletion of the homeodomain). To verify production and activity of the full Gax protein produced by the expression construct, we transduced HUVECs with vectors expressing Flag-hugax for 24 hours (Experimental Procedures) and then harvested the cells for protein for Western blot using anti-Flag antibody. Gax expression by this vector induced p21WAF1/CIP1 expression by two-fold (Figure 1B), consistent with our previous findings (57). In addition, expression of Gax, even when driven by adenoviral vectors, did not alter detectably the level of p53 (not shown), also consistent with previous observations in fibroblasts that Gax can still induce p21WAF1/CIP1 in p53−/− cells (37). Protein expression for the remaining deletion constructs was then verified by Western blot using anti-Flag antibodies (Figure 1C).
Figure 1. Gax deletions.
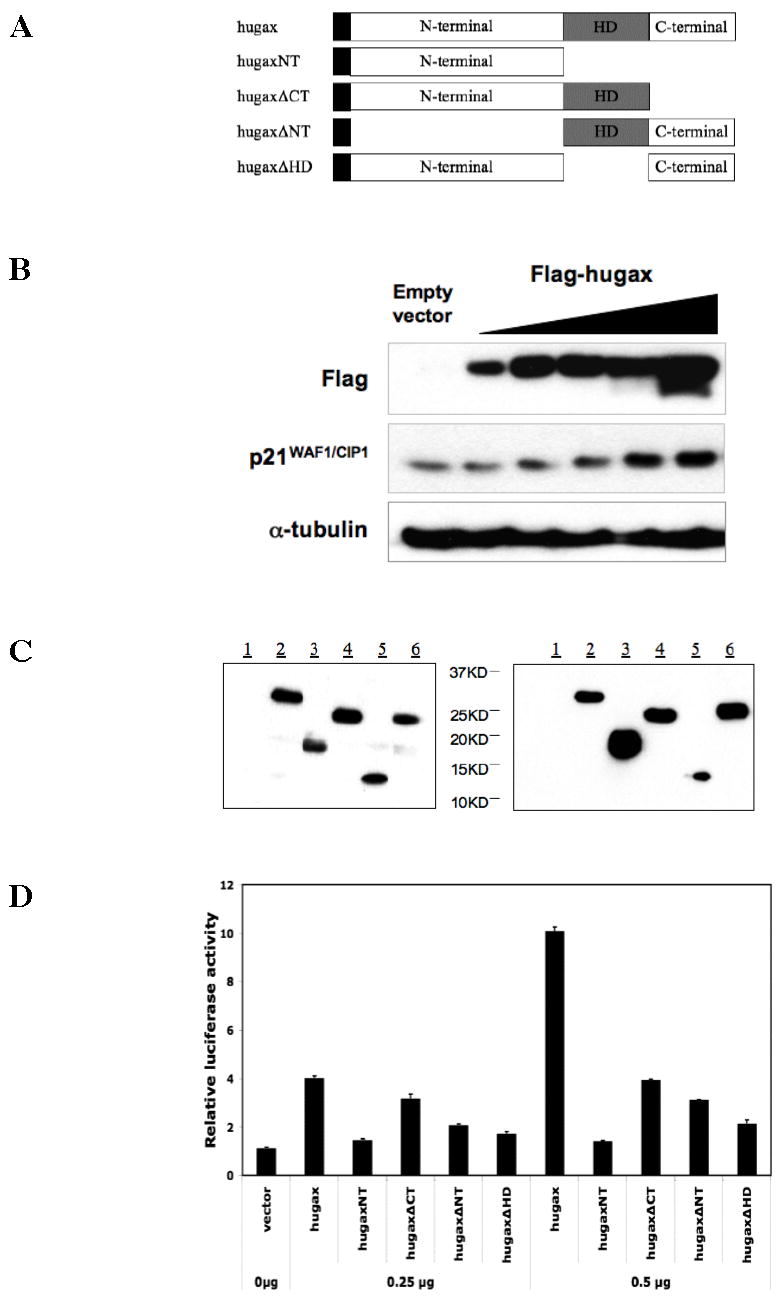
A. Plasmid construction. Gax deletions were constructed as described in the Experimental Procedures section according to the strategy outlined in this figure. Constructs were made in which either the homedomain, N-terminal domain, or C-terminal domains were deleted. See text for more details. B. Protein expression and function by full length Gax construct. HUVECs were transfected with the the pCDNA3.1 construct containing the full-length human Gax cDNA tagged with Flag and then harvested for Western blot 24 hours later. Western blots were carried out using anti-Flag and anti-p21WAF1/CIP1 antibodies. Strong Gax expression was observed, and p21WAF1/CIP1 was induced, as previously observed with the adenoviral constructs (20,37). C. Protein expression by Gax deletion constructs. HUVECs were transfected with the completed pCDNA3.1 and LZRSΔ Gax deletion constructs as in B and then incubated for 24 hours in standard medium, after which they were harvested for protein for Western blot. Western blot was carried out with anti-Flag antibody. (Legend: 1. Empty vector control; 2. Flag-hugax; 3. Flag-hugaxNT; 4. Flag-hugaxΔCT; 5. Flag-hugaxΔNT; 6. Flag-hugaxΔHD.) D. The effect of Gax deletions on transactivation of the p21WAF1/CIP1 promoter. The various pCDNA3.1-Gax constructs were cotransfected with a p21WAF1/CIP1 promoter-Luciferase construct previously used to demonstrate that Gax transactivates the p21WAF1/CIP1 promoter. Deleting the homeodomain abolishes transactivation. Similarly, deleting the N-terminal domain dramatically decreases transactivation, whereas deleting the C-terminal domain has only a minor effect. Each experiment was performed at least three times.
Finally, to determine which domains of the Gax protein are involved in activating p21WAF1/CIP1 expression, we cotransfected our Gax deletion constructs with a reporter construct containing the 2.4 kb of the p21WAF1/CIP1 promoter immediately upstream of the transcriptional start site coupled to a Luciferase reporter (p21-Luciferase) (37,51). We observed that, as expected, deletion of the homeodomain completely abolished the ability of Gax to transactivate the p21WAF1/CIP1 promoter (Figure 1D). In contrast, deleting the C-terminal domain decreased, but did not abolish, transactivation of the p21WAF1/CIP1 promoter, but deleting the N-terminal domain had a stronger effect, decreasing the ability of Gax to transactivate the p21WAF1/CIP1 promoter almost as much as deleting the homeodomain (Figure 1D).
Deletion of the CAX repeat coding for a polyhistidine/glutamine region abolishes transactivation
Having observed the effect of deleting the N-terminal domain of the Gax protein on transactivation of the p21WAF1/CIP1 promoter, we noted that contained within the N-terminal domain of Gax is a (CAX)n repeated motif, also known as an opa or M repeat (58). This motif is frequently found in developmentally regulated genes in Drosophila. It is also found in other homeobox genes, such as HOXA1 (59), Dfd (60), and Antp and believed to be an important transcriptional regulatory domain (61). In Gax, an opa repeat near the N-terminus of the protein codes for an 18 amino acid polyhistidine/glutamate tract consisting of twelve straight histidine residues followed by six residues consisting of four glutamates and two histidines (32). We wished to determine whether this domain functioned in the trans-activation of the p21WAF1/CIP1 promoter (Figure 2A). To this end, we constructed another Gax deletion construct, this time lacking only the opa repeat and tested its ability to transactivate the p21WAF1/CIP1 promoter by cotransfecting pCDNA3.1-hugaxΔCAX with the p21-Luciferase reporter construct. Deleting the CAX repeat completely abolished the ability of Gax to activate the p21WAF1/CIP1 promoter (Figure 2B), indicating the importance of this motif for transcriptional activation by Gax.
Figure 2. Function of the CAX/polyhistidine (opa) repeat.
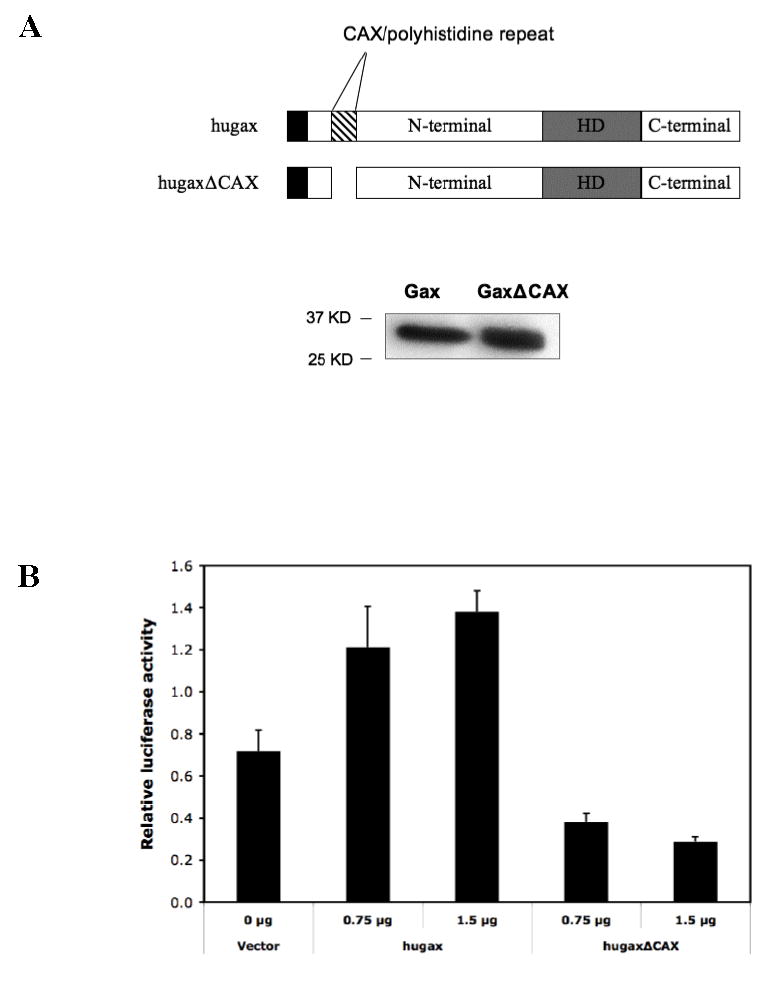
A. Deletion of the CAX/polyhistidine repeat. Expression of the truncated lacking the 18 amino acid CAX repeat is verified by Western blot. B. Effect of deleting the CAX/polyhistidine repeat on transactivation of the p21WAF1/CIP1 promoter by Gax. We compared the ability of full length Gax and GaxΔCAX constructs to transactivate the p21WAF1/CIP1 promoter. Deleting the CAX repeat dramatically decreased transactivation. Each experiment was performed at least three times.
Gax binds to a sequence 15 kb upstream of the p21WAF1/CIP1 start codon
Because we did not know a priori where Gax binds in the p21WAF1/CIP1 promoter, we performed chromatin immunoprecipitation (ChIP) using primers designed to sample the chromatin at regular intervals using 200 bp amplicons, beginning at the start codon and proceeding to approximately 15 kb upstream (Figure 3A). Surprisingly, the first sequence to which Gax could bind that we identified was located approximately 15 kb upstream of the p21WAF1/CIP1 start codon (sequence A, Figure 3, A and B) and was not within the 2.4 kb p21WAF1/CIP1 (51) promoter. We compared the level of sequence A to all other sequences using QRT-PCR normalized to α-actin sequence and found considerable enrichment for sequence A in the Flag immunoprecipitate. None of the other sequences was enriched in the chromatin, nor was Sequence A detected above any of the other sequences when ChIP was carried out with vectors expressing Gax without the Flag tag (Figure 3C). These results confirm that there is an in vivo binding site for Gax approximately 15 kb upstream from the transcriptional start site of the p21WAF1/CIP1 gene.
Figure 3. Chromatin immunoprecipitation.
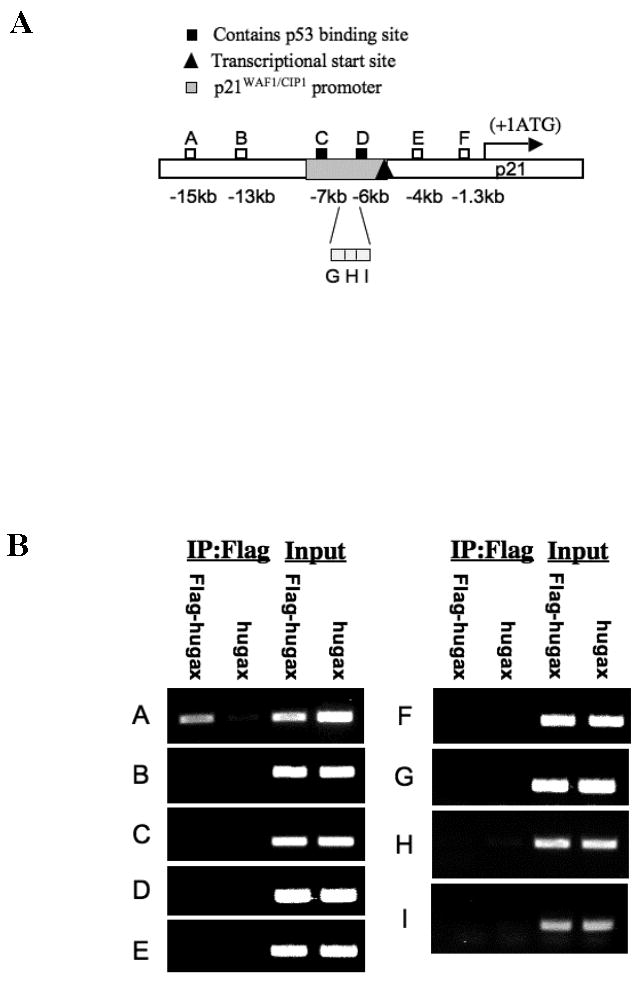
A. Design of initial primer sets for chromatin immunoprecipitation assay. B. Chromatin immunoprecipitation with the initial set of nine primer pairs. HUVECs were transduced with pCDNA3.1 constructs expressing either hugax or Flag-hugax and then subjected to ChIP assay as described in Experimental Procedures. Only primer pair A amplified a fragment that had been bound to chromatin, Fragment A. C. Quantitative real time PCR. The immunoprecipitates from the ChIP assay were subjected to quantitative real time PCR with the primer pairs used and normalized to β-actin. Only Fragment A was amplified at a ratio of greater than one compared to β-actin (p<0.05, two-way ANOVA). D. Chromatin immunoprecipitation with Gax deletion constructs. ChIP assays were carried out with HUVECs transduced with Gax deletion constructs using primer pair A. Enrichment of chromatin for Fragment A was only observed with Gax deletions containing the Gax homeodomain. Each experiment was repeated at least three times.
Given these findings, we next wished to verify that Gax binds this upstream chromatin sequence through its DNA-binding homeodomain. Consequently, we repeated the ChIP assay; only this time we used the Flag-tagged Gax deletion constructs that we had made initially (Figure 1A) and then performed quantitative real time PCR as before (Figure 3C), to determine which of these constructs produced an immunoprecipitate enriched in Sequence A. We found that none of the constructs that lacked the Gax homeodomain (hugax-NT, hugaxΔHD) resulted in enrichment for Sequence A, whereas constructs containing the Gax homeodomain (wild-type hugax, hugaxΔNT, and hugaxΔCT) all did (Figure 3D). These results indicate that Gax binds to this upstream chromatin sequence (Sequence A) through its homeodomain.
Identification of multiple ATTA-containing core binding sites for Gax
Because sequence A is 200 bp long, we wished to identify where in sequence A Gax binds. To this end, we designed several overlapping probes for use in electrophoretic mobility shift assays (Table 2 and Figure 4A), to determine which sequence was bound by Gax. We found that A6 strongly bound Gax in EMSAs using recombinant Gax protein (Figure 4B), and supershifts using anti-Gax antibody (40) demonstrated that the Gax protein was bound in this complex (Figure 4C). The sequence of A6 contained an AT-rich sequence, with ATTACAATTA at its core. Because this resembles the DNA binding sites of other homeodomain proteins (62), we systematically mutated residues beginning one residue to the 5′ and 3′ end of this core sequence (labeled A6Mt1 through A6Mt11) and repeated the EMSAs. All of the mutations resulted in a significant decrease in Gax binding, but mutating residues in the second ATTA in essence eliminated Gax binding altogether. Indeed, in particular, mutating the first T (A6Mt9) abolished binding completely (Figure 4D).
Figure 4. Identification of the 15 kb upstream binding site for Gax.
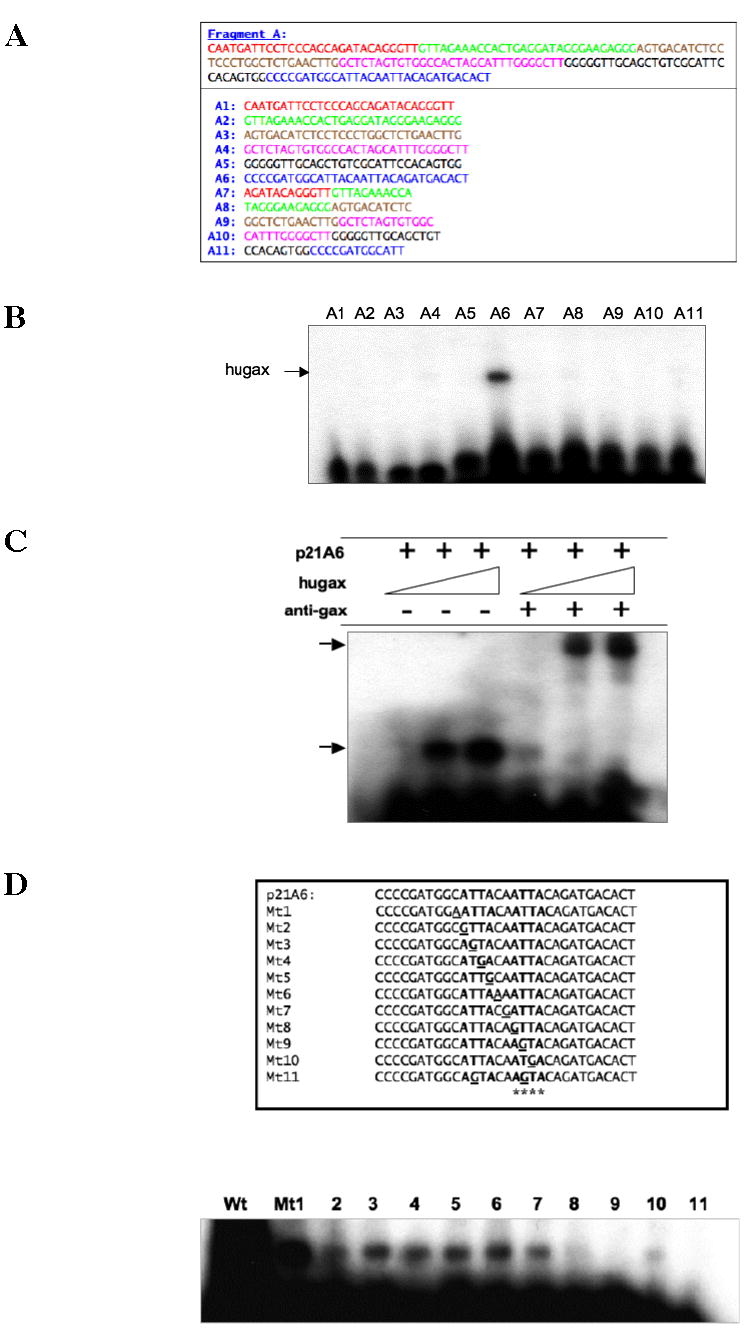
A. Probes for electrophoretic mobility shift assay. Overlapping probes were designed to span the entire length (200 bp) of Fragment A as shown. B. Electrophoretic mobility shift assays. The various probes from A were end-labeled with γ-32P-dATP and then subjected to EMSA using nuclear extracts from HUVECs transduced with full length Gax expression construct. Only Fragment A6 produced a band shift. C. Supershift of binding activity to Fragment A6 using anti-Gax antibody. The nuclear extracts in B were subjected to supershifts using fragment A6 and anti-Gax antibody (40). A marked supershift was observed. D. Site-directed mutagenesis of the AT-rich core of Fragment A6. Individual nucleotides in the ATTACAATTA core of the putative Gax binding site in Fragment A6 were mutated, end-labeled, and subjected to EMSA, as described in Experimental Procedures. Each experiment was performed at least three times.
To study the functional consequences of altering the sequence of this AT-rich site, we constructed three expression vectors (Figure 5A). One (p21A6-Luc) contained the A6 sequence inserted upstream of a minimal promoter and a Luciferase reporter. The others contained A6Mt1 upstream of Luciferase (p21A6Mt1-Luc), because this mutation resulted in the least diminution of Gax binding compared to control, and A6Mt11 (p21A6Mt11-Luc), because this mutant completely abolished Gax binding (Figure 4D). In cotransfection experiments, we observed that Gax effectively trans-activated the reporter containing sequence A6 (p21A6-Luc). In contrast, neither mutant (p21A6Mt1-Luc or p21A6Mt11-Luc) could be trans-activated by Gax (Figure 5B). From this, coupled with our initial ChIP results, we conclude that the AT-rich site that we have identified at -15 kb is likely to be an important regulatory element contributing to the activation of p21WAF1/CIP1 expression by Gax and may be an enhancer.
Figure 5. Effect of mutating the 15 kb upstream binding site for Gax on transactivation by Fragment A.
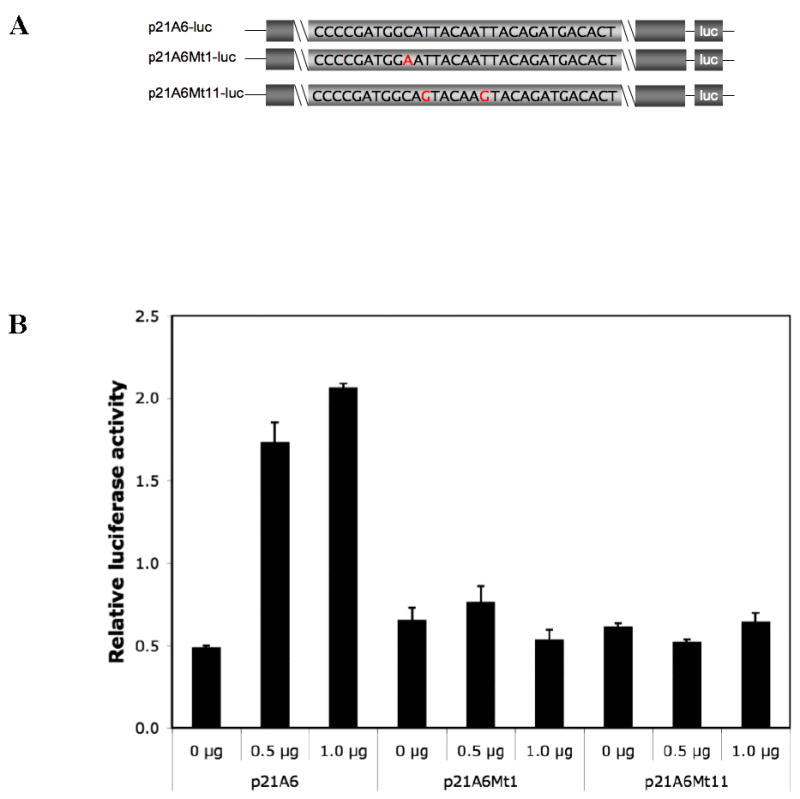
A. Promoter constructs. The A6 fragments subjected to site-directed mutagenesis were inserted upstream of Luciferase in a promoter/reporter construct as described in Experimental Procedures. B. Effect of mutations in A6 on transactivation by Gax. The constructs described in A were cotransfected with pCDNA3.1-Flag-hugax at different ratios. Both the A6Mt1 and A6Mt11 abolished transactivation, even though A6Mt1 did not abolish binding, as shown in Figure 6D. Each experiment was performed at least three times.
Next, we asked whether other AT-rich sequences resembling the AT-rich sequence in A6 exist in the p21WAF1/CIP1 chromatin exist and could also be bound and transactivated by Gax. Four additional such sequences were identified in the 15 kb upstream chromatin previously surveyed, two near Fragment B (dubbed B1 and B2), one in Fragment C (dubbed C1), and one near Fragment F (dubbed F1). All were able to bind Gax in vitro in electrophoretic mobility shift assays (Figure 6A). Because it was the only such sequence in the 2.4 kb p21WAF1/CIP1 promoter (Figure 6A), we were most interested in determining whether sequence C from the original ChIP contributed to regulation of p21WAF1/CIP1 promoter activity. Consequently, we deleted the two ATTA regions in the 200 bp fragment C (Figure 3A), inserted it upstream from Luciferase as above, and then compared its ability to drive Luciferase expression with that of unmodified fragment C itself (p21C-Luciferase), finding that fragment C was able to drive Luciferase expression. However, deleting the ATTA sequences in fragment C (p21C-mut-Luciferase) nearly completely abolished Luciferase activity compared to fragment C (Figure 6B). These results suggest that this AT-rich site is also active in regulating Gax expression.
Figure 6. Identification of an additional AT-rich sequence to which Gax can bind and activate transcription.
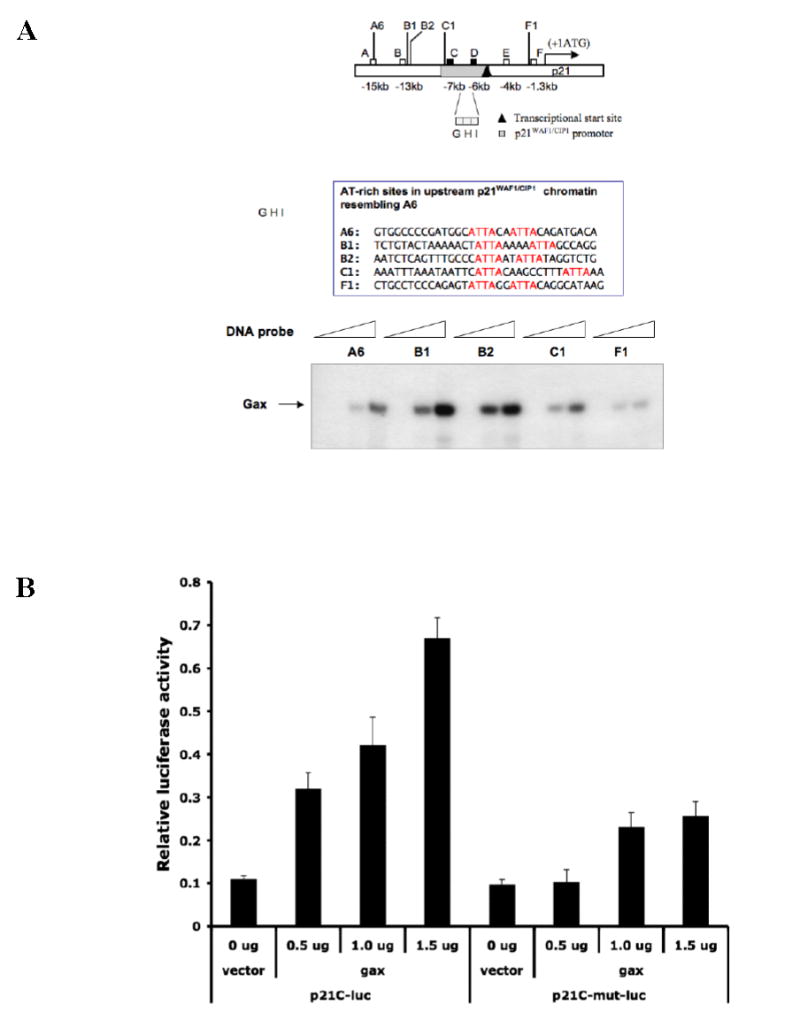
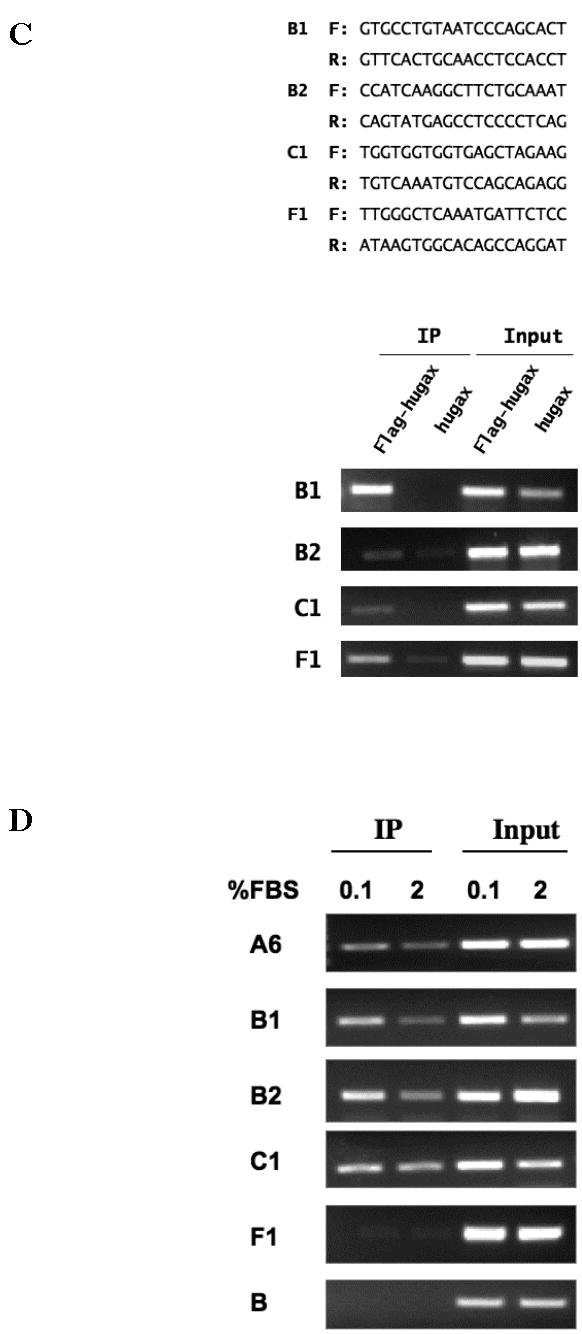
A. Additional AT-rich sequences in the upstream chromatin of p21WAF1/CIP1. Additional AT-rich sequences containing ATTA repeats resembling the one identified by ChIP were located on the upstream chromatin of p21WAF1/CIP1. EMSAs were then performed using probes containing these sequences, as well as the sequence identified by ChIP (sequence A6) as a Gax binding site. B. Effect deleting the ATTA repeat in the core p21WAF1/CIP1 promoter. The AT-rich sequence containing the ATTA was deleted from sequence C and then the wild type (p21C) and mutated sequences (p21C-mut) extended to 1.5 kb using PCR and placed upstream of Luciferase in the pGL3 plasmid to produce p21C-Luciferase and p21C-mut-Luciferase. These constructs were then cotransfected with pCDNA3.1-Flag-hugax at different ratios as described in Experimental Procedures. Deleting the sequence containing two ATTA elements nearly completely abolished transactivation of p21C-Luciferase by Gax. C. ChIP assays for additional ATTA-containing sequences identified in the p21WAF1/CIP1 chromatin. ChIP assay was carried out using the same methodology as in Figure 4B, using the primer sets listed, which were targeted for sequences B1, B2, C1, and F1 (see text and Figure 6A for descriptions), and exogenously expressed Flag-hugax. D. ChIP assay for binding of endogenous Gax. ChIP assays were carried out on untransduced HUVECs in either normal growth medium (2% FBS containing 5 ng/ml VEGF) or in low serum medium (0.1% FBS) using a polyclonal rabbit anti-Gax antibody. Sequence B was used as a negative control, as no enrichment for sequence bound to Gax was observed in the previous ChIP assay (Figure B4). Each experiment was performed at least three times.
Finally, to verify that the AT-rich sequences identified by comparison with sequence A6 could also bind Gax, we repeated the ChIP assay in Figure 3, this time using PCR primers sets designed to encompass sequences B1, B2, C1, and F1. In all cases, Gax binding to chromatin containing these sequences was detected (Figure 6C). Finally, because all of our ChIP assays had been performed using HUVECs expressing Gax via an exogenous vector, we wished to determine whether endogenously expressed Gax binds the putative Gax binding sites identified thus far as well. We therefore repeated the ChIP assays with the primer sets for sequences A6, B1, B2, C1, and F1, only this time in HUVECs not expressing exogenous Gax and using a polyclonal rabbit antibody against Gax (40). In for all of these sequences, we were able to detect enrichment of chromatin containing these sequences in the Gax immunoprecipitate, with a slight decrease in Gax binding to some of the sequences in HUVECs incubated in 2% serum plus 5 ng/ml VEGF when compared with HUVECs incubated in 0.1% serum (Figure 6D, sequences B1 and B2). In contrast, using fragment B from our original ChIP assay (which contains none of the AT-rich sequences identified and did not enrich in the ChIP assay; see Figure 3), we observed no enrichment using anti-Gax antibody (Figure 6D).
Effect of deletions of the homeodomain or the CAX repeat on Gax function
We have previously reported that Gax inhibits proliferation in VSMCs and that this effect depends upon p21WAF1/CIP1, as shown by observation that p21−/− fibroblasts are not susceptible to Gax-induced cell cycle arrest (37). Consequently, we wished to determine the functional consequences of silencing endogenous Gax expression on p21WAF1/CIP1 expression in ECs and the ability of Gax truncates to induce cell cycle arrest. First, we examined Gax and p21WAF1/CIP1 expression in HUVECs incubated in either normal growth medium (containing 2% FBS and 5 ng/ml VEGF) or low serum conditions (0.1% FBS, no VEGF). In the presence of serum, both Gax and p21WAF1/CIP1 are coordinately downregulated (Figure 7A). Next, we transduced HUVECs with an shRNA adenoviral vector targeted against Gax that has been reported to downregulate Gax by 40–50% in brain microvascular endothelial cells (Ad.shMeox2/Gax) (52) and compared Gax, p53, and p21WAF1/CIP1 expression at various MOI (Figure 7B). As was the case with overexpression of Gax (not shown), p53 levels were not affected by shRNA-mediated blockade of Gax at any MOI used. At an MOI=400, Gax protein levels, however, were decreased by 39.4% as measured by densitometry normalized to α-tubulin, consistent with previous results using this vector, and p21WAF1/CIP1 was downregulated by 59.5% (Figure 7B).
Figure 7. Gax expression causes G0/G1 cell cycle arrest in HUVECs associated with downregulation of p21WAF1/CIP1.
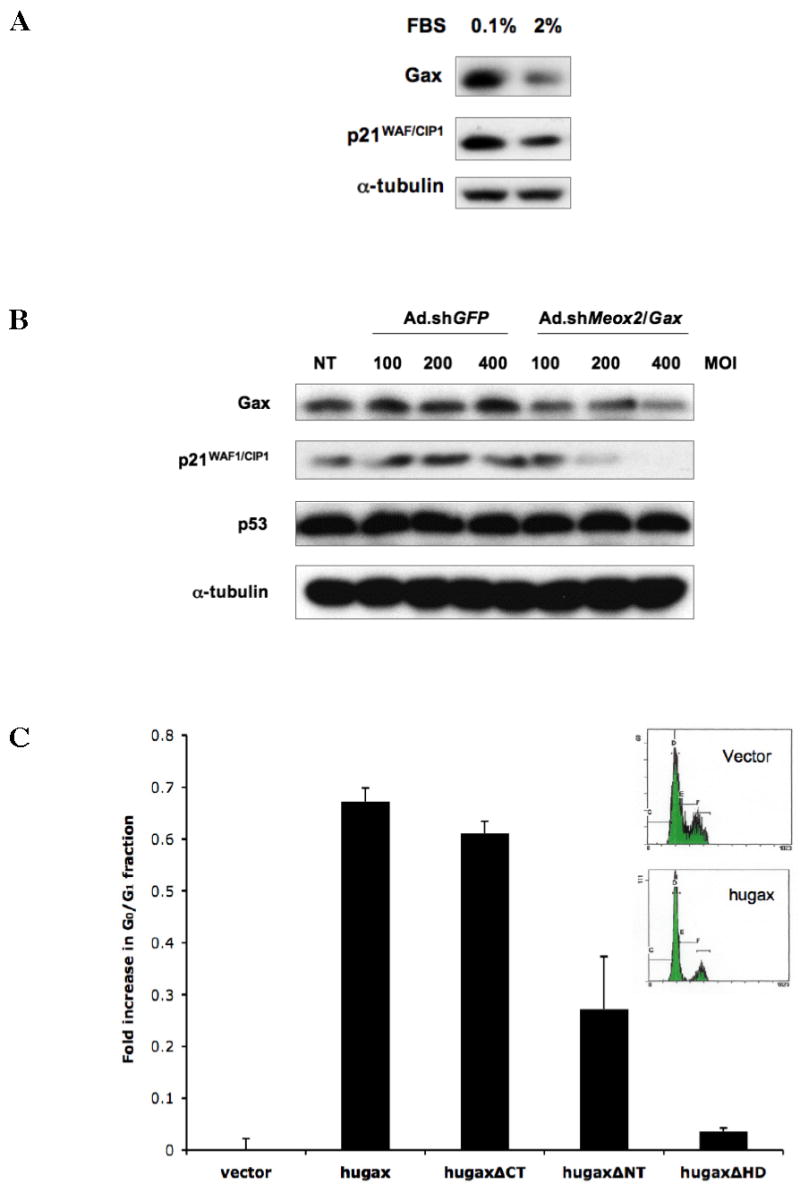
A. Gax and p21WAF1/CIP1 are coordinately regulated by serum. Sparsely plated HUVECs were incubated in either 0.1% serum or 2% serum supplemented with 5 ng/ml VEGF for 24 hours, after which they were harvested for Western blot. Gax was downregulated 50%, and p21WAF1/CIP1 was downregulated 35%, as determined by densitometry. B. Inhibition of Gax with shRNA also downregulates p21WAF1/CIP1. HUVECs were transduced with either Ad.shGFP or Ad.shMeox2/Gax (52) at the MOIs indicated and then incubated overnight, after which protein was harvested for Western blotting with anti-Gax and anti-p21WAF1/CIP1 antibodies. Gax was downregulated by approximately 40% compared to contro, but p21 was downregulated by close to 90%, as measured by densitometry. No change in p53 levels were observed. The above experiment was repeated three times. C. Gax induces cell cycle arrest. HUVECs were incubated in low serum (0.1%) medium for 24 hrs, after which they were stimulated with 10% serum and 5 ng/ml VEGF for 24 hours and then harvested for flow cytometry. There was a marked increase in the G0/G1 fraction (p <0.05) and a concomitant decrease in the S-phase fraction. Each experiment was performed at least three times.
Finally, in order to correlate Gax-dependent upregulation of p21WAF1/CIP1 expression with function, we determined the effect of expressing Gax and its truncates on HUVEC cell cycle. Sparsely plated, randomly cycling HUVECs were transfected with Gax or its truncates using empty vector for a control, incubated overnight in normal growth medium, and then harvested for cell cycle analysis. From the 30–40% fraction of HUVECs that started in G0/G1 under these conditions, Gax expression significantly increased G0/G1 fraction by approximately 70% (Figure 7C). Deleting either the CAX repeat-containing N-terminal domain of Gax or the Gax homeodomain abrogated or greatly diminished the Gax-dependent increase in G0/G1 fraction, indicating that these domains are both critical for this effect. Taken together with previous results showing that Gax-induced cell cycle arrest does not occur in p21−/− fibroblasts (37), these results suggest that both the homeodomain and N-terminal domain are required for p21WAF1/CIP1 induction and subsequent cell cycle arrest in ECs.
DISCUSSION
Interactions between tumors and their surrounding stroma, particularly angiogenesis, are critical in regulating the growth and metastasis of tumors. One of the early phenotypic changes that ECs undergo during angiogenesis is reentry into the cell cycle. Thus, identifying the factors that regulate EC proliferation is critical to understanding the process of angiogenesis and to developing therapeutic strategies to block it. One such strategy is to target EC proliferation in response to proangiogenic factors, such as VEGF (8) or bFGF (63). Another strategy is to target EC proliferation as part of antiangiogenic therapy, often by targeting signaling pathways downstream from the binding of proangiogenic factors to cell surface receptors, including the transcriptional programs that they activate. Because of the importance of EC proliferation in angiogenesis, the purpose of this study was to elucidate in more detail how Gax induces p21WAF1/CIP1 expression and thereby induces EC cell cycle arrest. Specifically, we were interested in (1) mapping the major domains of the Gax protein that are important in regulating this activity and (2) identifying potential DNA binding sites responsible for activation of the p21WAF1/CIP1 promoter by Gax.
Our results indicate that the homeodomain is critical for the activation of p21WAF1/CIP1 expression by Gax. Moreover, the CAX (opa) (58) repeat in the N-terminal domain is also likely involved in transcriptional activation by Gax. This finding is compatible with the observation that the expansion of the CAX repeats of other homeodomain genes have been associated with neurodegenerative diseases and autism (59), even though the molecular function of the poly-amino acid stretches encoded by these repeats remains unclear. Our results suggest one possible function for this domain in at least one homeobox gene, Gax. Moreover, the hypothesis that cell cycle arrest due to Gax depends upon its ability to induce p21WAF1/CIP1 expression is supported by our observation that Gax truncates in which the homeodomain or the N-terminal domain containing the CAX repeat is deleted lose both the ability to induce p21WAF1/CIP1 and to induce G0/G1 cell cycle arrest. These results suggest an important role for Gax in regulating EC proliferation. We acknowledge that many of our results were obtained using exogenous vectors to drive the expression of Gax. However, the observation that silencing endogenous Gax with shRNA also results in the downregulation of p21WAF1/CIP1 (Figure 7B) suggests that this result is indeed physiologically relevant, as does the observation that the sequences identified can also bind endogenous Gax in the ChIP assay (Figure 6D).
We were also able to identify putative Gax binding sites in the chromatin upstream from the p21WAF1/CIP1 transcriptional start site. The results of this assay identified multiple ATTA-containing sequences to which Gax can bind. The first sequence identified (A6) strongly resembles a universal homeobox DNA consensus sequence [Figure 4 and (62,64)]. That the results of our experiments in which individual residues within this binding site were mutated agree with results showing the importance of a CAATTA core sequence in homeodomain-binding sites (62). Based on the identification of A6, we also identified other ATTA-containing sites in the upstream p21WAF1/CIP1 chromatin that can also be bound by Gax (Figure 6), and these also resembled homeobox gene binding sites. Of these, only one (sequence C1) is located within the 2.4 kb p21WAF1/CIP1 promoter, but both A6 and C1 can drive p21WAF1/CIP1 promoter activity in cotransfection experiments (Figures 5B and 6B).
The importance of homeobox genes in the regulation of endothelial cell phenotype during tumor-induced angiogenesis is becoming increasingly more apparent, with the recent descriptions of several homeobox genes that promote (15–19,21,22,52) or inhibit (20,23–26) the angiogenic phenotype. We have now shown that the homeobox gene Gax upregulates p21WAF1/CIP1 expression through at least two CAATTA-containing sites, a site in the p21WAF1/CIP1 promoter and a site 15 kb upstream from the transcriptional start site. Moreover, we have shown that it is the Gax homeodomain that is responsible for binding to this site and that the Gax polyhistidine (opa) repeat is involved in mediating transcriptional activation of the p21WAF1/CIP1 gene. Given that it is known that HOXA10 can bind to the p21WAF1/CIP1 promoter through the recruitment of its trimeric partners Pbx1 and Meis1 in myelomonocytic cells and thus induce differentiation (65), it is tempting to speculate that Gax may activate p21WAF1/CIP1 through a similar mechanism and/or be involved in the regulation of EC differentiation. Future studies will investigate these possibilities.
The results of the present study are consistent with our previously published data about Gax activity in vascular cells indicating that it inhibits VSMC and EC proliferation (20,25,32,37). However, we must note that recently Wu et al (52) reported that, in the cerebral vasculature, Gax appears to have more proangiogenic effects. In brain ECs, downstream effects include transcriptionally suppressing AFX1 forkhead-mediated apoptosis, and silencing of Gax results in reductions in brain capillary density and attenuation of the angiogenic response to hypoxia. Potential explanations for this difference could include cell type-specific differences in Gax activity or more complex function in regulating the state of EC differentiation than our current understanding. More work will be needed to determine which of these is responsible for apparently different activities of Gax in two different vascular beds or if it is a combination of the two.
Taken together, however, our data coupled with other studies suggest that Gax is an important regulator of EC proliferation and angiogenesis. Moreover, given how Gax inhibits NF-κB signaling, the identification of a putative Gax binding site responsible for the activation of p21WAF1/CIP1 expression will permit us to narrow down candidate chromatin sequences in NF-κB-dependent genes that might mediate the ability of Gax to block activation of target genes by NF-κB, as well as other Gax-dependent promoters. These studies will allow us to determine the molecular mechanisms by which Gax inhibits EC activation and angiogenesis, as well as suggesting potential strategies for inhibiting angiogenesis by modulating Gax activity.
Acknowledgments
The work described in this article was supported by the U. S. Department of Defense (DAMD17-02-1-0511 and DAMD17-03-1-0292) and the National Cancer Institute (NCI 1 R01 CA111344). We would also like to thank Dr. Arnold Rabson (UMDNJ-Robert Wood Johnson Medical School, New Brunswick, NJ) for his critical reading and helpful advice.
References
- 1.Boehm T, Folkman J, Browder T, O'Reilly MS. Nature. 1997;390(6658):404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Semin Oncol. 2002;29(6 Suppl 16):15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Folkman J. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 4.Bergers G, Benjamin LE. Nat Rev Cancer. 2003;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J. Cancer Biol Ther. 2003;2(4 Suppl 1):S127–133. [PubMed] [Google Scholar]
- 6.Cross MJ, Claesson-Welsh L. Trends Pharmacol Sci. 2001;22(4):201–207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 7.Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. Trends Biochem Sci. 2003;28(9):488–494. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- 8.Ferrara N, Gerber HP, LeCouter J. Nat Med. 2003;9(6):669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 9.Spencer JA, Major ML, Misra RP. Mol Cell Biol. 1999;19(6):3977–3988. doi: 10.1128/mcb.19.6.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Sukumar S. Cancer Biol Ther. 2003;2(5):524–525. doi: 10.4161/cbt.2.5.525. [DOI] [PubMed] [Google Scholar]
- 11.Ford HL. Cell Biol Int. 1998;22(6):397–400. doi: 10.1006/cbir.1998.0329. [DOI] [PubMed] [Google Scholar]
- 12.Gorski DH, Walsh K. Trends Cardiovasc Med. 2003;13(6):213–220. doi: 10.1016/s1050-1738(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 13.Krumlauf R. Cell. 1994;78(2):191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 14.McGinnis W, Krumlauf R. Cell. 1992;68:283–302. doi: 10.1016/0092-8674(92)90471-n. [DOI] [PubMed] [Google Scholar]
- 15.Boudreau N, Andrews C, Srebrow A, Ravanpay A, Cheresh DA. J Cell Biol. 1997;139(1):257–264. doi: 10.1083/jcb.139.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boudreau NJ, Varner JA. J Biol Chem. 2004;279:4862–4868. doi: 10.1074/jbc.M305190200. [DOI] [PubMed] [Google Scholar]
- 17.Bruhl T, Urbich C, Aicher D, Acker-Palmer A, Zeiher AM, Dimmeler S. Circ Res. 2004;94:743–751. doi: 10.1161/01.RES.0000120861.27064.09. [DOI] [PubMed] [Google Scholar]
- 18.Charboneau A, East L, Mulholland N, Rohde M, Boudreau N. Angiogenesis. 2005:1–8. doi: 10.1007/s10456-005-9016-7. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Xu B, Arderiu G, Hashimoto T, Young WL, Boudreau N, Yang GY. J Cereb Blood Flow Metab. 2004;24(11):1280–1287. doi: 10.1097/01.WCB.0000141770.09022.AB. [DOI] [PubMed] [Google Scholar]
- 20.Gorski DH, Leal AD. J Surg Res. 2003;111(1):91–99. doi: 10.1016/s0022-4804(03)00042-8. [DOI] [PubMed] [Google Scholar]
- 21.Myers C, Charboneau A, Boudreau N. J Cell Biol. 2000;148(2):343–351. doi: 10.1083/jcb.148.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mace KA, Hansen SL, Myers C, Young DM, Boudreau N. J Cell Sci. 2005;118(Pt 12):2567–2577. doi: 10.1242/jcs.02399. [DOI] [PubMed] [Google Scholar]
- 23.Myers C, Charboneau A, Cheung I, Hanks D, Boudreau N. Am J Pathol. 2002;161(6):2099–2109. doi: 10.1016/S0002-9440(10)64488-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa T, Abe M, Yamazaki T, Miyashita H, Niwa H, Kokubun S, Sato Y. Arterioscler Thromb Vasc Biol. 2003;23(2):231–237. doi: 10.1161/01.atv.0000052670.55321.87. [DOI] [PubMed] [Google Scholar]
- 25.Patel S, Leal AD, Gorski DH. Cancer Res. 2005;65(4):1414–1424. doi: 10.1158/0008-5472.CAN-04-3431. [DOI] [PubMed] [Google Scholar]
- 26.Rhoads K, Arderiu G, Charboneau A, Hansen SL, Hoffman W, Boudreau N. Lymphat Res Biol. 2005;3(4):240–252. doi: 10.1089/lrb.2005.3.240. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Chung S, Sukumar S. Mol Cell Biol. 2004;24(2):924–935. doi: 10.1128/MCB.24.2.924-935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Nature. 2000;405(6789):974–978. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- 29.Raman V, Tamori A, Vali M, Zeller K, Korz D, Sukumar S. J Biol Chem. 2000;275(34):26551–26555. doi: 10.1074/jbc.C000324200. [DOI] [PubMed] [Google Scholar]
- 30.Candia AF, Kovalik JP, Wright CV. Nucleic Acids Res. 1993;21(21):4982. doi: 10.1093/nar/21.21.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Candia AF, Wright CV. Mech Dev. 1995;52(1):27–36. doi: 10.1016/0925-4773(95)00384-d. [DOI] [PubMed] [Google Scholar]
- 32.Gorski DH, LePage DF, Patel CV, Copeland NG, Jenkins NA, Walsh K. Mol Cell Biol. 1993;13(6):3722–3733. doi: 10.1128/mcb.13.6.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LePage DF, Altomare DA, Testa JR, Walsh K. Genomics. 1994;24(3):535–540. doi: 10.1006/geno.1994.1663. [DOI] [PubMed] [Google Scholar]
- 34.Gorski DH, LePage DF, Walsh K. Biotechniques. 1994;16(5):856–865. [PubMed] [Google Scholar]
- 35.Maillard L, Van Belle E, Smith RC, Le Roux A, Denefle P, Steg G, Barry JJ, Branellec D, Isner JM, Walsh K. Cardiovasc Res. 1997;35(3):536–546. doi: 10.1016/s0008-6363(97)00147-8. [DOI] [PubMed] [Google Scholar]
- 36.Perlman H, Sata M, Le Roux A, Sedlak TW, Branellec D, Walsh K. EMBO J. 1998;17(13):3576–3586. doi: 10.1093/emboj/17.13.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith RC, Branellec D, Gorski DH, Guo K, Perlman H, Dedieu JF, Pastore C, Mahfoudi A, Denefle P, Isner JM, Walsh K. Genes Dev. 1997;11(13):1674–1689. doi: 10.1101/gad.11.13.1674. [DOI] [PubMed] [Google Scholar]
- 38.Witzenbichler B, Kureishi Y, Luo Z, Le Roux A, Branellec D, Walsh K. J Clin Invest. 1999;104(10):1469–1480. doi: 10.1172/JCI7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng JH, Yang Z, Xu J, Qiu ML, Lin KC. Hepatobiliary Pancreat Dis Int. 2006;5(2):242–245. [PubMed] [Google Scholar]
- 40.Skopicki HA, Lyons GE, Schatteman G, Smith RC, Andres V, Schirm S, Isner J, Walsh K. Circ Res. 1997;80(4):452–462. doi: 10.1161/01.res.80.4.452. [DOI] [PubMed] [Google Scholar]
- 41.Quinn LM, Johnson BV, Nicholl J, Sutherland GR, Kalionis B. Gene. 1997;187(1):55–61. doi: 10.1016/s0378-1119(96)00706-8. [DOI] [PubMed] [Google Scholar]
- 42.Murthi P, So M, Gude NM, Doherty VL, Brennecke SP, Kalionis B. Placenta. 2006 doi: 10.1016/j.placenta.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Candia AF, Wright CV. Int J Dev Biol. 1996;40(6):1179–1184. [PubMed] [Google Scholar]
- 44.Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY. J Biol Chem. 2001;276(10):7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 45.Shono T, Ono M, Izumi H, Jimi SI, Matsushima K, Okamoto T, Kohno K, Kuwano M. Mol Cell Biol. 1996;16(8):4231–4239. doi: 10.1128/mcb.16.8.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klein S, de Fougerolles AR, Blaikie P, Khan L, Pepe A, Green CD, Koteliansky V, Giancotti FG. Mol Cell Biol. 2002;22(16):5912–5922. doi: 10.1128/MCB.22.16.5912-5922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida A, Yoshida S, Ishibashi T, Kuwano M, Inomata H. Invest Ophthalmol Vis Sci. 1999;40(7):1624–1629. [PubMed] [Google Scholar]
- 48.Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. J Cell Biol. 1998;141(4):1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scatena M, Giachelli C. Trends Cardiovasc Med. 2002;12(2):83–88. doi: 10.1016/s1050-1738(01)00151-7. [DOI] [PubMed] [Google Scholar]
- 50.Kinsella TM, Nolan GP. Hum Gene Ther. 1996;7(12):1405–1413. doi: 10.1089/hum.1996.7.12-1405. [DOI] [PubMed] [Google Scholar]
- 51.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 52.Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, Brooks AI, Kanagala S, Rubio A, Sagare A, Liu D, Li F, Armstrong D, Gasiewicz T, Zidovetzki R, Song X, Hofman F, Zlokovic BV. Nat Med. 2005;11(9):959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- 53.Bustin SA. J Mol Endocrinol. 2000;25(2):169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 54.Pfaffl MW. Nucleic Acids Res. 2001;29(9):E45–45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gorski DH, Leal AD, Goydos JS. J Am Coll Surg. 2003;197(3):408–418. doi: 10.1016/S1072-7515(03)00388-0. [DOI] [PubMed] [Google Scholar]
- 56.Goydos JS, Gorski DH. Clin Cancer Res. 2003;9(16 Pt 1):5962–5967. [PubMed] [Google Scholar]
- 57.Gorski DH. Ann Surg Oncol. 2002;9:S42. [Google Scholar]
- 58.Wharton KA, Yedvobnick B, Finnerty VG, Artavanis-Tsakonas S. Cell. 1985;40:55–62. doi: 10.1016/0092-8674(85)90308-3. [DOI] [PubMed] [Google Scholar]
- 59.Paraguison RC, Higaki K, Sakamoto Y, Hashimoto O, Miyake N, Matsumoto H, Yamamoto K, Sasaki T, Kato N, Nanba E. Biochem Biophys Res Commun. 2005;336(4):1033–1039. doi: 10.1016/j.bbrc.2005.08.212. [DOI] [PubMed] [Google Scholar]
- 60.Regulski M, Harding K, Kostriken R, Karch F, Levine M, McGinnis W. Cell. 1985;43 (1):71–80. doi: 10.1016/0092-8674(85)90013-3. [DOI] [PubMed] [Google Scholar]
- 61.Schneuwly S, Kuroiwa A, Baumgartner P, Gehring WJ. Embo J. 1986;5(4):733–739. doi: 10.1002/j.1460-2075.1986.tb04275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kassis JA, Desplan C, Wright DK, O'Farrell PH. Mol Cell Biol. 1989;9(10):4304–4311. doi: 10.1128/mcb.9.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Straume O, Akslen LA. Am J Pathol. 2002;160(3):1009–1019. doi: 10.1016/S0002-9440(10)64922-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalionis B, O'Farrell PH. Mech Dev. 1993;43(1):57–70. doi: 10.1016/0925-4773(93)90023-q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bromleigh VC, Freedman LP. Genes Dev. 2000;14(20):2581–2586. doi: 10.1101/gad.817100. [DOI] [PMC free article] [PubMed] [Google Scholar]