

Summary
Formation of the apical surface and lumen is a fundamental, yet poorly understood, step in epithelial organ development. We show that PTEN localizes to the apical plasma membrane during epithelial morphogenesis to mediate the enrichment of PtdIns(4,5)P2 at this domain during cyst development in three dimensional culture. Ectopic PtdIns(4,5)P2 at the basolateral surface causes apical proteins to relocalize to the basolateral surface. Annexin 2 (Ax2) binds PtdIns(4,5)P2 and is recruited to the apical surface. Ax2 binds Cdc42, recruiting it to the apical surface. Cdc42 recruits aPKC to the apical surface. Loss of function of PTEN, Ax2, Cdc42 or aPKC prevents normal development of the apical surface and lumen. We conclude that the mechanism of PTEN, PtdIns(4,5)P2, Ax2, Cdc42 and aPKC controls apical plasma membrane and lumen formation.
Introduction
Most organs consist of epithelial tubes (Lubarsky and Krasnow, 2003). Despite diversity of developmental mechanism, tubular organs and related structures, such as alveoli and cysts, share a common organization with the apical (AP) surface lining the central lumen and a basolateral (BL) surface attached to adjoining cells and extracellular matrix (ECM). A key gap in our knowledge of how cells assemble into tubules is how the AP surface and lumen are formed.
Culture systems that recapitulate tube morphogenesis have been useful in understanding the mechanism of tubulogenesis. The three-dimensional (3D) Madin-Darby canine kidney (MDCK) cell system is an excellent model of epithelial morphogenesis in vitro (Debnath and Brugge, 2005; Lubarsky and Krasnow, 2003; O’Brien et al., 2002). MDCK cells embedded in a gel of ECM form cysts, spherical epithelial monolayers enclosing a central lumen (Montesano et al., 1991). Results from MDCK and other systems have led to a general model for tube morphogenesis (Lubarsky and Krasnow, 2003; O’Brien et al., 2002). In this model, the cell interprets an extracellular cue from the ECM and transduces it into a signal to generate the axis of polarity. Following this initial event the most crucial step is the formation of the central lumen and AP PM.
Cdc42 plays a central role in polarization from yeast to mammals. In metazoa one target of Cdc42 is Par6, part of the Par3/Par6/aPKC complex, a master regulator of polarity. Cdc42 is associated with formation of tight junctions (TJ) and the regulation of traffic to the plasma membrane (PM) (Kroschewski et al., 1999; Musch et al., 2001). However, activation of Rac1, but not Cdc42, is needed for epithelial TJ formation (Mertens et al., 2005). Despite this controversy, Cdc42 is activated upon cell-cell contact suggesting a role for this protein during epithelial morphogenesis (Kawakatsu et al., 2002; Kazmierczak et al., 2004). Interestingly, FDG1 a guanine nucleotide exchange factor (GEF) that regulates Cdc42 activity, is needed for lumen formation in vivo (Suzuki et al., 2001).
Here we elucidate a molecular mechanism central to the formation of the AP PM and lumen during morphogenesis in the 3D-MDCK system.
Results
PtdIns(4,5)P2 and PTEN are at the AP PM
To determine the localization of PtdIns(4,5)P2 in MDCK cysts, we stably expressed the PH domain of phospholipase Cδ1, a high affinity marker for PtdIns(4,5)P2 (Rescher et al., 2004), fused to GFP (PHD-GFP). PHD-GFP was distributed mainly to the AP PM of MDCK cysts, where it largely colocalized with the AP marker gp135/podocalyxin (Fig 1a, arrowheads), a smaller degree was found in the BL PM, with a distribution similar to that of actin (Fig S1a). Using PH-Akt-GFP, a probe for products of PI3K (PtdIns(3,4)P2 and PtdIns(3,4,5)P3 we refer to both collectively as PtdIns(3,4,5)P3), we found PtdIns(3,4,5)P3 highly enriched at the BL PM and absent from the AP PM of MDCK cysts in Matrigel (Fig S1b), confirming previous data in collagen (Yu et al., 2003).
Fig 1. PtdIns(4,5)P2 and PTEN localize to the AP PM in cysts.
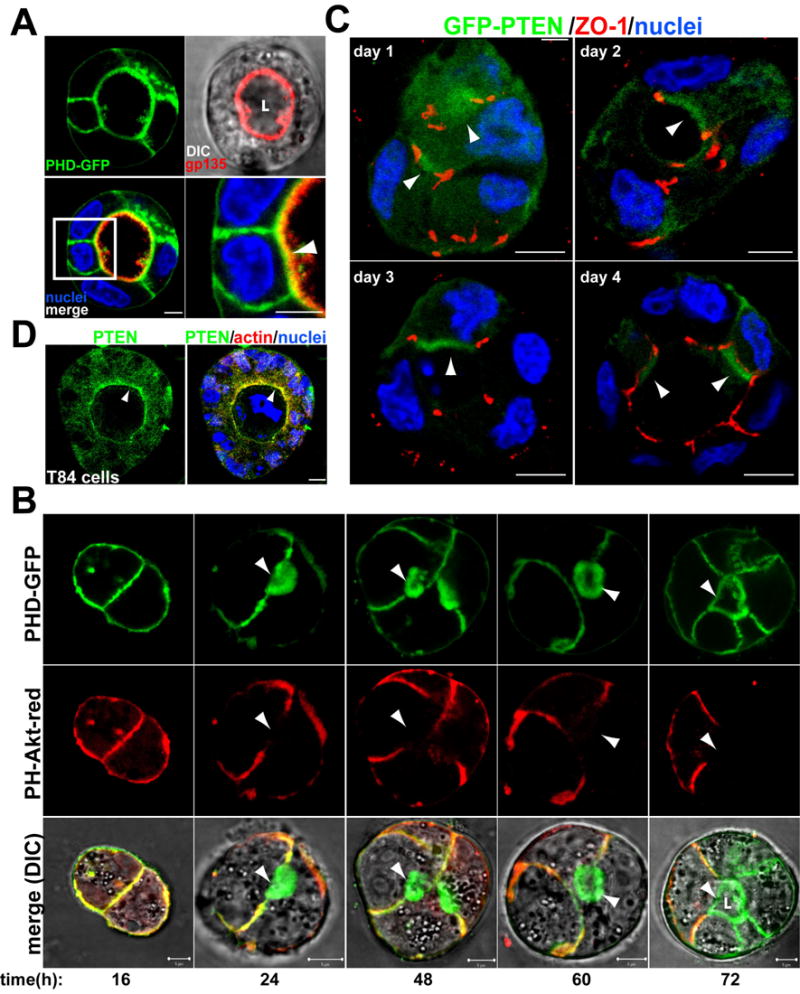
(A) PtdIns(4,5)P2 is enriched in the AP PM. MDCK PHD-GFP cysts were stained for nuclei (blue) and gp135 (red, upper-right panel merged with DIC). In all micrographs single confocal sections through the middle of cysts are shown and nuclei are stained blue unless otherwise indicated. Bottom-right panel shows a magnification of the indicated region of the merged panel (bottom left). Arrowhead indicates the colocalization of PHD-GFP and gp135 at the AP PM. All scale bars are 5 μm unless otherwise indicated.
(B) PtdIns(4,5)P2 segregates from PtdIns(3,4,5)P3 during cystogenesis. MDCK PHD-GFP (top row) and PH-Akt-GFP (middle row, merged with DIC in bottom row) were plated to form cysts for 4 days. Live cells were analyzed by confocal microscopy at 1, 2, 3 or 4d. In this and all other micrographs “L” indicates lumen.
(C) GFP-PTEN localizes to the AP PM. MDCK GFP-PTEN cells forming cysts at 1, 2, 3 and 4d. Cells were stained for ZO-1 (red). Arrowheads indicate GFP-PTEN at the AP PM.
(D) PTEN localizes to the AP PM in T84 human colon cysts. Endogenous PTEN could not be localized in MDCK cysts, but could be localized in T84 cysts. T84 human colon cells formed cysts for 8d; stained for PTEN (green), actin (red) and nuclei, and visualized. Arrowheads indicate PTEN at the AP PM.
We analyzed PH-Akt-dsRed and PHD-GFP localization during cystogenesis. At 16h when cysts still lacked lumens, PtdIns(3,4,5)P3 and PtdIns(4,5)P2 were colocalized to cell-cell and cell-ECM contacts (Fig 1b, left). As soon as lumens started to form (24–60h) and in mature cysts (72h) PtdIns(4,5)P2 became confined mainly to the AP PM, delimiting the newly-formed lumen, whereas PtdIns(3,4,5)P3 remained associated with cell-cell contacts (Fig 1b, arrowheads lumen). By contrast, PtdIns(3,4,5)P3 became exclusively BL (Fig 1b, right column). These data suggest that segregation of PtdIns(4,5)P2 and PtdIns(3,4,5)P3 to distinct PM domains may be important in AP PM and lumen formation.
Next we investigated the mechanism of enrichment of PtdIns(4,5)P2 and depletion of PtdIns(3,4,5)P3 in the AP PM. Phosphatase and tensin homolog on chromosome 10 (PTEN) is a lipid-phosphatase whose substrates are PtdIns(3,4,5)P3, and to a lesser extent PtdIns(3,4,)P2. PTEN thus antagonizes PI3K and increases the level of PtdIns(4,5)P2 by converting PtdIns(3,4,5)P3 to PtdIns(4,5)P2 (Maehama and Dixon, 1998). We determined the location of PTEN using MDCK cells expressing GFP-PTEN, which localizes similarly to endogenous PTEN (Lacalle et al., 2004). Initially, most GFP-PTEN was in the cytoplasm, although a small fraction was in the AP region surrounded by TJ (Fig 1c, day1, arrowheads). At later stages (day 2–4), GFP-PTEN was mainly in the AP PM, delimiting the lumen (Fig 1c). GFP-PTEN also associated with cell-cell junctions overlapping extensively with PtdIns(4,5)P2 (Fig 1c). We found the same localization of endogenous PTEN in human colon T84 cells (Fig 1d). Thus, PTEN is highly enriched in the AP PM, suggesting involvement in depletion of AP PtdIns(3,4,5)P3 and/or enrichment of AP PtdIns(4,5)P2.
Loss of PTEN function inhibits lumen formation
To test if PTEN is needed to form the AP PM and lumen, we depleted PTEN using siRNAs. Endogenous PTEN was reduced to 20–36% of control using 2 different siRNA heteroduplexes, PTEN1 and PTEN2 (Fig 2a). When siRNA transfected cells were plated to form cysts, most control cells formed normal lumens (91%) (Fig 2b), MDCK cysts with reduced PTEN formed cysts of similar size, but only 34% of them had normal lumens (with PTEN2 siRNA; 36% with PTEN1 siRNA). Instead, most cysts with reduced PTEN had multiple small lumens, though the BL marker β-catenin was localized normally (Fig 2b, left; quantization in 2c). Also, F-actin was greatly reduced in PTEN-depleted cysts (Fig 2b, right panels). To confirm the role of PTEN we used bpV(pic), an inhibitor of PTEN (Schmid et al., 2004). We observed a dose-dependent inhibition of lumen formation by bpV(pic) (Fig 2d). Then, we analyzed the effect of PTEN inhibition on the distribution of PtdIns(4,5)P2 and PtdIns(3,4,5)P3 during cystogenesis. Whereas in control cells PtdIns(4,5)P2 segregated from PtdIns(3,4,5)P3 in the AP PM, in bpV(pic) treated cells both PtdIns remained homogenously distributed in the PM, resulting in cysts with no central lumen (Fig 2e, arrows indicate PtdIns(4,5)P2 and PtdIns(3,4,5)P3; arrowheads indicate TJ). Thus PTEN is required for enrichment of PtdIns(4,5)P2 at the AP PM and concomitant removal of PtdIns(3,4,5)P3 from this domain. PTEN is also needed for AP PM and lumen formation.
Fig 2. Disruption of PTEN with siRNA or a specific inhibitor blocks central lumen formation.
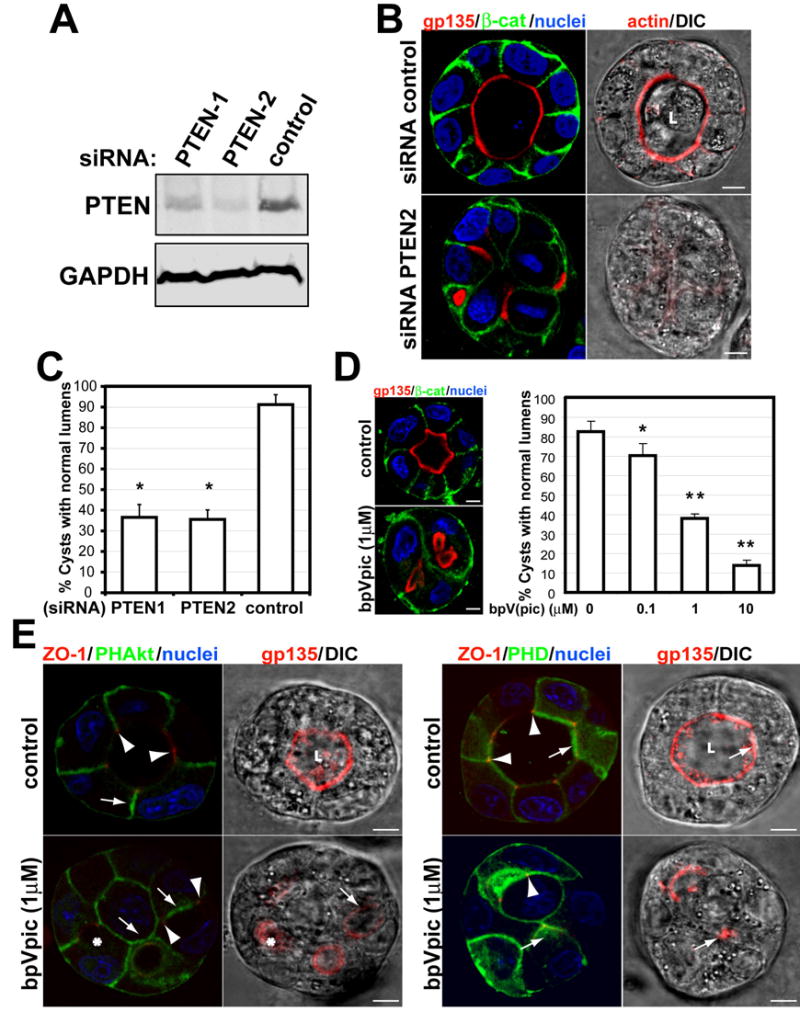
(A) Down-regulation of PTEN by siRNA. Total lysates of MDCK cells transfected with siRNAs PTEN-1 and PTEN-2 or with control siRNA formed cysts for 72h; then western blotted for PTEN and GAPDH.
(B) Effect of siRNA PTEN-2 on lumen formation. Cells were transfected with PTEN-2 (bottom row), or control (top row) siRNA and plated to form cysts for 72h. Cells were stained to detect gp135 (red), β-catenin (green), nuclei (left panels) and actin (red, right panels merged with DIC).
(C) Quantitation of cysts with normal lumens in cells transfected with control siRNA or specific siRNA PTEN-1, PTEN-2. Values are mean ±SD from 4 different experiments. N=100 cysts/experiment. *P<0.001.
(D) The PTEN inhibitor bpV(pic) disrupts lumen formation in a dose-dependent manner. MDCK cells forming cysts for 48h were treated with indicated concentrations of bpV(pic). After 48h cells were stained to detect gp135 (red), β-catenin (green), nuclei (blue)-left panels. Right panel is quantitation of cysts with normal lumens in cells treated with 0 (control), 0.1, 1 or 10μM bpV(pic). The values shown are mean ±SD from 3 different experiments. N=100/experiment. *P<0.005; **P<0.001.
(E) BpV(pic) inhibits segregation of phosphoinositides. PHD-GFP (right panels) or PH-Akt-GFP (left panels) cells formed cysts for 4d. The cells were stained to detect ZO-1 (red), nuclei (left panels) and gp135 (red, right panel merged with DIC).
Exogenous PtdIns(4,5)P2 targets AP proteins to the BL domain
Our results suggested that enrichment of PtdIns(4,5)P2 at the AP pole initiates formation of the AP PM and lumen. To test this, we delivered exogenous PtdIns(4,5)P2 to the BL PM of mature cysts. A similar strategy has been used to show the effect of exogenous PtdIns(3,4,5)p3 on neutrophil polarity (Weiner et al., 2002) and the role of PtdIns(3,4,5)p3 in expanding the BL surface of MDCK cells (Gassama-Diagne et al., 2006). Exogenous PtdIns(4,5)P2 induced dramatic shrinkage of the lumen and redistribution of PHD-GFP from the AP to the BL surface, often in a patchy distribution (Fig 3a). We also saw relocalization of the AP markers gp135 and ezrin, and the (TJ) component ZO-1 from the AP to the BL region over 30min (Fig 3b and S2a; arrows indicate ZO-1, gp135 and ezrin localization). Movement seems to occur initially by extension of the AP surface and projections of the lumen out to the cyst periphery, followed by collapse of the lumen. The area occupied by lateral markers shrank, with p58 and PtdIns(3,4,5)p3 ending up in small puncta at the periphery of the cysts at 30min, and eventually disappearing from the basal region (Fig 3c and S2b, arrowheads). This phenotype was not caused by the mere loss of polarity since depolarization induced by calcium depletion did not alter the localization of AP markers (Fig S2c, arrowheads)
Fig 3. Exogenous PtdIns(4,5)P2 relocalizes AP and TJ proteins to the BL PM. MDCK PHD-GFP cells forming cysts for 2d were incubated with histone (control) or PtdIns(4,5)P2-histone complexes (+PtdIns(4,5)P2) for up to 30min, fixed and visualized.
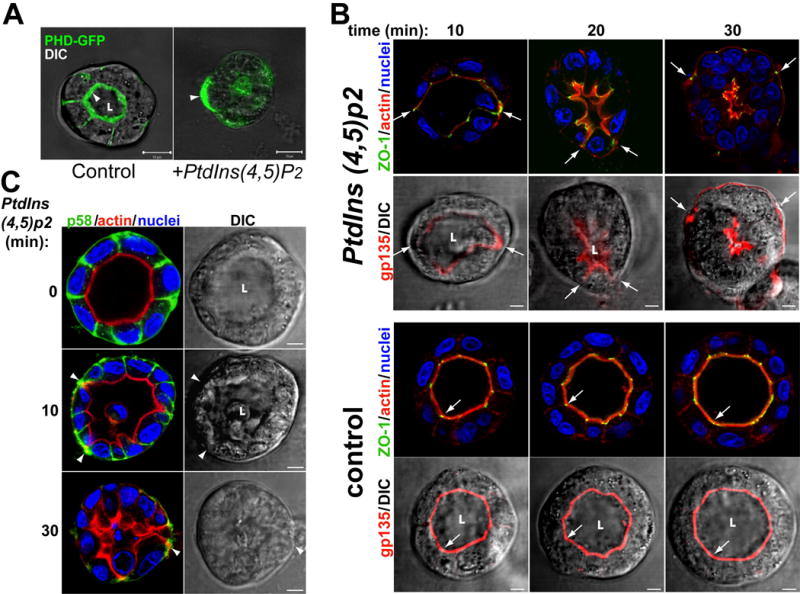
(A) Exogenous PtdIns(4,5)P2 relocalizes PHD-GFP from AP to the basal PM. Arrowheads indicate PHD-GFP enrichment at the AP (control) or basal PM (+PtdIns(4,5)P2). Scale bar 10 μm.
(B) Exogenous PtdIns(4,5)P2 relocalizes gp135, actin and ZO-1 from the AP to the basal PM. Top half of figure was treated with PtdIns(4,5)P2 for indicated time, while bottom half was control. Samples were stained for actin (red), ZO-1 (green) and nuclei in top rows; and gp135 (red) merged with DIC in bottom rows. Arrows indicate AP makers and ZO-1.
(C) Exogenous PtdIns(4,5)P2 disrupts lateral PM. The samples were stained for actin (red), p58 (green) and nuclei (left panels), or DIC (right panels). Arrowheads indicate p58 localization.
We conclude that ectopic insertion of exogenous PtdIns(4,5)P2 into the BL PM is sufficient to relocalize AP and TJ components to the periphery of the cyst, substantiating a key role for this lipid in constructing the AP PM.
Annexin 2 distributes to the AP PM in MDCK cysts
Annexin (Ax2) is targeted to the PM of epithelial cells by binding to PtdIns(4,5)P2 (Rescher et al., 2004). To localize Ax2 in cysts, we stably expressed GFP-Ax2, which was previously described to behave similarly to the endogenous protein (Merrifield et al., 2001). Ax2-GFP was detected mainly in the AP PM of cells in cysts outlining the lumen, with a very minor amount at the BL PM (Fig 4a). Ax2 overlapped extensively with actin (Fig 4a, arrow) suggesting association of Ax2 with the AP actin cytoskeleton. We next analyzed the localization of Ax2-GFP during cystogenesis. The pattern of localization for Ax2-GFP was identical to that observed for PtdIns(4,5)P2 (Fig S3a). Therefore, the similar distributions of PtdIns(4,5)P2 and Ax2 during cysts formation suggests that Ax2 and PtdIns(4,5)P2 might be involved in forming the cortical actin cytoskeleton and AP domain.
Fig 4. Ax2 localizes to the AP PM and is needed for lumen formation.
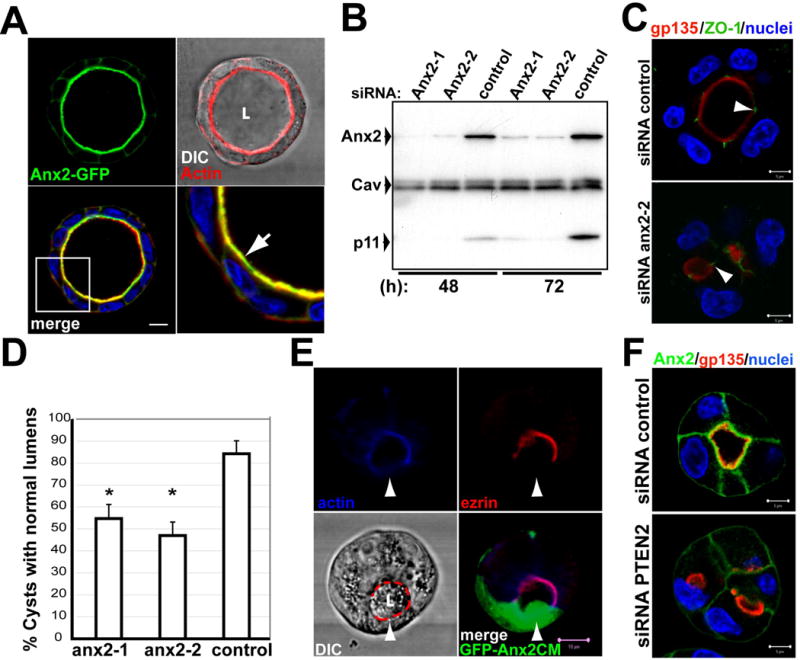
(A) MDCK Ax2-GFP cells were grown for 5 days to form mature cysts, and stained for actin (red) and nuclei (blue). Actin and DIC (top-right panel). Bottom-right panel shows a magnification of the region indicated with a white square in the merged panel (bottom left). Arrow indicates colocalization of Ax2-GFP and actin at the AP PM.
(B) Down-regulation of endogenous Ax2 and p11 by siRNA to Ax2. Total cell lysates of cysts (48 or 72h) treated with siRNAs Ax2-1 and Ax2-2 against canine Ax2, or control siRNA, were analyzed by western blot for Ax2, p11 and caveolin-1 as a control.
(C) Effect of siRNA Ax2-2 in lumen formation. MDCK cells were transfected with Ax2-2 (bottom panel), or control (top panel) siRNAs, and plated to form cysts for 48h. Cells were stained for gp135 (red), ZO-1 (green) and nuclei. Arrowheads indicate ZO-1.
(D) Quantitation of cysts with normal lumens in cells transfected with scramble (control) siRNA or siRNA Ax2-1 or Ax2-2. Values are mean ±SD from 4 different experiments. N=100/experiment. *P <0.001.
(E) Ax2CM disrupts the cortical actin cytoskeleton. Cells were infected with a tet-off regulated adenovirus encoding GFP-Ax2CM, and incubated with a low level of dox (1ng/ml) (GFP-Ax2CM). After 48h cells were fixed, stained for actin (blue) and ezrin (red), and analyzed by confocal microscopy. Arrowheads indicate disruption of actin cytoskeleton in GFP-Ax2 cell. Scale bar 10 μm.
(F) Down-regulation of PTEN by siRNA inhibits AP accumulation of Ax2. Cells expressing Ax2-GFP were transfected with siRNAs to PTEN or control siRNAs cysts formed for 48h, fixed and stained for gp135 (red) and nuclei (blue).
Loss of Ax2 inhibits lumen formation
To determine whether Ax2 is required for the formation of the central lumen we used siRNAs. By using 2 different siRNA heteroduplexes we reduced endogenous Ax2 levels >90% (Fig 4b). As shown before, the down-regulation of Ax2 in MDCK cells induced a drastic concomitant reduction of the Ax2 light chain, p11 (Puisieux et al., 1996). We analyzed the effect of depletion of Ax2 on cystogenesis. Cells were transfected with Ax2 siRNAs and plated to form cysts. Whereas 84% of control cysts had normal lumens (Fig 4c), MDCK cysts with reduced Ax2 formed cysts of similar size, but only 47% had normal lumens. Most cysts with depleted Ax2 had multiple small lumens with reduced actin (Fig 4c, S3b, and quantitation in 4d).
We confirmed the requirement of Ax2 in formation of the central lumen using a dominant negative Ax2, AnxCM (Merrifield et al., 2001). Expression of GFP-Ax2CM during cystogenesis inhibited formation of the central lumen (Fig S3c and S3d). Furthermore, when the lumens of the cysts were formed from a mixture of cells with high and low levels of GFP-Ax2CM, the cortical actin cytoskeleton was formed only in the cells with low levels of GFP-Ax2CM (Fig 4e). Also, ezrin and actin underlined the AP surface only of cells with low or no GFP-Ax2CM, whereas they were absent in the presence of GFP-Ax2CM (Fig 4e, arrowheads).
Next, we assessed the effects of delivering exogenous PtdIns(4,5)P2 to the BL PM on Ax2 localization. Exogenous PtdIns(4,5)P2 relocalized Ax2 from the AP to the BL PM (Fig S3e). Finally, we characterized the effect of reducing PTEN on localization of Ax2. Reduction of PTEN inhibited the accumulation of Ax2-GFP at the AP domain, rendering this protein homogenously distributed in the PM (Fig 4f). These results confirm that Ax2 in combination with PtdIns(4,5)P2 is required for the formation of the cortical actin cytoskeleton, AP PM and lumen.
Ax2 associates with Cdc42 at the AP PM
PtdIns(4,5)P2 leads to recruitment and activation of Cdc42 in Xenopus egg extracts (Ho et al., 2004). We hypothesized that Cdc42 might be directly involved in AP PM and lumen formation.
To determine the localization of Cdc42 we stably expressed GFP-Rac1 and GFP-Cdc42, which behave like the endogenous proteins in MDCK cells (Ehrlich et al., 2002). The expression of endogenous Rac1 and Cdc42 were reduced to compensate for expression of Rac1-GFP and Cdc42-GFP, respectively, in these stable lines (Fig 5a). Rac1-GFP was mostly localized to the cell-cell junctions at the BL PM (not shown). However, the localization of Cdc42-GFP was enriched at the AP PM, colocalizing extensively with actin (Fig 5b, arrowheads). To visualize active Cdc42 we used CBD-GFP, which detects activated Cdc42 (Nalbant et al., 2004). CBD binds specifically to Cdc42-GTP via the CRIB domain of WASp (aa 201–321). At day 1 (Fig 5c left) Rac1, Cdc42 and CBD-GFP localized to cell-cell and cell-ECM contacts (Fig 5c; arrows indicate Cdc42 and Rac1 at cell-cell contacts). However, as cysts started to form lumens and thereafter (days 2–5), the majority of CBD-GFP relocalized to the AP PM together with Cdc42, delimiting the newly-formed lumen (Fig 5c, days 2 and 5, middle and right panels, arrowheads indicate CBD-GFP and Cdc42 at the lumen). By contrast, Rac1 remained mostly at cell-cell contacts throughout cystogenesis. To confirm activation of Cdc42 during lumen formation, we pulled down active Rac1 and Cdc42 using the p21-binding domain of PAK 1 fused to GST (Benard et al., 1999). Cdc42-GTP increased significantly during cystogenesis (Fig S4a).
Fig 5. Ax2 binds to Cdc42 at the AP PM.
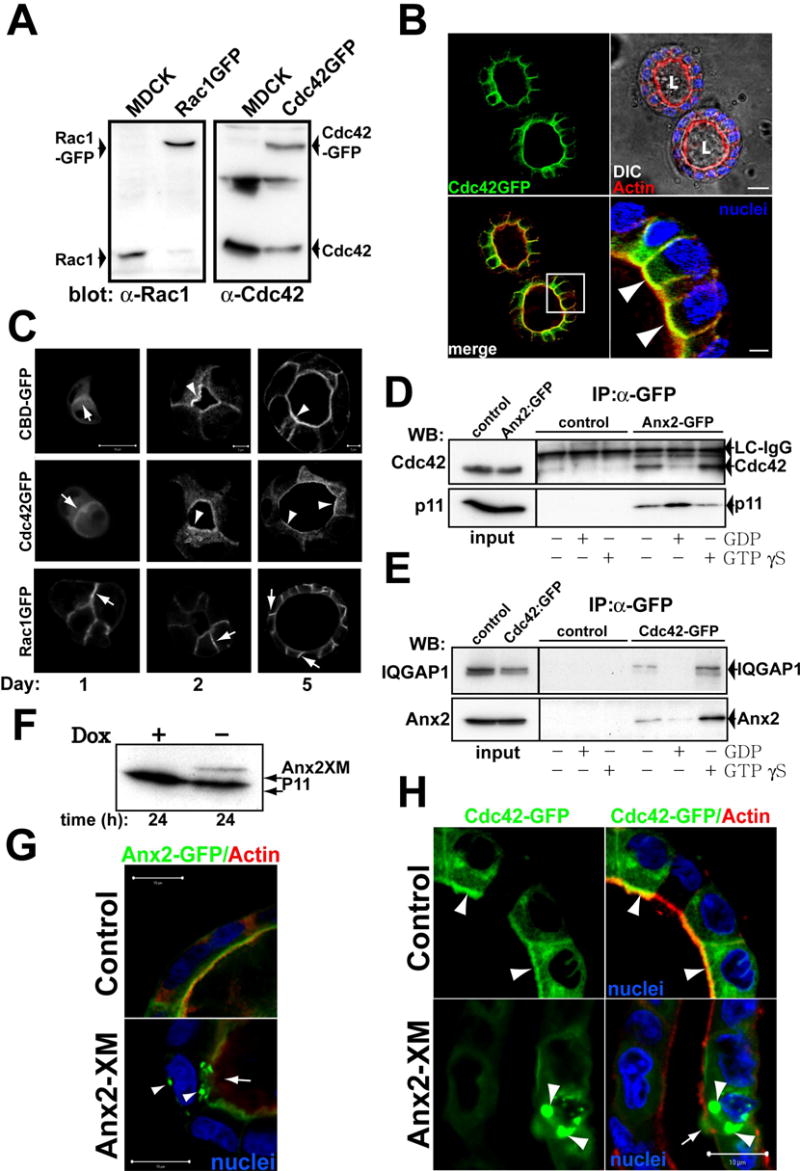
(A) Western blot of stable expression of GFP-Rac1 or GFP-Cdc42. Extracts from MDCK GFP-Cdc42, GFP-Rac1 or control cells were inmunoblotted with anti-Rac1 (left panel) and anti-Cdc42 (right panel) to detect endogenous and transfected proteins.
(B) GFP-Cdc42 distribution in mature cysts. GFP-Cdc42 cells were grown for 5d and stained for nuclei (blue) and actin (red). Bottom-right panel shows the magnification of the indicated region of the merged panel (bottom left). Arrowheads indicate colocalization of GFP-Cdc42 and actin at the AP PM. Scale bar, 10 μm in upper-right panel and 2 μm in the bottom-right panel.
(C) Distribution of CBD-GFP, GFP-Cdc42 and Rac1-GFP during cystogenesis. MDCK CBD-GFP (top row), GFP-Cdc42 (middle row) or GFP-Rac1 (bottom row) cells were grown for 1, 2 or 5d and visualized. Arrows indicate fluorescent proteins at cell-cell or cell-ECM junctions. Arrowheads indicate the localization of CBD-GFP and GFP-Cdc42 at the AP surface. Scale bars are 5 μm or 10 μm as indicated.
(D) Ax2-GFP interacts with endogenous Cdc42 in a GTP-dependent manner. MDCK Ax2-GFP or control cells in mature cysts were lysed and extracts were loaded with GDP or GTPγS. Extracts were immunoprecipitated with antibody against GFP and immunoblotted to analyze Cdc42 and p11. 3% of input or 30% of immunoprecipitated material were loaded on the gel. A band corresponding to the light chain (LC) of IgG was detected with the anti-Cdc42 antibodies and served as a loading control.
(E) Cdc42-GFP interacts with endogenous Ax2 in a GTP dependent manner. Cdc42-GFP or control cells in mature cysts were lysed and the extracts loaded with GDP or GTPγS. Extracts were immunoprecipitated using antibodies against GFP and immunoblotted to analyze Ax2 and IQGAP1. 3% of input and 30% of immunoprecipitated material were loaded in the gel.
(F–H) Effect of Tet-off inducible adenovirus-mediated expression of Ax2X on Ax2-GFP or Cdc42-GFP localization in cysts. MDCK Ax2-GFP (G) or Cdc42-GFP (H) cells formied cysts for 5d were infected with a tet off regulated adenovirus encoding Ax2XM and maintained in the presence (control) or absence (Ax2XM) of 20ng/ml of dox for 16h. Cells were lysed and extracts analyzed by western blot to detect Ax2XM (F). The cysts were and stained for actin (red) and nuclei (blue). Arrowheads indicate aggregates of Ax2-GFP or Cdc42-GFP. Arrows indicate disruption of the AP actin cytoskeleton. Scale bar 10 μm.
Phosphoinositides recruit Rho family GTPases to the PM through pleckstrin homology (PH) domains of GEFs (Ferguson et al., 1995). However, the PH domains of GEFs bind phospholipids with low affinity and little specificity, implying that these interactions are insufficient to specify PM localization of Rho proteins (Snyder et al., 2001). We hypothesized that the high affinity PtdIns(4,5)P2-binding protein Ax2 mediates the interaction between PtdIns(4,5)P2 and Cdc42. Ax2 binds to the active form of another Rho-family GTPase, Rac1 (Hansen et al., 2002). Endogenous Cdc42 co-precipitated with Ax2-GFP (Fig 5d, lane 4 of the IP blot). This interaction was increased in the presence of GTPγS (Fig 5d, lane 6). Ax2-GFP also interacted with endogenous Ax2 (not shown) and P11 (Fig 5d, bottom panel), indicating that this protein forms normal heterotetramers. The association of Ax2-GFP with p11, the ligand of Ax2, was stronger in the presence of GDP, while the co-precipitation of Cdc42 was reduced, suggesting competition of active Cdc42 and p11 for Ax2 binding (Fig 5d, lane 5 of the IP blot). As expected, endogenous Ax2 co-precipitated with Cdc42-GFP and this interaction was stabilized in the presence of GTPγS (Fig 5e, lanes 4 and 6, respectively). We also observed an interaction between Cdc42-GFP and IQGAP1 as described (Hart et al., 1996), which was stabilized by GTPγS, indicating normal behavior of Cdc42-GFP (Fig 5e, bottom).
To confirm the interaction of Cdc42 and Ax2 in vivo, we expressed transdominant Ax2, Ax2XM. Ax2XM is a chimera of the NH2 terminus (residues 1–18) of Ax2 fused to the NH2 terminus of p11. Expression of Ax2XM leads to the formation of cytosolic aggregates containing endogenous Ax2, p11 and the chimera (Harder and Gerke, 1993). Expression of Ax2XM aggregated Ax2-GFP in the cytosol (Fig 5g, arrowheads), and disrupted partially the AP actin cytoskeleton (Fig 5g, arrow) and, importantly, aggregated Cdc42-GFP in the cytosol (Fig 5h, arrowheads). By contrast, Ax2XM did not change the localization of Rac1-GFP (Fig S4b), or the BL protein β-catenin (Fig S4c). Thus Ax2 binds to Cdc42 in vivo and the presence of Ax2 in the AP PM is required for the proper AP localization of Cdc42.
Depletion of Cdc42 inhibits formation of the central lumen
We reduced the level of Cdc42 by 90% using siRNA (Fig 6a). Whereas most of control cysts formed normal lumens (75%), only 17% showed normal lumen formation in cysts with reduced Cdc42 (Fig 6b, quantitation in 6c, and 3D views of cysts in movies S1 and S2). >80% of cysts with reduced Cdc42 had multiple small lumens with decreased F-actin and the cortical actin-associated protein (Fig 6b), resembling the phenotype observed with reduction of PTEN or Ax2 (Figs. 2, 4). β-catenin remained at cell-cell contacts, indicating that the BL surface is relatively normal (Fig 6b, left). Thus functional disruption of Cdc42 induces accumulation of AP markers in lumens between cells and probably in large intracellular vesicles (Fig 6b, left panels). By contrast, reduction of Cdc42 did not affect polarity of MDCK cells in 2D monolayers (Fig S5a).
Fig 6. Cdc42 siRNA inhibits formation of central lumen.
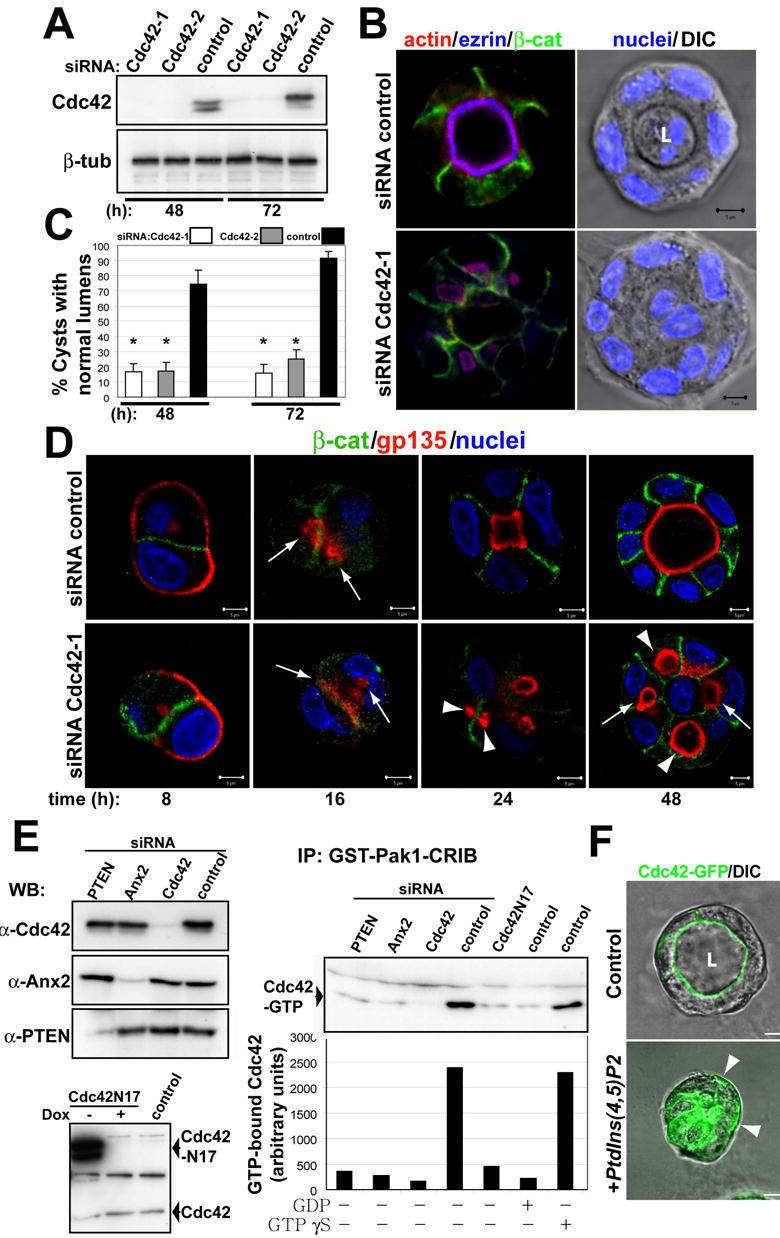
(A) Down-regulation of Cdc42 by siRNA. Cells were transfected with siRNAs Cdc42-1 and Cdc42-2 against canine Cdc42 or with control siRNA, allowed to form cysts for 48 or 72h, and then total cell lysates western blotted for Cdc42 and β-tubulin (control).
(B) Effect of Cdc42-1 siRNA on lumen formation. MDCK cells were transfected with Cdc42 siRNA (bottom panels) or siRNA control (top panels) and plated to form cysts for 48h. Cells were stained to detect actin (red), ezrin (blue), β-catenin (green, left panels) and nuclei (right panel merged with DIC). Single confocal sections through the middle of cysts are shown
(C) Quantitation of cysts with normal lumens in cells transfected with control siRNA (black bars), specific siRNA Cdc42-1 (white bars) or Cdc42-2 (grey bars). Values shown are mean ±SD from 4 different experiments. *P<0.001.
(D) Time course analysis of gp135 localization during lumen formation in control and Cdc42 depleted cells. Cells were transfected with Cdc42 siRNA-1 (bottom panels) or siRNA control (top panels) and plated to form cysts for 8, 16, 24 and 48h. Cells were stained to detect gp135 (red), β-catenin (green) and nuclei. Arrows indicate small intracellular vesicles at 16h and intercellular lumens at 48h. Arrowheads indicate intracellular lumens.
(E) siRNA-mediated reduction of PTEN and Ax2 inhibits activation of Cdc42. Cells were transfected with siRNAs Cdc42-1, Ax2-2, or PTEN2 or with control siRNAs; or infected with adenovirus expressing myc-tagged N17Cdc42. Total cell lysates of cysts at 48h were immunoblotted for Cdc42, Ax2 and PTEN, (top-left panel); or for Cdc42 (bottom-left panel). Extracts from these cells were pulled down with PBD-PAK1-GST. Total and GTP–bound Cdc42 was detected by immunoblotting with specific antibodies, and the ratios of GTP-bound to total protein quantified. Values shown are mean ±SD from 3 different experiments.
(F) Exogenous PtdIns(4,5)P2 delocalizes Cdc42-GFP from the AP PM. Cdc42-GFP cysts at 48h were incubated with PtdIns(4,5)P2-histone complexes for 30min (+PtdIns(4,5)p2) or histone alone (control), fixed and analyzed by confocal microscopy. Arrowheads indicate Cdc42-GFP enrichment at the basal PM.
To understand the role of Cdc42 in the formation of the AP PM and the lumen, we analyzed the effect of reducing Cdc42 on the localization of the AP marker gp135 during cystogenesis. Gp135 localized initially to external PM (Fig 6d, left panels), confirming the described distribution of this protein to free surfaces in non-polarized epithelial cells (Meder et al., 2005). Then, Gp135 relocalized to internal vesicles presumably by endocytosis. Cdc42 disruption did not affect this relocalization (Fig 6d, 16h panels, arrows). However, whereas in control cells these internal vesicles fused with the PM to form lumens between cells, in the cysts with reduced Cdc42, Gp135 accumulated in intracellular vesicles that did not fuse with the PM (Fig 6d, 24h panels, arrowheads). Since we reduced but did not eliminate Cdc42, eventually some of these vesicles fused with the PM, generating mature cysts with intracellular and small intercellular lumens (Fig 6d, right panels, arrows indicate intercellular and arrowheads indicate intracellular lumens). Another possibility for formation of the central lumen is that a single lumen forms by coalescence of multiple intercellular lumens. However, we observed that most cysts with single lumens formed without a preceding multiluminal intermediate (Fig 6d, upper panels, quantitation in Fig S5b). TJ were apparently unaffected, as indicated by the staining for ZO-1 (Fig S5c, arrowheads).
Exogenous PtdIns(4,5)P2 targets activated Cdc42 to the BL PM and reduces the central lumen
Our data suggest a model for lumen formation that is initiated by the PTEN-dependent enrichment of PtdIns(4,5)P2 at the AP pole of MDCK cells. PtdIns(4,5)P2 recruits Ax2, which in turn recruits Cdc42. Cdc42 is essential for formation of the AP PM and lumen.
To test this we analyzed the localization and activity of Cdc42 in cells depleted of PTEN or Ax2. We observed a drastic reduction of Cdc42 activity in the cysts with reduced levels of PTEN or Ax2, as measured by GST-Pak1-Crib pull-down (Fig 6e). Activated Cdc42 was reduced in cysts with reduced Cdc42 or expressing the dominant negative form of Cdc42, N17-Cdc42 (Fig 6e). Despite the effect on the localization of actin, gp135, ezrin and ZO-1 upon depletion of Cdc42, Ax2 or PTEN, we did not see alterations in total protein levels of these markers (Fig S6a). Confirming the biochemical results, we also observed a relocalization of Cdc42 from the plasma PM to the cytoplasm upon knock down of either PTEN or Ax2 (Fig S6b, and Fig S6c).
We next assessed the effects of delivering exogenous PtdIns(4,5)P2 to the BL PM on the localization and activity of Cdc42. Exogenous PtdIns(4,5)P2 induced the relocalization of Cdc42-GFP (Fig 6f, arrowheads) and CBD-GFP (Fig S6d) from the AP domain to the cytoplasm with enrichment at the basal portion of the cyst. Thus, activated Cdc42 followed a pattern of redistribution similar to that observed for PHD-GFP, Ax2, gp135, ezrin and ZO-1. The partial translocation of activated Cdc42 to the basal PM was associated with the formation of a partial abnormal actin cytoskeleton at the periphery of the cyst (Fig S6d, arrowheads). In sum these data show that PtdIns(4,5)P2-mediated AP enrichment of Ax2 targets activated Cdc42 to regulate the formation of the AP PM and lumen during epithelial morphogenesis.
Cdc42 targets aPKC/Par6 to the AP PM to form the lumen
The Rho-GTPase effector protein Par6 links Par3 and aPKC to activated Cdc42, forming a multimeric polarity-regulatory complex at the PM of MDCK cells (Joberty et al., 2000). We addressed the role of the Par3/Par6/aPKC complex in the formation of the AP PM and lumen in MDCK cysts. First we characterized the localization of members of this complex. Interestingly, whereas aPKC (Fig 7a, upper panels) and Par6 (not shown) distributed along the whole AP PM, Par3 was confined to the tight junctions (Fig 7a, bottom panels). This differential distribution of Par3 and Par6/aPKC was previously found in Drosophila (Harris and Peifer, 2005), and suggests that Cdc42/Par6/aPKC could have a specific role in the morphogenesis of the AP PM independent of Par3. Since activated Cdc42 is enriched at the AP PM, these data also suggest that the activity of Par6/aPKC at the AP PM is dependent on Cdc42.
Fig 7. Cdc42 targets aPKC to the AP plasma PM to form the central lumen.
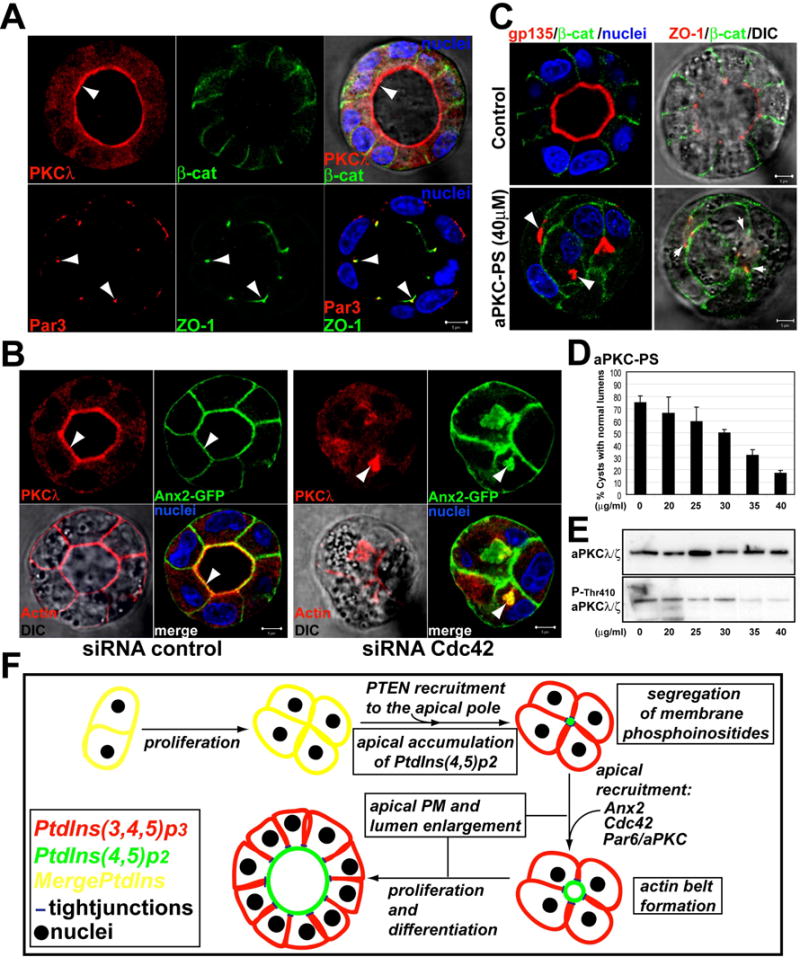
(A) aPKC and Par3 distribute differently in nature cysts. Cysts were stained for aPKCλ (top panels) or Par3 (bottom panels) (red, left panels), β-catenin or ZO-1 (green, middle panels), and for nuclei (merged with DIC, right panels). Arrowheads indicate aPKC at the AP PM, and colocalization of Par3 and ZO-1 at TJ.
(B) Reduction of Cdc42 induces intracellular accumulation of aPKC and Ax2. Ax2-GFP cells were transfected with Cdc42 or control siRNAs and plated to form cysts for 48h. Cells were stained for aPKC (red) and merged with Ax2-GFP and nuclei (bottom-right panels); and actin (red, left-bottom panel merged with DIC). Arrowheads indicate aPKC at the AP PM in controls cells (left panels), or to the intracellular vesicles in Cdc42 depleted cells (right panels).
(C) aPKC-PS disrupts lumen formation. MDCK cysts were treated with aPKC-PS (40μM) or not (control). Cells were stained to detect gp135 (red), β-catenin (green) and nuclei in left panels; or ZO-1 (red), β-catenin (green) and merged with DIC in right panels.
(D) The aPKC inhibitor aPKC-PS disrupts lumen formation in a dose-dependent manner. MDCK cysts were treated with indicated concentrations of aPKC-PS for 48h. Cells were then fixed, stained and quantified for lumen formation. Values shown are mean ±SD from 3 different experiments. N=100/experiment.
(E) aPKC-PS inhibits aPKC phosphorylation in a dose-dependent manner.
(F) Model: PtdIns(3,4,5)p3 (red) and PtdIns(4,5)p2 (green) colocalize in unpolarized MDCK cells (yellow). AP recruitment of PTEN induces the accumulation of PtdIns(4,5)p2 at the AP domain. PtdIns(4,5)p2 recruits Ax2, Cdc42 and Par6/aPKC to form the AP PM and lumen.
To test if the localization of aPKC at the AP PM depends on Cdc42, we characterized the distribution of aPKC in cells depleted of Cdc42. In cysts depleted of Cdc42 aPKC localized intracellularly and partially colocalized with Ax2-GFP on intracellular lumens (Fig 7b arrowhead). The localization of aPKC to the AP PM also depends on PTEN and Ax2, since the reduction of these proteins caused intracellular accumulation of aPKC (Fig S7a and S7b). Interestingly, we also found a partial intracellular accumulation of Ax2 in the cells with reduced Cdc42 (Fig 7b). By contrast, PtdIns(4,5)p2 remained associated with the plasma PM in the cells with reduced Cdc42 (not shown). These data suggest that whereas the enrichment of PtdIns(4,5)p2 at the AP PM might control this whole pathway, there is a potential positive feedback loop between Cdc42 and Ax2 to promote the localization of AP proteins at the lumen of mature cysts.
To address the role of the Par6/aPKC in AP PM and lumen formation, we used a myristoylated pseudosubstrate of aPKC-ζ (aPKC-PS) that specifically inhibits all aPKCs (Nunbhakdi-Craig et al., 2002). Treatment with aPKC-PS inhibited phosphorylation of aPKC, as well as formation of the AP PM and central lumen in a dose dependent manner (Fig 7c–e). In contrast, we saw a normal distribution of ZO-1, as previously described in MDCK monolayers (Fan et al., 2004). Finally, we found that exogenous PtdIns(4,5)P2 relocalized aPKC from the AP to the basal PM (Fig S7c, arrowheads). Thus, aPKC followed a pattern of redistribution similar to that of PHD-GFP, Ax2, activated Cdc42, gp135, ezrin and ZO-1.
In sum these data show that PTEN, PtdIns(4,5)P2, Ax2 and activated Cdc42 are required for the targeting and localization of Par6/aPKC to the AP domain to regulate the formation of the AP PM and lumen during epithelial morphogenesis.
Discussion
Formation of the AP surface and lumen is a central problem in understanding how epithelial tissues arrange themselves into tubes and other hollow structures, such as cysts. We have uncovered a molecular mechanism of AP surface and lumen formation (model in Fig 7f). PTEN is needed for segregation of PtdIns(4,5)p2 to the AP PM and PtdIns(3,4,5)p3 to the BL PM. AP PtdIns(4,5)p2 recruits Ax2, which in turn recruits Cdc42 to the AP PM, causing the organization of the subapical actin cytoskeleton and formation of the AP surface and lumen. Cdc42 binds and localizes the Par6/aPKC complex to the AP PM to promote establishment of polarity.
PtdIns(4,5)p2 is thus a key determinant of the AP surface. Similarly PtdIns(3,4,5)p3 is a key determinant of the BL surface (Gassama-Diagne et al., 2006). Together PtdIns(4,5)p2 and PtdIns(3,4,5)p3 play complementary roles in epithelial polarity. More generally, phosphoinositides have emerged as general determinants of membrane identity (Di Paolo and De Camilli, 2006). An advantage of epithelia is that we can insert exogenous phosphoinositides into limited ectopic locations. These gain-of-function experiments provide a direct test of the role of the lipid in specifying domain identity.
To exert its effects PtdIns(4,5)p2 interacts with Ax2, which clusters this lipid with high affinity and specificity. Ectopic PtdIns(4,5)p2 in the BL surface recruits Ax2 to the BL plasma PM. Although loss of Ax2 by RNAi prevents lumen formation, RNAi of Ax2 did not produce as strong a phenotype as expression of the DN Ax2-CM or RNAi of PTEN or Cdc42. One explanation could be the existence of ~20 annexin family members, which might have redundancy with each other. Indeed, Ax2KO mice are viable (Ling et al., 2004). Alternatively, even low levels of Ax2 might suffice to exert its function.
Cdc42 interacts with Ax2 in a GTP-dependent manner. Cdc42 is activated during cystogenesis. Most activated Cdc42 re-localizes from cell-cell contacts to the AP pole as lumens form. RNAi of Cdc42 caused malformation of the central lumen in cysts, but did not affect polarization of MDCK cells in 2D monolayers. This effect of Cdc42 depletion in cysts highlights the importance of using 3D models in analysis of lumen formation. Interestingly, we also saw an intracellular accumulation of Ax2 in the cells with reduced Cdc42 suggesting a potential positive feedback loop whereby Cdc42 and Ax2 each promote the localization of the other at the lumen of mature cysts. Ax2 might work by recruiting Cdc42 or a GEF for Cdc42, and this GEF may in turn activate Cdc42 at this location. On the other hand, active Cdc42 might promote the exocytosis of Ax2 and other AP proteins.
Formation of the AP surface and lumen has been suggested to be mediated by exocytosis of large intracellular vacuoles, termed vacuolar apical compartment (VAC) (Vega-Salas et al., 1987). Formation of endothelial lumens occurs by vacuolar exocytosis (Kamei et al., 2006). Cdc42 is needed for the exocytosis of secretory vesicles from neuroendocrine cells, apparently via rearrangement of the actin cytoskeleton and this may be analogous to the fusion of VACs or smaller vesicles with the AP surface (Malacombe et al., 2006). We saw accumulation of apparent VACs when Cdc42 was depleted. Similarly, DN Cdc42 blocks capillary lumen formation (Bayless and Davis, 2002). Perhaps during normal MDCK cyst lumen formation smaller vesicles are rapidly exocytosed to form the lumen. Inhibition of this by Cdc42 depletion may cause the accumulation of larger, more easily detected VACs. Indeed, this may be the defect underlying the phenotypes we observed with loss of function of PTEN, Ax2, Cdc42 or aPKC. Cdc42 is also needed for exit of AP and BL proteins from the TGN (Musch et al., 2001), so Cdc42 may act at multiple levels in the formation of the AP surface.
Localized active Cdc42 may promote formation of the AP surface and lumen by additional mechanisms. Active Cdc42 binds to Par6, a member of the Par3/Par6/aPKC complex that regulates TJ and polarity formation (Munro, 2006)). In Drosophila, Par6/aPKC function at the AP PM independently of Par3, which is associated with the junctional complex. Indeed, we have found that Par6/aPKC localizes at the AP PM of MDCK cysts independently of Par3, and that the disruption of aPKC function inhibits normal lumen formation. Mutation of zebrafish aPKCλ causes defects in lumen formation in the intestine (Horne-Badovinac et al., 2001). These data suggest the existence of two distinct Par complexes for the establishment of epithelial polarity; a complex of Par6/aPKC localized to the AP PM and involved in the formation of this domain; and a complex that also includes Par3, localized at the TJ and required for their formation.
We previously reported that inhibition of Rac1 or β1 integrin in cysts leads to inversion of polarity orientation and abnormal organization of laminin (O’Brien et al., 2001; Yu et al., 2005). Rac1, β1 integrin and laminin may be part of a pathway that determines orientation of the axis of polarity, while PTEN, PtdIns(4,5)p2, Ax2, Cdc42 and aPKC/Par6 are part of a pathway that controls formation of the AP surface and lumen. The Rac1/β1 integrin/laminin pathway might be upstream and/or parallel to the PTEN/PtdIns(4,5)p2/Ax2/Cdc42 pathway, and determines the location of the AP surface. Since activation of Rac1 at the primordial adhesions of epithelial cells controls the association and activation of the Par3-Par6-aPKC complex to induce TJ biogenesis and cell polarity (Mertens et al., 2005), one potential connection between these pathways might be the targeting of PTEN to the AP domain through its interaction with TJs. PTEN localizes to the adherens junctions in fly epithelium through its interaction with Bazooka/Par3 (von Stein et al., 2005). Here we show that PTEN is needed for AP PM and lumen formation during cyst development and that Par3 localizes specifically to the TJs in MDCK cyst. This observation is consistent with previous studies showing that the expression of dominant negative Par-3 cells disrupted MDCK cyst morphogenesis, causing the lack of a central lumen (Hurd et al., 2003).
Experimental Procedures
Reagents
1o antibodies were mouse (unless otherwise specified) against: β˜ tubulin (Chemicon); β-catenin (rabbit), myc (Sigma); GFP (Roche); anti-Rac1, Cdc42, Ax2, p11, anti-IQGAP1, Ezrin, GAPDH, aPKCλ and caveolin1 (rabbit) (BD Transduction Lab); PTEN (Cell Signaling); ASIP/Par3 (rabbit, Zymed); Phospho-Thr410-aPKC (rabbit, Santa Cruz); p58 (gift of K Matlin); ZO-1 (rat R40.76; gift from B Stevenson); gp135 (gift of George Ojakian). 2o antibodies used were highly cross-absorbed anti-mouse AlexaFluor 546 and anti-rabbit AlexaFluor 488 (Molecular Probes,), goat anti-rabbit or anti-mouse HRP (Jackson). Actin filaments were stained with AlexaFluor 488, 546 or 633 phalloidin (Molecular Probes). Nuclei were stained with Hoescht (Molecular Probes). C-Zeta Pseudosubstrate, myristoylated (aPKC-PS) inhibits aPKC (Biosource).
Cells
MDCK cells were grown as described (O’Brien et al., 2001; Yu et al., 2005). MDCK stably expressing Cdc42-GFP, Rac1-GFP, Ax2-GFP, PHD-GFP and PH-Akt-GFP (construct from T Balla, NIH), PH-Akt-dsRed (construct from H Bourne, UCSF), GFP-PTEN and CBD-GFP (from K. Hahn, U North Carolina) were made by cotransfection with blasticidin resistant gene. After 3 weeks in selective medium clones were isolated and analyzed by IF and WB.
To prepare cysts in Matrigel, cells were trypsinzed to a single cell suspension of 4 x 104 cells/ml in 2% Matrigel. 250 μl of cells in Matrigel were plated in 8 well coverglass chambers (Nalge Nunc) covered with Matrigel. Cells were fed every 2d and grown for 2-5 d until cysts with lumen formed.
Adenovirus
Preparation of recombinant adenoviruses encoding GFP-Ax2, GFP-Ax2CM, Ax2XM were obtained sub-cloning into pADtet, downstream of tetracycline-responsive elements. Construction of adenoviruses through CRE lox recombination and viruses were described (Altschuler et al., 1998). For infection, cysts were washed well with PBS+, and incubated for 10–15 min in trypsin (Ca2+ and Mg2+) at 37°C to disrupt Matrigel. After inhibiting trypsin with medium + serum, Matrigel-free cysts were infected with the adenovirus following the protocol described before for cells in monolayer.
Microscopy
Immunofluorescence of cysts was previously described (O’Brien et al., 2001; Yu et al., 2005). Cysts were analyzed on a Zeiss 510 LSM. Cysts with actin/gp135 staining at the interior surface and β-catenin facing the ECM were identified as normal lumens (interior AP pole). Cysts that had actin/gp135 either absent, in small multiple lumens or at the periphery were considered as abnormal lumens. Per condition >100 cysts/experiment were analyzed, SD calculated and statistical significance was by paired Student’s t- test.
Biochemistry
Preparation of lysates, immunoblotting and CoIP in cysts were described before (O’Brien et al., 2001; Yu et al., 2005), for more information see supplementary data.
GTPase Activation
1–4 day old cysts were lysed in 500 μl of 4oC 2x Gold lysis buffer (2% Triton X-100, 40 mM Tris-HCl, pH 7.5, 1000 mM NaCl, 20 mM MgCl2, 30% glycerol, 1 mM dithiothreitol, and EDTA-free protease inhibitors; Roche); then microcentrifuged at 15,000 rpm for 5 min. A 50-μl sample from the supernatant was set aside for determination of Cdc42 and Rac levels in the total lysate, and GTP loading on Cdc42 and Rac1 was determined with GST-Pak3-CRIB bead pulldown as described (Hansen and Nelson, 2001).
RNAi
25-nt siRNA duplexes targeting mRNA sequences of canine Cdc42, Ax2, and PTEN, were purchased from Invitrogen (details in supplemental experimental procedures). Sequences were submitted to BLAST search to ensure targeting specificity. The specificity of PTEN, Cdc42 and Ax2 down-regulations was further checked by Western blotting analysis. MDCK cells plated at low density (3 x 104/cm2) were transfected with 20 nM of either PTEN, Ax2 or Cdc42 siRNA duplex, or scrambled siRNA using Lipofectime 2000TM (Invitrogen) and incubated for 24h after transfection in the same plates. Then the cells were plated to form cyst for 2–5 days as described above confocal and western blot analysis.
PtdIns(4,5)P2 basal delivery
Basal delivery of PtdIns(4,5)P2 in cysts in matrigel was previously described (Gassama-Diagne et al., 2006). For more details see supplemental experimental procedures.
Supplementary Material
Acknowledgments
We thank H Bourne, CM Ruiz-Jarabo, MA Alonso and P Leroy for comments on the manuscript, and members of the Mostov lab for discussion. Supported by HFSP fellowship LT00426/2004-C to FM-B, Natl. Kidney Assoc to WY, and NIH grants to KM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschuler Y, Barbas SM, Terlecky LJ, Tang K, Hardy S, Mostov KE, Schmid SL. Redundant and distinct functions for dynamin-1 and dynamin-2 isoforms. J Cell Biol. 1998;143:1871–1881. doi: 10.1083/jcb.143.7.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayless KJ, Davis GE. The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J Cell Sci. 2002;115:1123–1136. doi: 10.1242/jcs.115.6.1123. [DOI] [PubMed] [Google Scholar]
- Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Ehrlich JS, Hansen MD, Nelson WJ. Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev Cell. 2002;3:259–270. doi: 10.1016/s1534-5807(02)00216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S, Hurd TW, Liu CJ, Straight SW, Weimbs T, Hurd EA, Domino SE, Margolis B. Polarity proteins control ciliogenesis via kinesin motor interactions. Curr Biol. 2004;14:1451–1461. doi: 10.1016/j.cub.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell. 1995;83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- Gassama-Diagne A, Yu W, ter Beest M, Martin-Belmonte F, Kierbel A, Engel J, Mostov K. Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat Cell Biol. 2006;8:963–970. doi: 10.1038/ncb1461. [DOI] [PubMed] [Google Scholar]
- Hansen MD, Ehrlich JS, Nelson WJ. Molecular mechanism for orienting membrane and actin dynamics to nascent cell-cell contacts in epithelial cells. J Biol Chem. 2002;277:45371–45376. doi: 10.1074/jbc.M207747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MD, Nelson WJ. Serum-activated assembly and membrane translocation of an endogenous Rac1:effector complex. Curr Biol. 2001;11:356–360. doi: 10.1016/s0960-9822(01)00091-4. [DOI] [PubMed] [Google Scholar]
- Harder T, Gerke V. The subcellular distribution of early endosomes is affected by the annexin II2p11(2) complex. J Cell Biol. 1993;123:1119–1132. doi: 10.1083/jcb.123.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TJ, Peifer M. The positioning and segregation of apical cues during epithelial polarity establishment in Drosophila. J Cell Biol. 2005;170:813–823. doi: 10.1083/jcb.200505127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, Callow MG, Souza B, Polakis P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. Embo J. 1996;15:2997–3005. [PMC free article] [PubMed] [Google Scholar]
- Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Horne-Badovinac S, Lin D, Waldron S, Schwarz M, Mbamalu G, Pawson T, Jan Y, Stainier DY, Abdelilah-Seyfried S. Positional cloning of heart and soul reveals multiple roles for PKC lambda in zebrafish organogenesis. Curr Biol. 2001;11:1492–1502. doi: 10.1016/s0960-9822(01)00458-4. [DOI] [PubMed] [Google Scholar]
- Hurd TW, Fan S, Liu CJ, Kweon HK, Hakansson K, Margolis B. Phosphorylation-dependent binding of 14-3-3 to the polarity protein Par3 regulates cell polarity in mammalian epithelia. Curr Biol. 2003;13:2082–2090. doi: 10.1016/j.cub.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- Kamei M, Saunders WB, Bayless KJ, Dye L, Davis GE, Weinstein BM. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442:453–456. doi: 10.1038/nature04923. [DOI] [PubMed] [Google Scholar]
- Kawakatsu T, Shimizu K, Honda T, Fukuhara T, Hoshino T, Takai Y. Trans-interactions of nectins induce formation of filopodia and Lamellipodia through the respective activation of Cdc42 and Rac small G proteins. J Biol Chem. 2002;277:50749–50755. doi: 10.1074/jbc.M209846200. [DOI] [PubMed] [Google Scholar]
- Kazmierczak BI, Mostov K, Engel JN. Epithelial cell polarity alters Rho-GTPase responses to Pseudomonas aeruginosa. Mol Biol Cell. 2004;15:411–419. doi: 10.1091/mbc.E03-08-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschewski R, Hall A, Mellman I. Cdc42 controls secretory and endocytic transport to the basolateral plasma membrane of MDCK cells. Nat Cell Biol. 1999;1:8–13. doi: 10.1038/8977. [DOI] [PubMed] [Google Scholar]
- Lacalle RA, Gomez-Mouton C, Barber DF, Jimenez-Baranda S, Mira E, Martinez AC, Carrera AC, Manes S. PTEN regulates motility but not directionality during leukocyte chemotaxis. J Cell Sci. 2004;117:6207–6215. doi: 10.1242/jcs.01545. [DOI] [PubMed] [Google Scholar]
- Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, Silverstein RL, Hempstead B, Mark WH, Hajjar KA. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest. 2004;113:38–48. doi: 10.1172/JCI200419684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Malacombe M, Ceridono M, Calco V, Chasserot-Golaz S, McPherson PS, Bader MF, Gasman S. Intersectin-1L nucleotide exchange factor regulates secretory granule exocytosis by activating Cdc42. Embo J. 2006;25:3494–3503. doi: 10.1038/sj.emboj.7601247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meder D, Shevchenko A, Simons K, Fullekrug J. Gp135/podocalyxin and NHERF-2 participate in the formation of a preapical domain during polarization of MDCK cells. J Cell Biol. 2005;168:303–313. doi: 10.1083/jcb.200407072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Rescher U, Almers W, Proust J, Gerke V, Sechi AS, Moss SE. Annexin 2 has an essential role in actin-based macropinocytic rocketing. Curr Biol. 2001;11:1136–1141. doi: 10.1016/s0960-9822(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Mertens AE, Rygiel TP, Olivo C, van der Kammen R, Collard JG. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J Cell Biol. 2005;170:1029–1037. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R, Schaller G, Orci L. Induction of epithelial tubular morphogenesis in vitro by fibroblast-derived soluble factors. Cell. 1991;66:697–711. doi: 10.1016/0092-8674(91)90115-f. [DOI] [PubMed] [Google Scholar]
- Munro EM. PAR proteins and the cytoskeleton: a marriage of equals. Curr Opin Cell Biol. 2006;18:86–94. doi: 10.1016/j.ceb.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Musch A, Cohen D, Kreitzer G, Rodriguez-Boulan E. cdc42 regulates the exit of apical and basolateral proteins from the trans-Golgi network. Embo J. 2001;20:2171–2179. doi: 10.1093/emboj/20.9.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM. Activation of endogenous Cdc42 visualized in living cells. Science. 2004;305:1615–1619. doi: 10.1126/science.1100367. [DOI] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, 3rd, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–978. doi: 10.1083/jcb.200206114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien LE, Jou TS, Pollack AL, Zhang Q, Hansen SH, Yurchenco P, Mostov KE. Rac1 orientates epithelial apical polarity through effects on basolateral laminin assembly. Nat Cell Biol. 2001;3:831–838. doi: 10.1038/ncb0901-831. [DOI] [PubMed] [Google Scholar]
- O’Brien LE, Zegers MM, Mostov KE. Opinion: Building epithelial architecture: insights from three-dimensional culture models. Nat Rev Mol Cell Biol. 2002;3:531–537. doi: 10.1038/nrm859. [DOI] [PubMed] [Google Scholar]
- Puisieux A, Ji J, Ozturk M. Annexin II up-regulates cellular levels of p11 protein by a post-translational mechanisms. Biochem J . 1996;313(Pt 1):51–55. doi: 10.1042/bj3130051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescher U, Ruhe D, Ludwig C, Zobiack N, Gerke V. Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes. J Cell Sci. 2004;117:3473–3480. doi: 10.1242/jcs.01208. [DOI] [PubMed] [Google Scholar]
- Schmid AC, Byrne RD, Vilar R, Woscholski R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004;566:35–38. doi: 10.1016/j.febslet.2004.03.102. [DOI] [PubMed] [Google Scholar]
- Snyder JT, Rossman KL, Baumeister MA, Pruitt WM, Siderovski DP, Der CJ, Lemmon MA, Sondek J. Quantitative analysis of the effect of phosphoinositide interactions on the function of Dbl family proteins. J Biol Chem. 2001;276:45868–45875. doi: 10.1074/jbc.M106731200. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Buechner M, Nishiwaki K, Hall DH, Nakanishi H, Takai Y, Hisamoto N, Matsumoto K. A putative GDP-GTP exchange factor is required for development of the excretory cell in Caenorhabditis elegans. EMBO Rep. 2001;2:530–535. doi: 10.1093/embo-reports/kve110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Salas DE, Salas PJ, Rodriguez-Boulan E. Modulation of the expression of an apical plasma membrane protein of Madin-Darby canine kidney epithelial cells: cell-cell interactions control the appearance of a novel intracellular storage compartment. J Cell Biol. 1987;104:1249–1259. doi: 10.1083/jcb.104.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Stein W, Ramrath A, Grimm A, Muller-Borg M, Wodarz A. Direct association of Bazooka/PAR-3 with the lipid phosphatase PTEN reveals a link between the PAR/aPKC complex and phosphoinositide signaling. Development. 2005;132:1675–1686. doi: 10.1242/dev.01720. [DOI] [PubMed] [Google Scholar]
- Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Datta A, Leroy P, O’Brien LE, Mak G, Jou TS, Matlin KS, Mostov KE, Zegers MM. Beta1-integrin orients epithelial polarity via Rac1 and laminin. Mol Biol Cell. 2005;16:433–445. doi: 10.1091/mbc.E04-05-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, O’Brien LE, Wang F, Bourne H, Mostov KE, Zegers MM. Hepatocyte growth factor switches orientation of polarity and mode of movement during morphogenesis of multicellular epithelial structures. Mol Biol Cell. 2003;14:748–763. doi: 10.1091/mbc.E02-06-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.